

Abstract
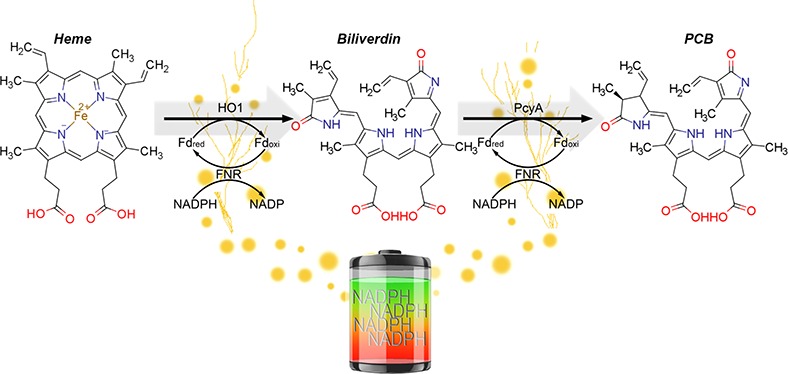
Transplanting metabolic reactions from one species into another has many uses as a research tool with applications ranging from optogenetics to crop production. Ferredoxin (Fd), the enzyme that most often supplies electrons to these reactions, is often overlooked when transplanting enzymes from one species to another because most cells already contain endogenous Fd. However, we have shown that the production of chromophores used in Phytochrome B (PhyB) optogenetics is greatly enhanced in mammalian cells by expressing bacterial and plant Fds with ferredoxin-NADP+ reductases (FNR). We delineated the rate limiting factors and found that the main metabolic precursor, heme, was not the primary limiting factor for producing either the cyanobacterial or plant chromophores, phycocyanobilin or phytochromobilin, respectively. In fact, Fd is limiting, followed by Fd+FNR and finally heme. Using these findings, we optimized the PCB production system and combined it with a tissue penetrating red/far-red sensing PhyB optogenetic gene switch in animal cells. We further characterized this system in several mammalian cell lines using red and far-red light. Importantly, we found that the light-switchable gene system remains active for several hours upon illumination, even with a short light pulse, and requires very small amounts of light for maximal activation. Boosting chromophore production by matching metabolic pathways with specific ferredoxin systems will enable the unparalleled use of the many PhyB optogenetic tools and has broader implications for optimizing synthetic metabolic pathways.
Keywords: optogenetics, phytochrome, phycocyanobilin, phytochromobilin, ferredoxin, FNR
An established research practice used in synthetic biology is the transplantation of metabolic reactions from one species to another, with wide-ranging potential applications including metabolic gene therapy,1,2 production of crops without fertilizer,3,4 and more fundamental applications in research, such as optogenetics. The exquisite temporal and spatial precision achieved through optogenetics have been used to develop an assortment of powerful analytical tools to control biological functions such as gene expression,5−10 neural activity,11,12 cell signaling,13 secretion,14 peroxisomal trafficking,15 and protein activity.16 Metabolically engineering cells to endogenously produce specific chromophores enables many optogenetic applications, including genetically encoded systems for optical control of genes.17 Many of the systems used and characterized for these applications utilize proteins that require red and far-red responsive phytobilin chromophores like phycocyanobilin (PCB) and phytochromobilin (PΦB). These molecules originate from phytochrome systems in cyanobacteria, algae, and plants, but are not naturally made in many fungal species, bacteria, or animal cells.18−21 Production of these chromophores requires biliverdin IX-alpha (BV), a degradation product of heme, and the enzymes phycocyanobilin:ferredoxin oxidoreductase (PcyA) or phytochromobilin:ferredoxin oxidoreductase (HY2), respectively (Figure 1A).22,23 Several groups produced PCB and PΦB in E. coli by expressing PcyA or HY2 along with heme oxygenase (HO1), without adding a ferredoxin (Fd) and ferredoxin-NADP+-reductase (FNR) reduction system from the same species as the PcyA or HY2 enzymes.24−29 Likewise, Müller et al. tested PCB production in mammalian cells by expressing cyanobacterial PcyA and HO1 in the mitochondria but did not cointroduce a cyanobacterial Fd-FNR system.11 Müller et al. reasoned that localizing PcyA and HO1 in the same cellular compartment where the chromophore precursor (heme) is produced would enhance PCB production.11 However, in addition to heme, HO1, PcyA, and HY2 also depend on Fd activity, leaving open the possibility that Fd and not heme was limiting.
Figure 1.

PCB and PφB production is limited by Fd+FNR in mammalian cells. (A) The metabolic pathway forPCB synthesis including the NADPH/FNR/Fd redox cascade (Heme: ChemSpider ID 4802, Bv: ChemSpider ID10628548, PCB: ChemSpider ID 16736730). (B) HEK293 cells were analyzed for phytobilin production using the plasmids shown. Phytobilin production was measured by covalent linkage to PhyB followed by immunoprecipitation with anti-HA, Zn-PAGE and Western blots. sPCYA and tPCYA produce PCB and aHY2 produces PΦB. Cells were either transfected with two ferredoxin-dependent enzymes (ho1 and pcyA or ho1 and HY2) alone (condition M2) or along with matching Fd+FNR (tpetF+tpetH) plasmids (condition M4). ho1 = heme oxygenase, pcyA = phycocyanobilin:ferredoxin oxidoreductase, HY2 = phytochromobilin:ferredoxin oxidoreductase, petF = ferredoxin, petH = ferredoxin:oxidoreductase/FNR, NE = No Enzymes, SYNP2 = Synechococcus PCC7002 and THEEB = Thermosynechococcus elongatus, ARATH = Arabidopsis thaliana, MTS = Mitochondrial Targeting Sequence, P2A = 2A self-cleaving peptide, IRES = Internal Ribosome Entry Site, NLS = Nuclear Localization Sequence, DBD = DNA Binding Domain.
Most cells already contain endogenous Fd; therefore, researchers have not typically considered it when transplanting enzymes from one species to another. However, Beale et al. and Frankenberg et al. demonstrated that Fd activity on PcyA from Anabaena sp. PCC 7120 varies greatly depending on the species Fd comes from.24,30 Similarly, mammalian Fds have also been shown to be highly specific to their target enzymes, suggesting that Fd and/or FNR may be limiting for chromophore production in mammalian cells.31,32 Consequently, to increase production of molecules like PCB for optogenetic uses in animal cells, we investigated the limiting factors for the PCB and PΦB production in mammalian cells.
To evaluate the rate-limiting reactants for endogenous chromophore production, we systematically tested each component of the biosynthetic pathway, including Fd and FNR. We showed that Fd+FNR is the primary rate-limiting component, followed by heme. The increased PCB production found with the addition of Fd+FNR was further improved by testing different stoichiometric expression levels of each enzyme. Endogenous PCB production was greatly increased compared to previous approaches17 that did not consider metabolic engineering with Fd+FNR systems.
To demonstrate the utility of increased chromophore production for optogenetic applications, we chose a PhyB-based optogenetic system, which utilizes PCB and has been used to control a wide array of biological processes. Since the light sensitivity of PhyB is proportional to the amount of chromophore in the cell, to apply PhyB optogenetic tools in transgenic animal models, it will be essential to genetically encode a high level of chromophore production. Able to produce significantly more chromophore than before,17 we fully genetically encoded the red/far-red PhyB-PIF3 two-hybrid gene switch for the first time.
