

Riboflavin transporter deficiency (formerly known as Brown-Vialetto-Van Laere [BVVL] or Fazio-Londe syndrome) is a neurodegenerative disorder characterized by progressive bulbar palsy with sensorineural deafness or bulbar hereditary neuropathy.1 It is caused by mutations in the riboflavin transporter genes SLC52A2 (RFVT2) or SLC52A3 (RFVT3).2,3 It is a rare syndrome with approximately 70 cases reported worldwide, with molecular diagnoses of RFVT2 or RFVT3.1,4 We have previously described the first Brazilian family with a clinical diagnosis of BVVL.5
In this report, we extend the clinical spectrum associated with this family and describe a new mutation related to the metabolism of riboflavin.
Case report
The proband was a previously healthy woman aged 34 years, who presented with hearing and vision loss in the last 6 months (figure, A). She was disturbed by facial pain, numbness in the left hemiface, difficulty moving her tongue, dysphagia, weight loss, and bilateral foot drop.
Figure. Family pedigree and clinical improvement with riboflavin treatment.
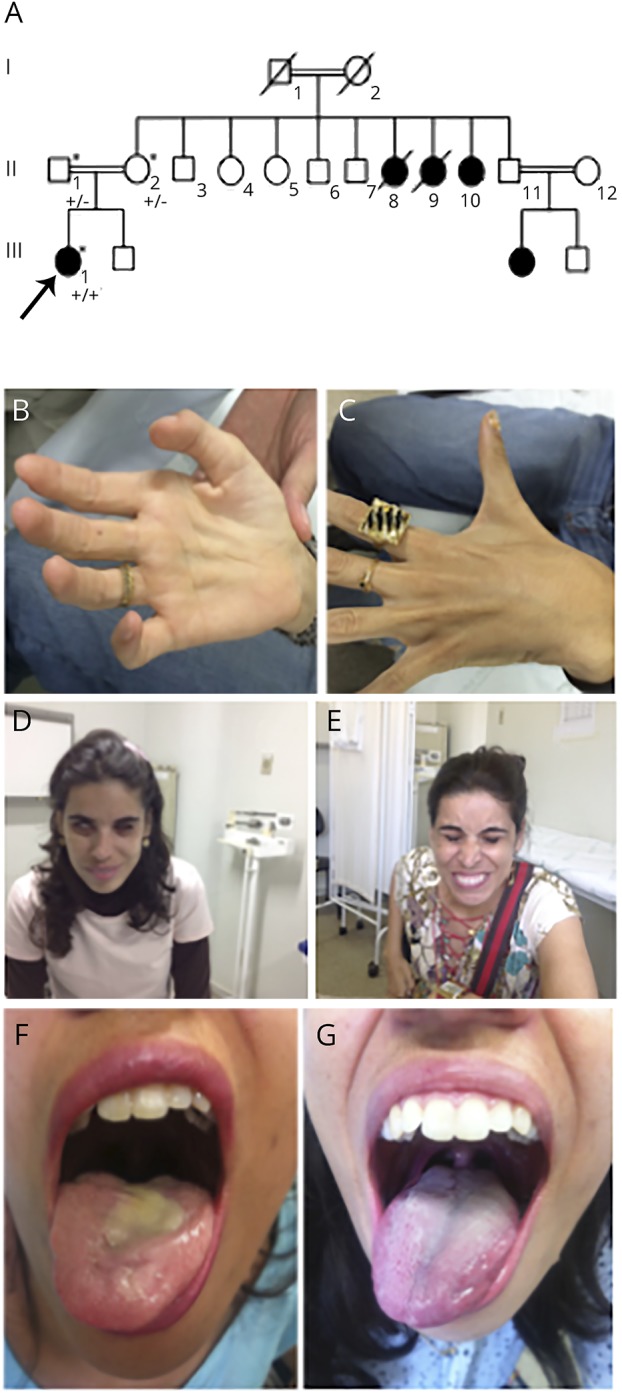
(A) The proband is marked with an arrow. This is a family member recently studied, while subjects II.8, II.9, II.10, and III.3 were previously described in Arq Neuropsiquiatr 2007;65(1):32–35. Squares represent males, and circles represent females. Black symbols represent affected family members. The proband's mother and father were found to be heterozygous for the SLC52A3 insertion (+/−). The mutation was found in homozygosity in the proband (+/+) and her aunt. (B and C) Proband's interosseal atrophy. (D and E) Improvement in facial weakness and (F and G) tongue atrophy after initiation of 1,800 mg of riboflavin.
Examination demonstrated bilateral optic atrophy, normal ocular movements, bilateral facial paresis, atrophic tongue, and flaccid dysarthria. Reflexes were brisk except for ankle reflexes that were absent. Plantar responses were indifferent. All sensory modalities were normal. Strength was globally diminished with important distal impairment and foot drop. As dysphagia and dyspnea progressed, a feeding tube was placed and noninvasive ventilation support was initiated. At that time, she was quadriplegic and could not walk.
Electroneuromyography demonstrated cervical and acute lumbar denervation, with chronic neurogenic changes. Audiologic evaluation demonstrated neurosensorial loss.
The patient was the eldest sibling of a consanguineous marriage. She had 3 maternal aunts, also sisters from a consanguineous marriage, with a probable diagnosis of BVVL syndrome.5
The patient was started on empiric treatment with riboflavin (1,800 mg per day), and within 6 months of therapy, she could walk with a cane; the feeding tube and noninvasive ventilation were withdrawn (figure, B–G). Electroneuromyography was performed after B2 treatment and demonstrated low CMAP amplitudes and persistence of recent denervation (figure e-1, http://links.lww.com/NXG/A17).
