

Abstract
Objective
To describe the clinico-radiological phenotype of 3 patients harboring a homozygous novel AP4M1 pathogenic mutation.
Methods
The 3 patients from an inbred family who exhibited early-onset developmental delay, tetraparesis, juvenile motor function deterioration, and intellectual deficiency were investigated by magnetic brain imaging using T1-weighted, T2-weighted, T2*-weighted, fluid-attenuated inversion recovery, susceptibility weighted imaging (SWI) sequences. Whole-exome sequencing was performed on the 3 patients.
Results
In the 3 patients, brain imaging identified the same pattern of bilateral SWI hyposignal of the globus pallidus, concordant with iron accumulation. A novel homozygous nonsense mutation was identified in AP4M1, segregating with the disease and leading to truncation of half of the adap domain of the protein.
Conclusions
Our results suggest that AP4M1 represents a new candidate gene that should be considered in the neurodegeneration with brain iron accumulation (NBIA) spectrum of disorders and highlight the intersections between hereditary spastic paraplegia and NBIA clinical presentations.
Hereditary spastic paraplegias (HSPs) are a heterogeneous group of neurodegenerative diseases clinically characterized by progressive lower extremity weakness and spasticity, which may be isolated (pure HSP) or combined with other neurologic or nonneurological signs (complex HSP).1,2 More than 70 genes have been implicated, emphasizing diverse molecular pathogenic mechanisms.3 In this respect, recessive mutations in genes encoding the different subunits of adaptor protein complex-4, (AP4B1, AP4M1, AP4E1, and AP4S1) have been identified in patients with complex HSP (SPG 47, 50, 51, and 52 respectively).4–8 The AP4-deficiency syndrome is characterized by progressive spasticity, microcephaly, intellectual deficiency, dysmorphic traits, and growth retardation,4–8 while epilepsy and peripheral neuropathy might be associated.4,9 Brain imaging phenotypes reported up to now are characterized by cerebral atrophy, asymmetric enlargement of lateral ventricles, white matter loss, and thin corpus callosum splenium.8–10 Thin and globoid hippocampal cortex9 and tortuosity of intraextracranial large vessels were also reported.4
Neurodegeneration with brain iron accumulation (NBIA), which is characterized by dystonia, parkinsonism, spasticity, and brain iron accumulation on MRI, represents another inherited group of neurodegenerative disorders, due to mutations in 10 genes, with molecular overlaps with HSP.11,12
Here, we report 3 patients from the same kindred who harbor a homozygous AP4M1 mutation. They exhibit the typical clinico-radiological phenotype of AP4-deficiency syndrome, but surprisingly associated with bilateral pallidal iron accumulation on brain imaging, thus establishing a link between AP4-related complex HSP and NBIA disorders.
Methods
Standard protocol approvals, registrations, and patient consents
The study was conducted in accordance with the Declaration of Helsinki and was approved by the local ethical committee. Written informed consent was obtained from the patients' legal representatives.
Whole-exome sequencing and brain imaging
Whole-exome sequencing (WES) was performed on the DNA from the 3 affected patients by Aros Ltd. Homozygous mutations common to the 3 patients were filtered progressively for their frequency (<1%), alteration of the open reading frame (frameshift, splicing, missense, and nonsense mutations), and ultimately for their localization in the homozygous regions common to the 3 patients. Sanger sequencing allowed for their confirmation and segregation study in the family.
CT was performed on a 64-section CT scanner (Discovery750 HD; GE Health care, Milwaukee, WI). MRIs were acquired on a 1.5-T system (AVENTO; Siemens medical solutions, Erlangen, Germany) as follows: axial slices T2-weighted, T2*-weighted, fluid-attenuated inversion recovery, susceptibility weighted imaging (SWI) sequences, and sagittal slices T1-weighted sequences.