A genetically encoded PhyB-PIF3 system with PCB production is particularly significant because when bound to PhyB, chromophores such as PCB: (i) are extremely sensitive to light (high absorbance/extinction coefficient), (ii) have a long-lived activation state, ranging from tens of minutes to hours,33 (iii) are reversible upon illumination with a specific wavelength of far-red light,14 and (iv) respond to wavelengths optimal for tissue penetration. The reversibility of this system with far-red light allows for additional spatial control by enabling suppression of gene activity with far-red light in specific locations.34 After adapting the PhyB-PIF3 system from Shimizu Sato et al.(5) for mammalian cells, we found that it can induce gene expression by several hundred fold, it is reversible with a stable “on state” in the order of hours, and it requires very low amounts of red light for maximum activation (calculated to be below the equivalent of 40 nW/cm2 of continuous light for full activation over 24 h). Since genetically encoding the system maintains a constant supply of chromophore, we were also able to find that the light intensity required for maximal gene activation depends on the duration of illumination.
More generally than optogenetics, there are numerous biomolecules produced in bacteria and plants that are Fd-dependent. Matching the Fd species to a biosynthetic production pathway makes possible the metabolism of many other classes of molecules such as lipids, sterols, luciferins, quinones, carotenoids, nitrates/nitrogen, and sulfites not normally produced in those cells.3,35−40 Increasing production of these classes of molecules can improve agriculture, increase the production of pharmaceuticals, and enable other tools for synthetic biology.
Results and Discussion
Regulation of PCB Production in Mammalian Cells by Fd, FNR, and Heme
Given that previous studies have shown that PCB production can be limited by heme, Fd or FNR,25,30 we tested limiting factors of PCB production in mammalian cells using combinations of these components in excess. Zinc-PAGE PhyB immunoprecipitation assays in Human Embryonic Kidney (HEK293) cells were used to test PCB production with metabolic enzymes from two species: Synechococcus sp. PCC 7002 (SYNP2/sPcyA) or Thermosynechococcus elongatus (THEEB/tPcyA). We tested PCB production under two conditions, either mitochondrial-HO1+PcyA (M2) or mitochondrial-HO1+PcyA+Fd+FNR (M4), (Figure 1B). When either species of HO1+PcyA enzymes were expressed, we detected low levels of PCB (Figure 1B, M2). However, when all four enzymes HO1+PcyA+Fd+FNR (M4) were expressed, we observed a striking increase in PCB levels (Figure 1B), which agrees with recent findings from Uda et al.(41) To exclude the possibility that this was specific to cyanobacterial enzymes, we also produced the plant chromophore PΦB, by replacing the cyanobacterial PcyA with a plant homologue Arabidopsis HY2. PcyA and HY2 showed the same Fd+FNR dependence (Figure 1B, M2-asHY2versusM4-asHY2). It is noteworthy that the Fd+FNR-dependent increase in PΦB production was still observed when plant HY2 was used along with cyanobacterial HO1/Fd/FNR. We chose SYNP2 Fd+FNR for recycling HY2 because SYNP2 Fd was more similar than THEEB Fd in amino acid sequence identity to Arabidopsis Fds and specifically the major ferredoxin that recycles HY2 in Arabidopsis (Table S1).42 However, PΦB production may be further increased by employing Arabidopsis Fd+FNR enzymes. It may be possible to predict compatibility of a transplanted ferredoxin-dependent pathway to the host cells Fd based on sequence similarity as shown in Table S1. These findings show that excess Fd+FNR activity can increase PCB or PΦB production in mammalian cells (Figure 1B).
Next, we delineated the limiting factors for the endogenous production of chromophores in mammalian cells. We decided to test PCB production in both the cytoplasm and mitochondria because the endogenous ferredoxin system of mammalian cells is localized in the mitochondria; therefore, we considered the cytoplasmic enzyme localization as a condition with negligible endogenous Fd+FNR activity. We show in Figure 2A that expression of cytoplasmic-PcyA+HO1 (C2) is not sufficient to produce significant levels of PCB (lane 3 vs lane 2). When cytoplasmic-PcyA+HO1 was cotransfected along with cytoplasmic Fd+FNR (C4) higher, but statistically nonsignificant levels of PCB were detected (lane 3 vs 4, p > 0.05). Similarly, when PcyA+HO1 were localized to the mitochondria (M2), very low levels of PCB were detected (lane 5). However, when PcyA+HO1 and Fd+FNR were all localized to the mitochondria (M4), PCB production was significantly increased when compared to PcyA+HO1 only (M2) (lane 5 vs 6, p < 0.001). These findings were corroborated by imaging PhyB-bound PCB using the Cy-5 channel (blue) (Figure S1). These results demonstrate that the Fd+FNR system is the primary limiting factor of the PCB production pathway in mammalian mitochondria, but it is not sufficient for high levels of PCB production when expressed in the cytoplasm.
Figure 2.

Order of rate limiting factors of PCB production in mammalian cells. (A,B) HEK293 cells were analyzed for PCB production using the plasmids shown. PCB production was measured by covalent linkage to PhyB followed by immunoprecipitation with anti-HA, Zn-PAGE and Western blots. (A) PCB production was compared with excess (+heme) and without (−heme), using the cytoplasmic expression of pcyA+ho1 alone (condition C2) or with cytoplasmic pcyA+ho1+fd+fnr (condition C4); mitochondrial expression of pcyA+ho1 alone (condition M2) or with mitochondrial pcyA+ho1+fd+fnr (condition M4) (n = 4). (B) Cells were either transfected with two ferredoxin-dependent enzymes alone, ho1 and pcyA (condition M2), or along with a matching fd:tpetF (condition M3) or along with matching fd+fnr:tpetF + tpetH (condition M4) (n = 4). ho1 = heme oxygenase, pcyA = phycocyanobilin:ferredoxin oxidoreductase, HY2 = phytochromobilin:ferredoxin oxidoreductase, petF = ferredoxin/fd, petH = ferredoxin:oxidoreductase/fnr, NE = No Enzymes, SYNP2 = Synechococcus PCC7002 and THEEB = Thermosynechococcus elongatus, ARATH= Arabidopsis thaliana, IRES = Internal Ribosome Entry Site, NLS = Nuclear Localization Sequence, MTS = Mitochondrial Targeting Sequence, P2A = 2A self-cleaving peptide, DBD = DNA Binding Domain. One-way ANOVA with Bonferroni post-test was used to calculate p values using GraphPad Prism 5.01. (*) = p < 0.05, (**) = p < 0.01, (***) = p < 0.001. Error bars = Standard Deviation. n = independent experiments.
Since heme is a metabolic precursor in the PCB production pathway, we systematically tested if it was limiting for PCB production in either the cytoplasm or in the mitochondria. We hypothesized that if heme was a limiting factor for PCB production in the cytoplasm, then the addition of excess heme would increase production. While a faint band was visible in C2+heme (Figure 2A lane 9), it was indistinguishable from cells transfected with PhyB and no enzymes and given excess heme (Figure 2A lane 8). However, excess heme significantly increased levels of PCB production in the C4 condition (lanes 4 and 10, p < 0.01). In addition, we found that Fd+FNR was limiting when comparing C2+heme to C4+heme (lanes 9 and 10, p < 0.01). This demonstrates that heme is the limiting factor for PCB production when an excess of Fd+FNR is present in the cytoplasm. Importantly, PCB production was not influenced by excess heme when enzymes were localized to the mitochondria (M4–heme and M4+heme, lanes 6 and 12). This confirms that Fd+FNR is primarily limiting in both the cytoplasm and the mitochondria and that heme is secondarily limiting only in the cytoplasm.