Methods
The genetic study of the patient and both parents was conducted after written informed consent was obtained. Whole-exome sequencing (WES) was performed in the index case, and whole-genome genotyping (WGG) was used to identify large tracts of homozygosity in the trio, given the apparent autosomal recessive pattern of inheritance of the disease in the family and the presence of several loops of consanguinity. The 3 samples were genotyped at the whole-genome level using Illumina HumanOmniExpress Bead chips, and results were analyzed with GenomeStudio.
Results
By analyzing the variability identified by WES in genes previously known to cause riboflavin transporter deficiency, we identified a novel homozygous insertion in SLC52A3 (ENST00000217254(SLC52A3):c.1232_1233insCTACGCTTCCCTCCCGGCCCCGCAGGTGGCCTCGTGGGTGCTTTTCAGCGGCTGCCTCAG; p.Ser411_Tyr412insTyrAlaSerLeuProAlaProGlnValAlaSerTrpValLeuPheSerGlyCysLeuSer). The mutation was confirmed to be present in homozygosity in the index and was found in heterozygosity in both parents using Sanger sequencing. In addition, WGG revealed a large (1.5 Mb) homozygous region encompassing the SLC52A3 locus (chromosome 20: 643,919–2,146,580 Mb) that was not present in either parent. Consequently, we tested the phenotypically affected aunt (II.10), and she presented the mutation in homozygosity (figure e-2, http://links.lww.com/NXG/A18).
The mutation found is neither present in homozygosity in population databases of genetic variability (ExAC and gnomAD), nor have we identified it in our in-house sequencing data from healthy controls and other diseases (n > 6,000).
Discussion
Both SCL52A3 and SCL52A2 are riboflavin transporters. Riboflavin is a precursor of flavin mononucleotide and flavin adenine dinucleotide, which are important cofactors for energy metabolism. Since the description of these 2 genes related to riboflavin transporter deficiency, the perspective about this disease has changed significantly.2,4,6 However, the mechanisms by which the disrupted proteins lead to the disease are still to be fully understood.
Motor neuron disease, neurosensorial deafness, optic neuropathy, and the involvement of cranial nerves are common in both genotypes.4,7 Nevertheless, some phenotypic differences have been described: RFVT2 mutations present predominantly upper limb weakness and sensory ataxia; such findings are not commonly seen in patients with RFVT3 mutations.7
Here, we report a Brazilian patient with late-onset and uncharacteristic acute and severe presentation, demonstrating some phenotypic heterogeneity within a family.5 The mutation, a homozygous insertion of 60 bp in SCL52A3, has not been previously described as the cause of riboflavin transporter deficiency. So far, response to riboflavin therapy was documented in 11 patients harboring mutations in RFVT3. Of them, 9 patients demonstrated some response and 2 remained stable.4,6 Some authors argue that response tends to be better and more rapid when earlier treatment is started. Riboflavin dose reposition is unknown, and treatment, although generally efficient, is empirical. In addition, there is still no evidence to reassure that treatment would prevent the occurrence of symptoms indefinitely. Despite all this, clinicians might be aware of this potentially treatable condition and initiate riboflavin supplementation as soon as diagnosis is suspected.
Acknowledgment
The authors acknowledge the patients who participated in this study.
Author contributions
Sarah Camargos: study concept, design, acquisition of data, and study supervision. Rita Guerreiro: study concept, design, and critical revision of the manuscript for important intellectual content. Jose Bras: analysis and interpretation of the data. Luis Sergio Mageste: analysis and interpretation of the data.
Study funding
No targeted funding reported.
Disclosure
S. Camargos has served on the scientific advisory board of Teva Pharmaceuticals and has received travel funding/speaker honoraria from Roche, Teva Pharmaceuticals, and Aché Pharmaceuticals. R. Guerreiro serves on the editorial boards of Science Matters, the American Journal of Neurodegenerative Disease, and the Journal of Parkinson's Disease and has received research support from Alzheimer's Research UK and Alzheimer's Society. J. Bras serves on the editorial board of the Journal of Parkinson's Disease and has received research support from Alzheimer's Society. L.S. Mageste reports no disclosures. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Bosch AM, Stroek K, Abeling NG, Waterham HR, Ijlst L, Wanders RJ. The Brown-Vialetto-Van Laere and Fazio Londe syndrome revisited: natural history, genetics, treatment and future perspectives. Orphanet J Rare Dis 2012;7:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Green P, Wiseman M, Crow YJ, et al. Brown-Vialetto-Van Laere syndrome, a ponto-bulbar palsy with deafness, is caused by mutations in c20orf54. Am J Hum Genet 2010;86:485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson JO, Gibbs JR, Megarbane A, et al. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain 2012;135:2875–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaeger B, Bosch AM. Clinical presentation and outcome of riboflavin transporter deficiency: mini review after five years of experience. J Inherit Metab Dis 2016;39:559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malheiros JA, Camargos ST, Oliveira JT, Cardoso FE. A Brazilian family with Brown-Vialetto-Van Laere syndrome with autosomal recessive inheritance. Arq Neuropsiquiatr 2007;65:32–35. [DOI] [PubMed] [Google Scholar]
- 6.Bashford JA, Chowdhury FA, Shaw CE. Remarkable motor recovery after riboflavin therapy in adult-onset Brown-Vialetto-Van Laere syndrome. Pract Neurol 2017;17:53–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foley AR, Menezes MP, Pandraud A, et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain 2014;137:44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]