Results
Clinical data
The clinical features of the 3 patients originating from a large consanguineous Moroccan family (figure 1A) are described in table. Psychomotor retardation with spasticity of the 4 limbs was noticed early in life. Clinical examination from the first year showed spastic tetraplegia, with pyramidal tract signs and equinovarus. Patients IV-2 and IV-5 sat unaided at 7 months; patient IV-2 was able to crawl at 2 years but never managed to walk; her sister IV-5 could walk short distances with unsteady spastic gait from the age of 5 years. Patient IV-7 sat unaided at 11 months of age and walked at 3 years, with a broad-based unsteady gait. The patients exhibited stable severe mental deficiency, without behavioral disturbance. Motor achievements progressively deteriorated at adolescence, with loss of the highest motor skills, but without additional cognitive decline; from that time, bradykinesia, hypomimy, drooling, and athetoid movements of the hands were also noticed. Patients IV-5 and IV-7 displayed short stature. Dysmorphic features (figure 2, Aa, Ba, Ca) were also present. The 3 patients needed assistance to most common daily living activities.
Figure 1. Identification of a novel AP4M1 mutation.

(A) Pedigree showing the segregation of the AP4M1 c.916C>T (p.R306X) mutation in the family; black symbols indicate affected patients. (B) Electrophoregrams showing the wild-type (top), the homozygous mutated (middle), and heterozygous (bottom) sequence of AP4M1. (C) Localization of AP4M1 mutations in the protein: the structure of the AP4M1 protein (domain and amino acid positions) is described with all the pathogenic missense mutations5,14 (green), nonsense mutation9,15 (violet), frameshift4,13 (pink flag) mutations reported to date (in black), and in the present cases (in red). HM = homozygous; HT = heterozygous; WT = wild type.
Table.
Clinical features of 3 AP4M1 individuals
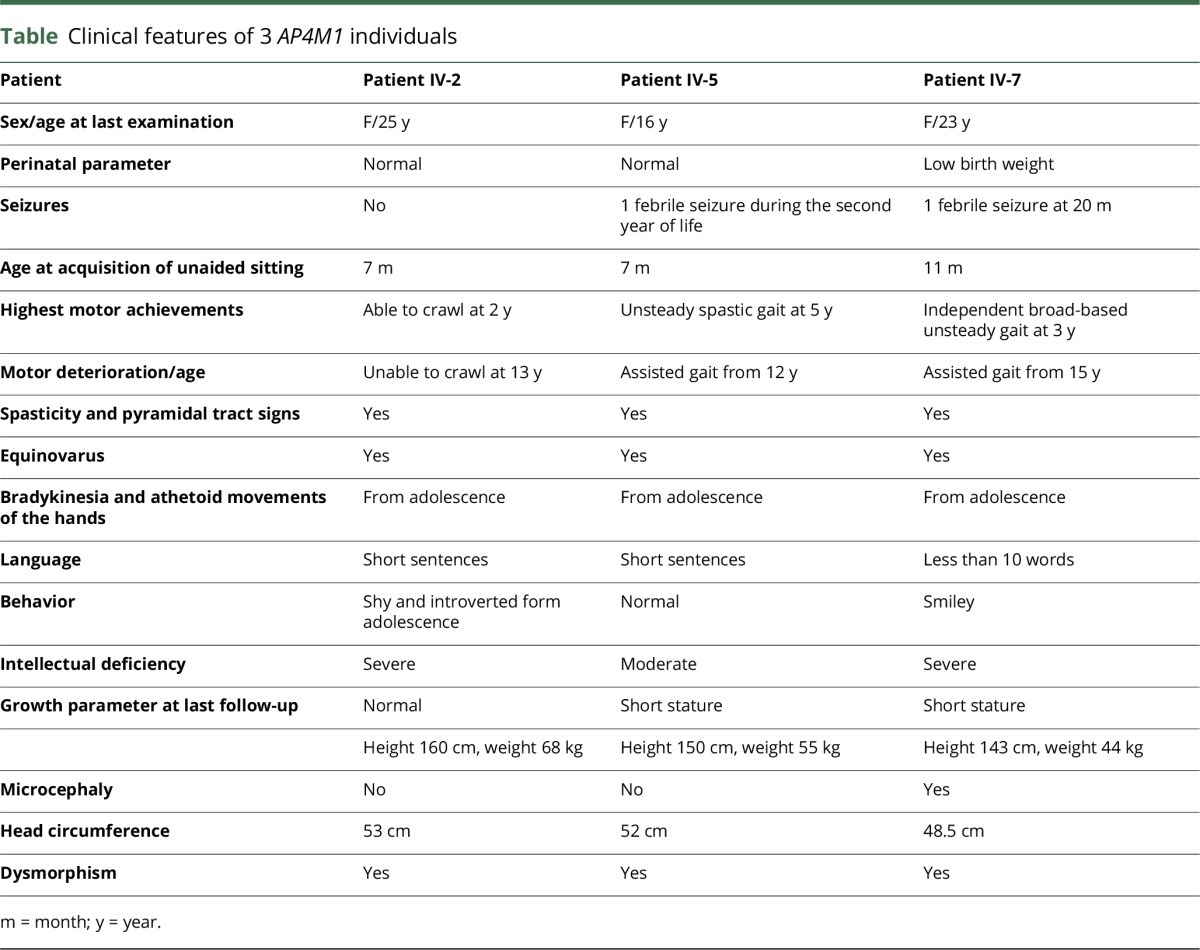
Figure 2. Facial appearance and brain imaging.