To further investigate the PCB production dependence on Fd, we transfected cells with two, three or all four enzymes in the pathway: PcyA-HO1 (M2), PcyA+HO1+Fd (M3), or PcyA+HO1+Fd+FNR (M4), along with PhyB for all conditions (Figure 2B). We show in Figure 2B that the addition of Fd to PcyA+HO1 (M3) significantly increased PCB production compared to PcyA+HO1 alone (M2) (p < 0.05). Importantly, Fd+FNR (M4) produces significantly more PCB than adding Fd alone (p < 0.01), demonstrating that for maximum PCB production both Fd and FNR are required.
While we considered testing the overexpression of the host cell’s Fd+FNR, there are noteworthy advantages to using orthogonal Fd+FNR matching the species of the transplanted metabolic pathway. The mammalian Fd+FNR may be able to reduce BV bound to PcyA but only at a fraction of the rate of the cyanobacterial Fd+FNR. The required overexpression needed for the host cell’s system to perform at the same production rate would therefore more likely disturb the cell’s metabolism. Using an orthogonal system would be more efficient and would also less likely interact with the host cell’s metabolic proteins. Matching the orthogonal enzyme species thus allows for minimal perturbation of the normal host cell physiology and at the same time maximize production rates.
Effects of PcyA, HO1 and Fd+FNR Stoichiometry on PCB Production Levels
Okada et al.(43) demonstrated that Fd forms stable complexes with both HO1 and PcyA. Therefore, we hypothesized that PCB production may be further optimized through enzyme stoichiometry. We transfected separate PcyA+HO1 and Fd+FNR plasmids at different ratios and observed that PCB production was highly dependent on the ratio between PcyA+HO1 and Fd+FNR (Figure 3A). Considering this, to serve as a quantitative guide for optimizing PCB production, we developed computational models of this pathway using coupled ordinary differential equations (model details in Supporting Information). We tested the enzyme stoichiometry using a functional PhyB-PIF3 luciferase gene expression system adapted from Shimizu Sato et al.(5) (Figure 3B). First, we used optimized versions of the PhyB-PIF3 switch, including optimizing DNA binding domains (Figure S2), activation domains (Figure S3), and reporter constructs (Figure S4). Next, the stoichiometry was tested by transfecting different ratios of the PcyA+HO1 and Fd+FNR plasmids and illuminating the cells with red light for 24 h (timeline of illumination as shown in Figure S3A), followed by a luciferase assay to compare gene induction levels. We found that gene activation levels were also highly dependent on enzyme stoichiometry, with only the 17:1 PcyA+HO1:Fd+FNR showing any measurable response to light (Figure 3C and 3D, p < 0.01). This demonstrates how chromophore levels influence the performance of PhyB optogenetic systems.
Figure 3.

Stoichiometry of PCB production constructs. (A) PCB production assay comparing plasmid ratios of pcyA+ho1 to fd+fnr using the plasmids shown. Transfection ratios are indicated in boxes below the Western blot. PCB production was measured by covalent linkage to PhyB followed by immunoprecipitation with anti-HA, Zn-PAGE and Western blots. (B) Schematic of the PhyB-PIF3 light switch. PhyB is fused to a DNA Binding Domain (DBD) and bound to a light-sensitive chromophore (PCB). The PhyB-DBD fusion remains bound to the UAS promoter. PIF3 is fused to an Activation Domain (AD). Upon absorption of a red photon (660 nm), PhyB changes conformation and recruits PIF3 to the promoter region. The AD fused to PIF3 then activates the gene downstream of the promoter. Upon absorption of a far-red photon (735 nm), PhyB changes conformation that leads to PIF3 unbinding, removing the AD from the promoter, shutting the downstream gene off. (C) Plasmid maps for endogenous PCB production and PhyB-PIF3 light switchable promoter. (D) Luciferase gene activation levels using endogenously produced PCB with several ratios of pcyA+ho1:petF+petH (n = 3). (E) Three construct designs consisting of all four biosynthetic enzymes on a single plasmid and a single plasmid for PIF3 and PhyB. (F) Testing gene activation comparing single plasmid biosynthetic plasmids (n = 7). ho1 = heme oxygenase, pcyA = Phycocyanobilin:ferredoxin oxidoreductase, petF = ferredoxin, petH = ferredoxin:oxidoreductase/FNR, MTS = Mitochondrial Targeting Sequence, P2A = 2A self-cleaving peptide, NLS = Nuclear Localization Sequence, IRES = Internal Ribosome Entry Site, AD = Activation Domain, DBD = DNA Binding Domain, R/FR = Red light/Far-red light. Error bars = Standard Deviation, (*) = p < 0.05, (**) = p < 0.01. Statistics were calculated using one-way ANOVA with Bonferroni post-test using GraphPad Prism 5.01. n = individual experiments.
Mammalian PhyB-PIF Gene Switch Using Endogenously Produced PCB
After identifying the requirements for high levels of endogenous PCB production, we sought to encode all four biosynthetic enzymes on a single plasmid. Our original four enzyme plasmid (pPKm-245) contained all PCB biosynthetic enzymes separated by P2A sequences to achieve a 1:1:1:1 expression level of each enzyme.44 However, the results in Figures 3A–D suggested that PCB production could be further optimized by modifying the plasmid’s expression stoichiometry. To this end, we replaced one of the P2A sequences with an Internal Ribosomal Entry Site (IRES), which typically gives 1 order of magnitude lower expression to the gene following the IRES sequence.45−47 The plasmid pPKm-244 was generated by placing an IRES between pcyA and Fd, leading to higher PcyA-HO1 levels and lower Fd+FNR levels (Figure 3E). We also constructed a plasmid, pPKm-248, containing HO1, Fd, and FNR all placed after the IRES sequence. This plasmid results in minimized heme oxygenase and Fd+FNR activity while keeping higher levels of PcyA (Figure 3E). Using the experiment timeline in Figure S3A, we found that lowering HO1 and Fd+FNR levels with the pPKm-248 plasmid produced 1.8-fold (p < 0.05) and 2.2-fold (p < 0.01) higher gene activation levels than pPKm-244 and pPKm-245 respectively (Figure 3F). In addition to producing more PCB, lower expression of HO1, Fd and FNR should provide maximal PCB levels with minimal interference in the host cells metabolism.
Light Sensitivity of the Mammalian PhyB-PIF3 Gene Switch Using Endogenously Produced PCB
PhyB-PIF optogenetic systems in animal cells have mostly been characterized in conditions where PCB is added externally. However, PCB degrades rapidly in cell culture media,7 which affects PhyB’s light sensitivity over long time spans.48 Since our constructs enable constant endogenous production of PCB, we sought to test the light sensitivity of the PhyB-PIF3 switch (pPKm-230) with the endogenously produced chromophore. We illuminated transfected cells with the activating red light, at different intensities for 24 h, and found that light intensities of 1.00 μmol/m2/s, 0.1 μmol/m2/s, and 0.01 μmol/m2/s achieved similar high levels of gene activation (Figure 4B and 4D). In contrast, transfected cells illuminated with a light intensity of 0.001 μmol/m2/s had a significantly lower gene response (p < 0.05). Since the system is bistable,33 we reasoned that activating with intensities between 1.0 and 0.01 μmol/m2/s, which activate the system over a long time span (24 h), may not represent saturating amounts of light for shorter illumination times.49 To test this hypothesis, we characterized the gene switch using these same light intensities, but with a single 1 min pulse of red light (Figure 4C and 4E). Unlike the 24-h illumination experiment, we found that when we illuminated the cells with red light for 1 min, light intensities of 0.1 μmol/m2/s and 0.01 μmol/m2/s had a significantly lower gene response than an intensity of 1.0 μmol/m2/s (p < 0.001). This finding highlight that for characterizing these light responsive bistable proteins, we should consider both the light intensity and duration of illumination. For example, our results using 0.1 μmol/m2/s and 0.01 μmol/m2/s show that those intensities are not saturating with a 1 min pulse, but those same intensities induce saturating activation levels over 24 h (Figure 4D and 4E). This is expected from a system that is bistable with a long-lived activation state,49 inactive molecules not activated in the first minute will be activated later if light is continuously applied, eventually activating all of the light-sensitive molecules.
Figure 4.

Light sensitivity of the genetically encoded PhyB-PIF3 switch. (A) Plasmids optimized for an endogenous PhyB-PIF3 light switchable promoter. (B) Pulsing program for 24-h illumination experiments. (C) Pulsing program for 1 min illumination experiments. (D) Gene response to a 24-h pulse with several light intensities (n = 4). (E) Gene response to a 1 min pulse with several light intensities (n = 4). (F) Gene activation responses using 1 μmol/m2/sec or 0.1 μmol/m2/sec of continuous light compared with using 0.1 μmol light at different pulse intervals for 24 h (n = 3). The blue stars indicate the minimal light dose for saturating activation using 24-h illuminations. (G) Pulsing program for testing the duration of activation. Pulsing was done as in B. (H) Gene response to pulsing at increasing intervals. Cells were pulsed for 1 min using 1 μmol/m2/sec 660 nm light, followed by darkness for the indicated times for a total of 24 h (n = 5). The blue arrows indicate the minimal light dose for saturating activation using 24 h illuminations. (I) Total light flux during 24 h period of illumination for experiments in Figure 4D and Figure 4H. Cont. = continuous illumination, 1 min/4 min = 1 min red light, 4 min darkness, 1 min/9 min = 1 min red light, 9 min darkness, 1 min/29 min = 1 min red light, 29 min darkness. ho1 = heme oxygenase, pcyA = Phycocyanobilin:ferredoxin oxidoreductase, petF = ferredoxin, petH = ferredoxin:oxidoreductase/FNR IRES = Internal Ribosome Entry Site, MTS = Mitochondrial Targeting Sequence, NLS = Nuclear Localization Sequence, P2A = 2A self-cleaving peptide, AD = Activation Domain, DBD = DNA Binding Domain, R/FR = Red light/Far-red light. Error bars = Standard Deviation, (*) = p < 0.05, (***) = p < 0.001. Statistics were calculated using one-way ANOVA with Bonferroni post-test using GraphPad Prism 5.01. n = individual experiments.
Endogenous Mammalian PhyB-PIF3 Gene Switch Bistability and Reversibility with Far-Red Light
We further tested the light sensitivity and bistability by shining activating red light at different pulse intervals (Figure 4F). As controls, we illuminated HEK293 cells with continuous 1.0 μmol/m2/s or 0.1 μmol/m2/s red light for 24 h and found they reach similar levels of gene activation. In addition to continuous illumination, we utilized alternating light/dark cycles composed of 1 min of red light and 4, 9, or 29 min of darkness (1 min/4 min, 1 min/9 min, 1 min/29 min respectively) for 24 h. Continuous red light at 0.1 μmol/m2/s, as well as the 1 min/4 min and 1 min/9 min conditions, did not produce statistically different activation levels (Figure 4F). In contrast, the condition with 0.1 μmol/m2/s of red light pulsed at 1 min/29 min had significantly lower activation levels than continuous light and pulsed light in the 1 min/4 min and 1 min/9 min conditions (Figure 4F, p < 0.05). Because the 1 min/9 min (blue star) condition has one-tenth the number of photons as 0.1 μmol/m2/s in total photon flux, it is equivalent in the number of photons to 0.01 μmol/m2/s of continuous illumination or 183 nW/cm2 for 660 nm red light. This agrees with the result where the same total amount of light is applied continuously, suggesting that the activation state of PhyB is much longer than the 9 min dark interval (Figure 4D and 4F).
Interestingly, we also found that cells containing the PhyB-PIF3 system had a slightly higher level of gene activation in the darkness than cells in the presence of far-red light, potentially due to the bistability of the protein (Figure 4F). Thermodynamically, in darkness, a mixed population of species (Pf and Pfr forms) is the expected nature of a bistable molecule, since some PhyB molecules can spontaneously switch to the “activated state”. Therefore, the proportion of activated PhyB molecules should be higher in darkness than when PhyB is illuminated with a deactivating far-red light.
Since pulsing the light on a minute time scale achieved similar levels of activation as continuous light (Figure 4F), we decided to test the duration of the activated state of PCB bound PhyB (PhyB·PCB) by increasing the spacing between red light pulses as shown in Figure 4G. Our results show similar levels of gene activation for red light pulses delivered for 1 min every 8, 6, 4, 2, 1 h, and a half hour at 1 μmol/m2/s (Figure 4H). However, a pulse delivered every 12 h (a total of two pulses in the 24 h period) produced significantly lower gene activation than the pulses delivered in the shorter intervals (Figure 4H). It is possible that those two pulses in the 24-h period delivered too little total amount of light to fully activate the system (Figure 4I). However, this data still supports that the switch effectively stays “on” for at least 8 h following a 1 min pulse of 1 μmol/m2/s of red light (Figure 4H, blue arrow). In terms of total light delivery (μmol/m2), the 1 min pulses every 8 h using 1.0 μmol/m2/s is effectively equivalent to the number of photons with continuous light at 0.0021 μmol/m2/s or 38nW/cm2 for 660 nm light, which is a strikingly small amount of light and speaks to the high sensitivity of this system.
One hallmark of PhyB based optogenetic switches is their conformational reversibility upon absorption of another photon of a different wavelength.33 While the ability for PCB bound PhyB (PhyB·PCB) to isomerize upon red light absorption and reverse upon far-red light absorption has been previously shown,5 whether the PhyB(1–621)-DBD and PIF3(1–524)-AD interaction was reversible by far-red light when expressed in mammalian cells has not been tested.13,50 To test the reversibility of the switch, we exposed HEK293 cells, transfected with the PhyB-PIF3 switch and endogenously producing PCB constructs (Figure 5A), to either 24 h of red light, 12 h of red light followed by 12 h of darkness, or 12 h of red light followed by 12 h of far-red light (Figure 5B). Luciferase expression was significantly lower in cells shifted into darkness after 12 h of continuous red light than cells exposed to 24 h of light (p < 0.05), indicating PhyB reversed to its inactive state once red-light illumination ended. Compared to switching from red light to darkness, switching from red to far-red light showed significantly lower luciferase expression, indicating that the far-red light inactivated the gene switch (red box, p < 0.05). This result indicates that after red light activation, the switch remains on for some time in the darkness and that it can be switched off with far-red light. This finding has important implications for the switch’s ability to control genes since it shows that the gene expression levels can be titrated temporally by timing the duration of red light or by red light followed by far-red light. Thus, this system can be used for spatial control by patterning red and far-red light for targeted localization of gene activation.34
Figure 5.