(A) Patient IV-2, 25 years. (B) Patient IV-5, 16 years. (C) Patient IV-7, 23 years. (D) Control, 20 years. (A.a, B.a, and C.a) Photographs showing dysmorphism, with bulbous nose, broad nasal bridge, coarse features, and wide-open mouth. (A.b, B.b, and C.b) Axial T2-weighted sequence showing asymmetric ventriculomegaly (black stars) and mild hypointense aspect of the globus pallidus (arrows). Diffuse brain atrophy and loss of white matter without signal abnormality, particularly in occipito parietal areas, are also observed. (A.c, B.c, C.c, and D) Axial susceptibility weighted imaging sequence showing bilateral intense hyposignal of the globus pallidus, significantly predominant in their medial part (arrows, A.c, B.c, and C.c) compared with the control (D).
The following investigations were normal: electromyoneurography recording, cardiac ultrasound scan, visual and auditory evoked potentials, fundus examination, karyotype analysis on lymphocytes (cases IV-2, IV-5 and IV-7), PANK2 and PLA2G6 Sanger sequencing (patient IV-7), and analyses of mitochondrial enzymatic activities on a muscle sample (patient IV-2).
Genetic results
Comparison of WES results performed for patients IV-2, IV-5, and IV-7 revealed 3 homozygous regions, 1 on chromosome 7 (5.7 Mb) and 2 on chromosome 9 (2.25 and 1.31 Mb). A total of 14,753 exonic variants were common to the 3 patients, and by progressively filtering them, we identified 4,974 homozygous variants, among which 2,546 were nonsynonymous, frameshift, splicing, or stop variants. Further filtering for damaging variants with a frequency lower than 1% identified 3 mutations in the AP4M1, HRNR, and NPIPL3 genes, but only the 1 in AP4M1 was located in chromosome 7, in 1 of the 3 homozygous regions.
This c.916C>T mutation (rs369459721) is leading to a premature stop codon (p.R306X), truncating the last 147 residues of the protein (figure 1, B and C). It has a global allelic frequency of 2.4 × 10−5 in the ExAC and a frequency of 3.0 × 10−5 in Non-Finnish European and 9.3 × 10−5 in African, while it was not encountered in the rest of the world.
Analysis of the homozygous variants located in the 10 known NBIA genes revealed 2 common variants, located in CP (rs701753) and PANK (rs3737084), but they were not damaging, had a frequency higher than 1%, and were located away from the 3 homozygous regions.
Brain imaging
Brain MRI of the 3 patients showed global cerebral atrophy, white matter loss, asymmetric ventriculomegaly (figure 2, B, E, and H), and thinning of the splenium of the corpus callosum (data not shown J). T1 sequences showed an isointense pattern of the globus pallidus (data not shown). T2 sequences revealed symmetric mild hypointensity of the globus pallidus, which was significantly accentuated on SWI sequences (figure 2, Ab, Ac, Bb, Bc, Cb, Cc, D). Patient IV-7's CT was normal (data not shown).