PhyB-PIF3 light switch bistability and reversibility with far-red light and performance in several cell types. (A) Plasmids optimized for an endogenous PhyB-PIF3 light switchable promoter. (B) Testing the reversibility of the PhyB-PIF3 light-switchable promoter in mammalian cells. Cells were in darkness, illuminated with 735 nm far-red light, 660 nm red light for 24 h, or with 12 h or red light followed by darkness or followed by far-red light (n = 3). (C) Testing the PhyB-PIF3 light switch in four different cell types. Cells were transfected, then illuminated with red light for 24 h as shown in Figure 3C (n = 4). ho1 = heme oxygenase, pcyA = phycocyanobilin:ferredoxin oxidoreductase, petF = ferredoxin, petH = ferredoxin:oxidoreductase/fnr, IRES = Internal Ribosome Entry Site, MTS = Mitochondrial Targeting Sequence, NLS = Nuclear Localization Sequence, P2A = 2A self-cleaving peptide, AD = Activation Domain, DBD = DNA Binding Domain, R/FR = Red light/Far-red light. Error bars = s.d., (*) = p < 0.05, Statistics were calculated using one-way ANOVA with Bonferroni post-test using GraphPad Prism 5.01. n = individual experiments.
Genetically Encoded PhyB-PIF3 Gene Switch in Several Mammalian Cell Lines
We also tested the PhyB-PIF3 gene switch performance in different cell types containing endogenously produced PCB. We transfected HEK293, hepatocellular carcinoma (HUH-7), HeLa, and mouse fibroblasts (3T3) cells with the PhyB-PIF3 gene switch and HO1+PcyA+Fd+FNR plasmids (pPKm-230 and pPKm-248, respectively). We used 1 μmol/m2/s of red light illumination in a cycle composed of 1 min pulses of red light followed by 4 min of darkness, for a duration of 24 h (Figure 5C). The PhyB-PIF3 switch with endogenously produced PCB activated luciferase about 280-fold in HEK293 cells, 70-fold in HUH-7 cells, 300-fold in HeLa cells and 440-fold in 3T3 cells. These findings show that the system is effective in producing PCB and activating different mammalian cell types. While we have highly optimized the PhyB-PIF3 light switch with endogenously produced PCB, there are several ways to customize the levels of activation or leakiness to tailor it to specific cell types and applications. For example, different activation or repression domains could be used (Figure S3). In addition, there are still other permutations of gene fusions that can be tested in future studies that may further enhance this system, such as using a DBD on the N-terminus of PhyB or optimizing linker sequences. Using a stronger or tissue-specific promoter to drive expression of PCB or PΦB biosynthetic enzymes may also lead to higher activation levels or can restrict light sensitivity to specific cell types.51 As presented in this research, using wavelengths that are optimal for tissue penetration,10,12 the PhyB(1–621)-PIF3 gene switch with endogenously produced PCB is among the most light-sensitive optogenetic switches.
Summary
We have shown that the Fd+FNR system is the rate-limiting factor for the production of the chromophores PCB and PΦB in the mitochondria of mammalian cells, and is limited by the Fd+FNR system followed by heme in the cytoplasm. The ability to produce PCB and PΦB with PcyA and HY2, respectively, suggests that matching reduction systems that efficiently supply electrons to a metabolic pathway can also enhance the production of other bilins and other classes of molecules. This finding creates new opportunities for engineering synthetic systems to produce these chromophores, along with many other molecules. This has potential industrial applications in decreasing costs of crop production, producing plant molecules in microbes, or delivering therapeutic molecules via genetically encoded pathways.
Genetically encoding endogenous production of chromophores like PCB also enables the use of several existing and compatible optogenetic tools to regulate cell signaling,13,52 cell migration,13 or protein localization13 without the addition of exogenous chemicals. This makes possible the use of PhyB when constant levels of PCB are required, facilitating potential in vivo applications, or when the addition of PCB to samples is not practical (such as when samples are in a sealed container or for long illumination times). This study achieves the long-sought goals in optogenetics of enabling high-level production of the chromophores PCB and PΦB in mammalian cells and demonstrates a more general method for efficiently producing molecules from one species in another.
Methods
Zinc-PAGE-Immunoprecipitation Assays
Protein G PLUS-Agarose (ThermoFisher, 22851) beads were prepared by adding 200 μg anti-HA (clone HA-7, Sigma H9658) into 2 mL 25% agarose. After overnight binding at 4 °C, unbound anti-HA was washed off four times with 1× Phosphate-buffered Saline (PBS, pH 7.4, ThermoFisher, 10010023). For each 6-well plate, 500 000 HEK293 cells (ATCC, CRL-1573) were transfected using 2.5 μg DNA and 6 μL of Lipofectamine 2000 per well (ThermoFisher Scientific, 11668019). For heme experiments, media or media containing 10 μM heme (Frontier Scientific, H651–9), was exchanged 18 h after transfection and again 43 h after transfection. Heme was dissolved at 10 mM in 100 mM NaOH and sterile filtered with a 0.22 μM filter (Millipore, SLGP033RS). Cells were then harvested with RIPA buffer (1% Triton X-100, 0.5% Sodium Deoxycholate, 25 mM Tris pH 8.0, 150 mM NaCl, 0.10% SDS and 2.5 mM EDTA, and 2× protease inhibitors (Sigma, P8340–1 ML)), immediately placed on ice, sonicated briefly and then centrifuged for 30 min at 21 000g. BCA assays (ThermoFisher Scientific, 23225) were used to determine the protein concentration of resulting supernatant/lysates. Equal masses for each protein sample were diluted with two parts of cold PBS, then loaded onto Protein G PLUS-Agarose beads containing anti-HA (preparation above), for overnight binding while mixing at 4 °C. Next beads were washed and boiled in sample buffer (30% glycerol, 10% SDS, 300 mM Tris pH 6.8, 0.03% Bromophenol Blue, 179 mM 2-Mercaptoethanol). After loading and running the samples in a SDS-PAGE gel, the gels were incubated in SDS-PAGE Running Buffer (25 mM Tris, 192 mM glycine, 0.1% SDS) containing 10 mM Zinc Acetate for 10 min prior to imaging in a Fluorochem E (Protein Simple). Gels were then transferred onto nitrocellulose and probed with the primary antibody anti-HA 1:5000 (Sigma, clone HA-7, H9658), and by Goat anti-Mouse secondary antibody 1:5000 (ThermoFisher, 32230). Western blots were imaged in a Fluorochem E (Protein Simple). Gel bands were quantified using the FIJI (ImageJ) gel analysis tool.53
Imaging PCB Production
HEK293 cells (ATCC, CRL-1573), plated at 100 000 cells per well in a 24-well plate, were transfected 24 h after plating on polylysine (Sigma P6407–5 mg) coated coverslips in each well. 43 h later, the media was exchanged with fresh media or media+5 μM PCB (Frontier Scientific, P14137) for the NE+PCB control. One hour later, cells were rinsed in 1× PBS and then fixed in 4% Paraformaldehyde in 1× PBS for 10 min. Cells were then washed with 1× PBS before incubating in permeabilization buffer (5% BSA + 0.3% TritonX-100 in PBS) for 30 min, followed by incubating with primary antibodies, anti-FLAG mouse monoclonal 1:1000 (Sigma, F3165) and polyclonal anti-HA rabbit 1:500 (Santa Cruz, Y-11) in antibody buffer (2% BSA + 0.2% TritonX-100 in PBS) at 4 °C overnight. Next coverslips were rinsed twice and washed three times in 1× PBS and then incubated in antibody buffer containing goat anti-mouse AlexaFluor 488 1:1000 (ThermoFisher, A11001), and goat anti-rabbit AlexaFluor 568 1:1000 (ThermoFisher, A11011). Coverslips were rinsed and washed again, then mounted with Fluoromount-G (SouthernBiotech, 0100–20). Images were taken using a DeltaVision RT Deconvolution Microscope.