Discussion
We identified a homozygous nonsense mutation in AP4M1 in 3 women from the same inbred family by WES. This R306X mutation deletes the last 147 residues of the protein, truncating half of the adap domain, an effect similar to that reported in 2 other families who harbored a stop codon truncating the AP4M1 protein at positions 318 and 338.9 Until now, only 5 different AP4M1 mutations have been reported in 7 families with a common clinical presentation4,5,9,13–15 (figure 1C). The 3 patients from our study share the same clinical phenotype with variable severity, consisting in early-onset developmental delay, tetraparesis, juvenile motor function deterioration, intellectual deficiency, athetoid upper limb movements, bradykinesia, and mild dysmorphism, which fits with the previously described AP4-deficiency syndrome. Even if the bilateral pallidal hyposignal is mild on T2 sequences and could be interpretated as physiologic iron accumulation at this age, the substantial hyposignal on SWI is totally unusual in patients of the same age. These findings, correlated with the absence of hypersignal on T1-weighted imaging or CT hyperdensities in the patients, are strongly suggestive of brain iron overload.
Iron deposits have not been previously reported in patients with AP4-deficiency syndrome. Nevertheless, magnetic susceptibility sequences, which can confirm the presence of iron, have not been performed in most of the reported cases; therefore, this feature might have been underdiagnosed. A search for homozygous mutations common to the 3 patients in the 10 published NBIA genes revealed 2 variants located in PANK and CP, but their frequency and the absence of pathogenicity were somehow incompatible with their involvement as modifier mutations switching HSP clinical presentation to NBIA.
Of interest, a patient with AP4E1 mutations, whose brain MRI showed bilateral T2-hypointensity of the globus pallidus, has already been described.8 This peculiar finding, although not discussed in the article, strongly suggests iron accumulation in this AP4E1 patient, as in our 3 AP4M1 patients.
The pathophysiology of HSP involves many cellular pathways as cellular transport, nucleotide metabolism, and synapse and axon developments, providing a causative link between HSP and other neurodegenerative diseases.3,16 Overlaps between HSP and NBIA are well known, as already reported for cases with mutations in FA2H and C19orf12 genes (SPG 35 and 43, respectively).11,17 The AP-4 complex is a heterotetramer ubiquitously expressed in the CNS early in the embryologic and postnatal development and is implicated in vesicle formation, post-Golgi protein trafficking, and sorting processes.18 Eventually, AP-4 dysfunction might affect autophagy by disrupting the early steps of endosomal formation, a process shared with Kufor-Rabeb disease and beta-propeller protein-associated neurodegeneration, 2 forms of NBIA related to ATP13A2 and WDR45 genes, respectively.11,12
Moreover, NBIA disorders are probably underdiagnosed, and the evolution of technologies and practices in radiology leads to the identification of many new candidate genes through the incorporation of susceptibility weighted sequences more frequently in the brain imaging protocols.19 Our study has limitations, especially because of the small sample size.
Nevertheless, according to our findings in AP4M1 mutated patients, we recommend that brain MRI with susceptibility weighted sequences be included in the brain imaging protocol for patients with suspected HSP and AP4-deficiency syndrome to collect a larger group of patients, and we propose that mutations in AP4 genes be considered and screened in a subset of patients with NBIA spectrum disorders.
Acknowledgment
The authors acknowledge the patients and their families for participating in the study. They thank La Fonation Maladies Rares et Retina France. They are indebted to the INSERM and CNRS and the Montpellier University for their institutional supports. They thank the Region Pays de la Loire, Angers Loire-Métropole, University of Angers, and University Hospital of Angers for their support to the PREMMI project. They are indebted to Dr. Lagavulin for helpful discussions.
Glossary
- HSP
hereditary spastic paraplegia
- NBIA
neurodegeneration with brain iron accumulation
- SWI
susceptibility weighted imaging
- WES
whole-exome sequencing
Author contributions
Design or conceptualization of the study: Agathe Roubertie, Christian P. Hamel, and Guy Lenaers. Analysis or interpretation of the data: Charles-Joris Roux, Nicolas Leboucq, Gael Manes, Majida Charif, Bernard Echenne, Cyril Goizet, Claire Guissart, Pierre Meyer, Cecilia Marelli, François Rivier, Lydie Burglen, and Rita Horvath. Drafting or revising the manuscript for intellectual content: Agathe Roubertie, Charles-Joris Roux, Cyril Goizet, and Guy Lenaers.
Study funding
No targeted funding reported.