Cell Culture, Transfection, Light Induction and Reporter Gene Assays
Human Embryonic Kidney 293 cells (HEK293, ATCC CRL-1573) were cultivated in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, 11965–092) supplemented with 10% fetal bovine serum (FBS, Omega Scientific, FB-02) and 100 U/mL of penicillin and 0.1 mg/mL of streptomycin (Gibco, 11548876). All cells were cultured under 5% CO2 at 37 °C. Cells were seeded at 100 000 HEK293 cells per well in 24-well plates, 24 h before transfection. Transfection of plasmids was achieved through lipofection following the manufacturer’s instructions and protocol (Lipofectamine 2000, ThermoFisher, 11668019). For each transfection reaction, a total of 0.5 μg of plasmid DNA was combined with specific plasmid ratios for each experiment as detailed in Table S2. A construct with Renilla luciferase reporter plasmid DNA was included as an internal transfection control in all transfections. The culture medium was replaced with fresh medium 24 h after transfection and the plates were placed inside black boxes (Hammond Manufacturing Company, 1591ESBK) for the remainder of the experimental procedure. For conditions where external PCB is added, 15 μM of PCB (Frontier Scientific, P14137) from a 20 mM stock dissolved in DMSO (Santa Cruz Biotechnology, sc-202581) was supplemented in fresh medium 24 h after transfection (Figure S2A).
Light induction was programmed to start 12 h after medium replacement. Each black box was equipped with a circuit consisting of six red LEDs (660 nm, Thorlabs, M660L3), except for the dark boxes and far-red boxes which had no LEDs or a single far-red LED (735 nm, Thorlabs, M735L2), respectively. In addition, each black box circuit was designed to allow for fine adjustment of light intensity (circuitry is shown Figure S5), from 0.0008 to 200 μmol/m2/s. Light intensity was measured in μW at the cell level, converted to μmol/m2/s (light sensor area = 63.6 mm2), and adjusted for each experiment design using Sper Scientific Direct’s Laser Power Meter (SSD, 8400). Detailed information on wavelengths, illumination intensity, and duration used for each experimental procedure and data shown are detailed in Table S6. Pulse duration and total illumination times were electronically controlled via a LabVIEW computer driving an Arduino microprocessor and custom-made circuits (see Supporting Information).
Luciferase Activity Assay
Luciferase assays were carried out using the Dual-Luciferase Assay system (Promega, PRE1960), and following the manufacturer’s protocol. Cells were lysed immediately after removing from the incubator using the manufacturer’s instructions. Firefly and Renilla Luciferase activities were measured from cell lysates using the luminometer module of the Infinite 200 PRO multimode reader (Tecan). Results of luciferase activity assays are expressed as a ratio of firefly luciferase (Fluc) activity to Renilla luciferase (Rluc) activity.
Illumination Circuits and Software
The light control system employs an Arduino Uno and a light intensity control circuit (Figure S6) driven by a user interface developed in LabVIEW (National Instruments) to control each box’s LED intensity (Figure S5). This system is ideal for precise timing and light-intensity control of each experimental box while allowing for user-determined experimental start delay, illumination frequencies, and control of the total duration of the experiment. Supporting Information contains a full description of the illumination apparatus, user interface, and circuitry.
Kinetic Model
Using PySB,54 we generated an in silico model to describe the biochemical interactions among the enzymes that compose the hypothesized PCB-production pathway, as seen in Figure 1A. The quantitative mathematical model was parametrized (Table S4) by experimental data and uses ordinary differential equations to describe the changes in the concentration of the molecular components of the reaction. We probed the proposed model directly as proposed in the literature and similar pathways published.17,25,43 We complement this work showing the model’s agreement with the tested pathway, demonstrating how heme, Fd, and FNR are rate limiting factors for the production of PCB, as confirmed experimentally in Figures 1, 2 and 3. A full description of the kinetic model can be found in Supporting Information.
Marvin
Marvin was used for drawing and displaying chemical structures in Figure 1A and Graphical Abstract, Marvin 17.28.0, 2017, ChemAxon (http://www.chemaxon.com).
Acknowledgments
We acknowledge Wilfried Weber for his kind gift of plasmids (pKM-087, pKM-022, and pMZ-802), William McGinnis for his kind gift of the Fluc minimal promoter, Steven P. Briggs for the MTAD construct, Peter Quail for PhyB and PIF3 genes, and Alexey Veraksa for helping initiate this project with P.K. Supported by the Kavli Institute for Brain and Mind at UC San Diego and the Salk Institute, National Science Foundation through the NSF Center for Science of Information under Grant CCF-0939370, NIH Grant NS060847, UCSD School of Medicine Microscopy Core Grant P30 NS047101. P.K. was supported by Training in Multiscale Analysis of Biological Structure and Function, Training Grant: NIH grant T32 EB009380 and by the UCSD Cellular and Molecular Genetics Training Program through an institutional grant from the National Institute of General Medicine (T32 GM007240). M.C. was a fellow of the AAUW (American Association of University Women) 2015 International Fellowship.
Glossary
Abbreviations
- PCB
phycocyanobilin
- PΦB
phytochromobilin
- Fd
ferredoxin
- FNR
ferredoxin-NADP+-reductase
- Fd+FNR
endogenous oxidation–reduction system containing Fd and FNR
- PcyA
PcyAphycocyanobilin:phycocyanobilin:ferredoxin oxidoreductase
- HY2
phytochromobilin:ferredoxin oxidoreductase
- BV
biliverdin IX-alpha
- HO1
heme oxygenase
- PhyB
phytochrome B
- PIF
phytochrome-interacting factor
- PhyB·PCB
PCB-bound PhyB
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.7b00413.
Illumination circuits and software; Kinetic model development and parametrization; Table S1: Similarity tables for ferredoxin and ferredoxin-dependent bilin reductases; Table S2: Plasmids used in this study; Table S3: Transfection and illumination details for of the study’s figures; Table S4: Parameters for the model; Figure S1: Imaging PCB production in mammalian cells; Figure S2: Optimizing PhyB and PIF light switches for mammalian cells; Figure S3: Optimizing PhyB and PIF gene switch; Figure S4: Comparing reporter constructs; Figure S5: Illumination setup; Figure S6: Circuit design for LED illumination; Figure S7: Model results (Initial Heme and Fd activation levels); Figure S8: Model results (Fd dependence and Heme dependence) (PDF)
Author Contributions
¶ P.K. and M.C. contributed equally to this work. P.K. conceived the original concept of mitochondrial targeting for connecting PCYA-HO1 for Fd-FNR and using it to genetically encode the PhyB-PIF3 gene switch. P.K., M.C., N.H., and T.P.C. initiated this project. M.C., W.T., L.N., and V.J.H. performed light induction experiments. P.K., M.C., N.H., V.P., C.S., L.E.D., A.H., and L.N., cloned the plasmids. M.C. modeled the metabolic pathways. M.C. and V.J.H. designed and built illumination apparatus and software. P.K. ran Zn-PAGE experiments; P.K. and L.E.D. imaged PCB in cells. G.P. provided expertise on experimental design and critical feedback. P.K., M.C., W.T., T.P.C., N.H., and G.P. wrote the manuscript with input from other authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Gaspar H. B.; Cooray S.; Gilmour K. C.; Parsley K. L.; Zhang F.; Adams S.; Bjorkegren E.; Bayford J.; Brown L.; Davies E. G.; Veys P.; Fairbanks L.; Bordon V.; Petropoulou T.; Kinnon C.; Thrasher A. J. (2011) Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci. Transl. Med. 3, 97ra80. 10.1126/scitranslmed.3002716. [DOI] [PubMed] [Google Scholar]
- Zhou X. Y.; Morreau H.; Rottier R.; Davis D.; Bonten E.; Gillemans N.; Wenger D.; Grosveld F. G.; Doherty P.; Suzuki K.; Grosveld G. C.; d Azzo A. (1995) Mouse model for the lysosomal disorder galactosialidosis and correction of the phenotype with overexpressing erythroid precursor cells. Genes Dev. 9, 2623–2634. 10.1101/gad.9.21.2623. [DOI] [PubMed] [Google Scholar]
- Burén S.; Young E. M.; Sweeny E. A.; Lopez-Torrejón G.; Veldhuizen M.; Voigt C. A.; Rubio L. M. (2017) Formation of Nitrogenase NifDK Tetramers in the Mitochondria of Saccharomyces cerevisiae. ACS Synth. Biol. 6, 1043–1055. 10.1021/acssynbio.6b00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani D.; DellaPenna D. (1998) Elevating the vitamin E content of plants through metabolic engineering. Science 282, 2098–2100. 10.1126/science.282.5396.2098. [DOI] [PubMed] [Google Scholar]
- Shimizu-Sato S.; Huq E.; Tepperman J. M.; Quail P. H. (2002) A light-switchable gene promoter system. Nat. Biotechnol. 20, 1041–1044. 10.1038/nbt734. [DOI] [PubMed] [Google Scholar]
- Wang X.; Chen X.; Yang Y. (2012) Spatiotemporal control of gene expression by a light-switchable transgene system. Nat. Methods 9, 266–269. 10.1038/nmeth.1892. [DOI] [PubMed] [Google Scholar]
- Müller K.; Engesser R.; Metzger S.; Schulz S.; Kämpf M. M.; Busacker M.; Steinberg T.; Tomakidi P.; Ehrbar M.; Nagy F.; Timmer J.; Zubriggen M. D.; Weber W. (2013) A red/far-red light-responsive bi-stable toggle switch to control gene expression in mammalian cells. Nucleic Acids Res. 41, e77. 10.1093/nar/gkt002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak G. P.; Strickland D.; Vrana J. D.; Tucker C. L. (2014) Benchmarking of optical dimerizer systems. ACS Synth. Biol. 3, 832–838. 10.1021/sb500291r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folcher M.; Oesterle S.; Zwicky K.; Thekkottil T.; Heymoz J.; Hohmann M.; Christen M.; Daoud El-Baba M.; Buchmann P.; Fussenegger M. (2014) Mind-controlled transgene expression by a wireless-powered optogenetic designer cell implant. Nat. Commun. 5, 5392. 10.1038/ncomms6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaberniuk A. A.; Shemetov A. A.; Verkhusha V. V. (2016) A bacterial phytochrome-based optogenetic system controllable with near-infrared light. Nat. Methods 13, 591–597. 10.1038/nmeth.3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden E. S.; Zhang F.; Bamberg E.; Nagel G.; Deisseroth K. (2005) Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268. 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- Lin J. Y.; Knutsen P. M.; Muller A.; Kleinfeld D.; Tsien R. Y. (2013) ReaChR: a red-shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation. Nat. Neurosci. 16, 1499–1508. 10.1038/nn.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levskaya A.; Weiner O. D.; Lim W. A.; Voigt C. A. (2009) Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 461, 997–1001. 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.; Gibson E. S.; Kennedy M. J. (2013) A light-triggered protein secretion system. J. Cell Biol. 201, 631–640. 10.1083/jcb.201210119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiltoir J. I.; Strickland D.; Glotzer M.; Tucker C. L. (2016) Optical control of peroxisomal trafficking. ACS Synth. Biol. 5, 554–560. 10.1021/acssynbio.5b00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. X.; Chung H. K.; Lam A. J.; Lin M. Z. (2012) Optical control of protein activity by fluorescent protein domains. Science 338, 810–814. 10.1126/science.1226854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller K.; Engesser R.; Timmer J.; Nagy F.; Zurbriggen M. D.; Weber W. (2013) Synthesis of phycocyanobilin in mammalian cells. Chem. Commun. (Cambridge, U. K.) 49, 8970–8972. 10.1039/c3cc45065a. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Romero J.; Hedtke M.; Kastner C.; Müller S.; Fischer R. (2010) Fungi, hidden in soil or up in the air: light makes a difference. Annu. Rev. Microbiol. 64, 585–610. 10.1146/annurev.micro.112408.134000. [DOI] [PubMed] [Google Scholar]
- Karniol B.; Wagner J. R.; Walker J. M.; Vierstra R. D. (2005) Phylogenetic analysis of the phytochrome superfamily reveals distinct microbial subfamilies of photoreceptors. Biochem. J. 392, 103–116. 10.1042/BJ20050826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auldridge M. E.; Forest K. T. (2011) Bacterial phytochromes: more than meets the light. Crit. Rev. Biochem. Mol. Biol. 46, 67–88. 10.3109/10409238.2010.546389. [DOI] [PubMed] [Google Scholar]
- Rockwell N. C.; Su Y. S.; Lagarias J. C. (2006) Phytochrome structure and signaling mechanisms. Annu. Rev. Plant Biol. 57, 837–858. 10.1146/annurev.arplant.56.032604.144208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankenberg N.; Mukougawa K.; Kohchi T.; Lagarias J. C. (2001) Functional genomic analysis of the HY2 family of ferredoxin-dependent bilin reductases from oxygenic photosynthetic organisms. Plant Cell 13, 965–978. 10.2307/3871353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohchi T.; Mukougawa K.; Frankenberg N.; Masuda M.; Yokota A.; Lagarias J. C. (2001) The Arabidopsis HY2 gene encodes phytochromobilin synthase, a ferredoxin-dependent biliverdin reductase. Plant Cell 13, 425–436. 10.2307/3871286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beale S. I. (1993) Biosynthesis of phycobilins. Chem. Rev. 93, 785–802. 10.1021/cr00018a008. [DOI] [Google Scholar]