Disclosure
A. Roubertie, N. Hieu, C.-J. Roux, N. Leboucq, G. Manes, M. Charif, and B. Echenne report no disclosures. C. Goizet has served on the scientific advisory boards of Amicus Therapeutics and Sanofi Genzyme and has received travel funding/speaker honoraria from Sanofi Genzyme, Amicus Therapeutics, and Shire. C. Guissart and P. Meyer report no disclosures. C. Marelli has received travel funding/speaker honoraria from Actelion Pharmaceuticals. F. Rivier, L. Burglen, and R. Horvath report no disclosures. C.P. Hamel is deceased; disclosures are not included for this author. G. Lenaers reports no disclosures. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Finsterer J, Löscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci 2012;318:1–18. [DOI] [PubMed] [Google Scholar]
- 2.Fink JK. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol 2013;126:307–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kara E, Tucci A, Manzoni C, et al. . Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain 2016;139:1904–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verkerk AJ, Schot R, Dumee B, et al. . AR ticle mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am J Hum Genet 2009;85:40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jameel M, Klar J, Tariq M, et al. . A novel AP4M1 mutation in autosomal recessive cerebral palsy syndrome and clinical expansion of AP-4 deficiency. BMC Med Genet 2014;15:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jamra RA, Philippe O, Raas-Rothschild A, et al. . Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am J Hum Genet 2011;88:788–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abdollahpour H, Alawi M, Kortüm F, et al. . An AP4B1 frameshift mutation in siblings with intellectual disability and spastic tetraplegia further delineates the AP-4 deficiency syndrome. Eur J Hum Genet 2014;23:256–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moreno-De-Luca A, Helmers SL, Mao H, et al. . Adaptor protein complex-4 (AP-4) deficiency causes a novel autosomal recessive cerebral palsy syndrome with microcephaly and intellectual disability. J Med Genet 2011;48:141–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tüysüz B, Bilguvar K, Koçer N, et al. . Autosomal recessive spastic tetraplegia caused by AP4M1and AP4B1gene mutation: expansion of the facial and neuroimaging features. Am J Med Genet A 2014;164:1677–1685. [DOI] [PubMed] [Google Scholar]
- 10.Blumkin L, Lerman-Sagie T, Lev D, Yosovich K, Leshinsky-Silver E. A new locus (SPG47) maps to 1p13.2-1p12 in an Arabic family with complicated autosomal recessive hereditary spastic paraplegia and thin corpus callosum. J Neurol Sci 2011;305:67–70. [DOI] [PubMed] [Google Scholar]
- 11.Arber CE, Li A, Houlden H, Wray S. Review: insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: unifying theories. Neuropathol Appl Neurobiol 2015;42:220–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer E, Kurian MA, Hayflick SJ. Neurodegeneration with brain iron accumulation: genetic diversity and pathophysiological mechanisms. Annu Rev Genomics Hum Genet 2015;16:257–279. [DOI] [PubMed] [Google Scholar]
- 13.Langouët M, Siquier-Pernet K, Sanquer S, et al. . Contiguous mutation syndrome in the era of high-throughput sequencing. Mol Genet Genomic Med 2015;3:215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Najmabadi H, Hu H, Garshasbi M, et al. . Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011;478:57–63. [DOI] [PubMed] [Google Scholar]
- 15.Duerinckx S, Verhelst H, Perazzolo C, et al. . Severe congenital microcephaly with AP4M1 mutation, a case report. BMC Med Genet 2017;18:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novarino G, Fenstermaker AG, Zaki MS, et al. . Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014;343:506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landouré G, Zhu PP, Lourenco CM, et al. . Hereditary spastic paraplegia type 43 (SPG43) is caused by mutation in C19orf12. Hum Mutat 2013;34:1357–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simmen T, Höning S, Icking A, Tikkanen R, Hunziker W. AP-4 binds basolateral signals and participates in basolateral sorting in epithelial MDCK cells. Nat Cell Biol 2002;4:154–159. [DOI] [PubMed] [Google Scholar]
- 19.Herebian D, Alhaddad B, Seibt A, et al. . Coexisting variants in OSTM1 and MANEAL cause a complex neurodegenerative disorder with NBIA-like brain abnormalities. Eur J Hum Genet 2017;25:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]