- Gambetta G. A.; Lagarias J. C. (2001) Genetic engineering of phytochrome biosynthesis in bacteria. Proc. Natl. Acad. Sci. U. S. A. 98, 10566–10571. 10.1073/pnas.191375198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooley A. J.; Cai Y. A.; Glazer A. N. (2001) Biosynthesis of a fluorescent cyanobacterial C-phycocyanin holo-alpha subunit in a heterologous host. Proc. Natl. Acad. Sci. U. S. A. 98, 10560–10565. 10.1073/pnas.181340998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukougawa K.; Kanamoto H.; Kobayashi T.; Yokota A.; Kohchi T. (2006) Metabolic engineering to produce phytochromes with phytochromobilin, phycocyanobilin, or phycoerythrobilin chromophore in Escherichia coli. FEBS Lett. 580, 1333–1338. 10.1016/j.febslet.2006.01.051. [DOI] [PubMed] [Google Scholar]
- Landgraf F. T.; Forreiter C.; Hurtado Picó A.; Lamparter T.; Hughes J. (2001) Recombinant holophytochrome inEscherichia coli. FEBS Lett. 508, 459–462. 10.1016/S0014-5793(01)02988-X. [DOI] [PubMed] [Google Scholar]
- Shin A. Y.; Han Y. J.; Song P. S.; Kim J. I. (2014) Expression of recombinant full-length plant phytochromes assembled with phytochromobilin in Pichia pastoris. FEBS Lett. 588, 2964–2970. 10.1016/j.febslet.2014.05.050. [DOI] [PubMed] [Google Scholar]
- Frankenberg N.; Lagarias J. C. (2003) Phycocyanobilin:ferredoxin oxidoreductase of Anabaena sp. PCC 7120. Biochemical and spectroscopic. J. Biol. Chem. 278, 9219–9226. 10.1074/jbc.M211643200. [DOI] [PubMed] [Google Scholar]
- Sheftel A. D.; Stehling O.; Pierik A. J.; Elsässer H. P.; Mühlenhoff U.; Webert H.; Hobler A.; Hannemann F.; Bernhardt R.; Lill R. (2010) Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 107, 11775–11780. 10.1073/pnas.1004250107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliverti A.; Pandini V.; Pennati A.; de Rosa M.; Zanetti G. (2008) Structural and functional diversity of ferredoxin-NADP(+) reductases. Arch. Biochem. Biophys. 474, 283–291. 10.1016/j.abb.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Smith R. W.; Helwig B.; Westphal A. H.; Pel E.; Hörner M.; Beyer H. M.; Samodelov S. L.; Weber W.; Zurbriggen M. D.; Borst J. W.; Fleck C. (2016) Unearthing the transition rates between photoreceptor conformers. BMC Syst. Biol. 10, 110. 10.1186/s12918-016-0368-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrian M.; Nijenhuis W.; Hoogstraaten R. I.; Willems J.; Kapitein L. C. (2017) A Phytochrome-Derived Photoswitch for Intracellular Transport. ACS Synth. Biol. 6, 1248–1256. 10.1021/acssynbio.6b00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke G.; Mulo P. (2013) Plant type ferredoxins and ferredoxin-dependent metabolism. Plant, Cell Environ. 36, 1071–1084. 10.1111/pce.12046. [DOI] [PubMed] [Google Scholar]
- Cahoon E. B.; Shanklin J. (2000) Substrate-dependent mutant complementation to select fatty acid desaturase variants for metabolic engineering of plant seed oils. Proc. Natl. Acad. Sci. U. S. A. 97, 12350–12355. 10.1073/pnas.210276297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curatti L.; Rubio L. M. (2014) Challenges to develop nitrogen-fixing cereals by direct nif-gene transfer. Plant Sci. 225, 130–137. 10.1016/j.plantsci.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Rekittke I.; Olkhova E.; Wiesner J.; Demmer U.; Warkentin E.; Jomaa H.; Ermler U. (2013) Structure of the (E)-4-hydroxy-3-methyl-but-2-enyl-diphosphate reductase from Plasmodium falciparum. FEBS Lett. 587, 3968–3972. 10.1016/j.febslet.2013.10.029. [DOI] [PubMed] [Google Scholar]
- Pinto R.; Harrison J. S.; Hsu T.; Jacobs W. R.; Leyh T. S. (2007) Sulfite reduction in mycobacteria. J. Bacteriol. 189, 6714–6722. 10.1128/JB.00487-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura-Sakakibara K.; Onda Y.; Ashikari T.; Tanaka Y.; Kusumi T.; Hase T. (2000) Analysis of reductant supply systems for ferredoxin-dependent sulfite reductase in photosynthetic and nonphotosynthetic organs of maize. Plant Physiol. 122, 887–894. 10.1104/pp.122.3.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uda Y.; Goto Y.; Oda S.; Kohchi T.; Matsuda M.; Aoki K. (2017) Efficient synthesis of phycocyanobilin in mammalian cells for optogenetic control of cell signaling. Proc. Natl. Acad. Sci. U. S. A. 114, 11962–11967. 10.1073/pnas.1707190114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu F. Y.; Chen Y. R.; Tu S. L. (2010) Electrostatic interaction of phytochromobilin synthase and ferredoxin for biosynthesis of phytochrome chromophore. J. Biol. Chem. 285, 5056–5065. 10.1074/jbc.M109.075747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K. (2009) HO1 and PcyA proteins involved in phycobilin biosynthesis form a 1:2 complex with ferredoxin-1 required for photosynthesis. FEBS Lett. 583, 1251–1256. 10.1016/j.febslet.2009.03.052. [DOI] [PubMed] [Google Scholar]
- Szymczak A. L.; Workman C. J.; Wang Y.; Vignali K. M.; Dilioglou S.; Vanin E. F.; Vignali D. A. (2004) Correction of multi-gene deficiency in vivo using a single “self-cleaving” 2A peptide-based retroviral vector. Nat. Biotechnol. 22, 589–594. 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- Licursi M.; Christian S. L.; Pongnopparat T.; Hirasawa K. (2011) In vitro and in vivo comparison of viral and cellular internal ribosome entry sites for bicistronic vector expression. Gene Ther. 18, 631–636. 10.1038/gt.2011.11. [DOI] [PubMed] [Google Scholar]
- Bochkov Y.; Palmenberg A. (2006) Translational efficiency of EMCV IRES in bicistronic vectors is dependent upon IRES sequence and gene location. BioTechniques 41, 283–292. 10.2144/000112243. [DOI] [PubMed] [Google Scholar]
- Mizuguchi H.; Xu Z.; Ishii-Watabe A.; Uchida E.; Hayakawa T. (2000) IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol. Ther. 1, 376–382. 10.1006/mthe.2000.0050. [DOI] [PubMed] [Google Scholar]
- Li J.; Li G.; Wang H.; Wang Deng X. (2011) Phytochrome signaling mechanisms. Arabidopsis Book 9, e0148. 10.1199/tab.0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattis J.; Tye K. M.; Ferenczi E. A.; Ramakrishnan C.; O’Shea D. J.; Prakash R.; Gunaydin L. A.; Hyun M.; Fenno L. E.; Gradinaru V.; Yizhar O.; Deisseroth K. (2011) Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat. Methods 9, 159–172. 10.1038/nmeth.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer H. M.; Juillot S.; Herbst K.; Samodelov S. L.; Müller K.; Schamel W. W.; Römer W.; Schäfer E.; Nagy F.; Strähle U.; Weber W.; Zurbriggen M. D. (2015) Red Light-Regulated Reversible Nuclear Localization of Proteins in Mammalian Cells and Zebrafish. ACS Synth. Biol. 4, 951–958. 10.1021/acssynbio.5b00004. [DOI] [PubMed] [Google Scholar]
- Qin J. Y.; Zhang L.; Clift K. L.; Hulur I.; Xiang A. P.; Ren B. Z.; Lahn B. T. (2010) Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS One 5, e10611. 10.1371/journal.pone.0010611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toettcher J. E.; Gong D.; Lim W. A.; Weiner O. D. (2011) Light-based feedback for controlling intracellular signaling dynamics. Nat. Methods 8, 837–839. 10.1038/nmeth.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J.; Arganda-Carreras I.; Frise E.; Kaynig V.; Longair M.; Pietzsch T.; Preibisch S.; Rueden C.; Saalfeld S.; Schmid B.; Tinevez J. Y.; White D. J.; Hartenstein V.; Eliceiri K.; Tomancak P.; Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez C. F.; Muhlich J. L.; Bachman J. A.; Sorger P. K. (2013) Programming biological models in Python using PySB. Mol. Syst. Biol. 9, 646. 10.1038/msb.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.