

Abstract
Introduction:
We aimed to assess the effect of anti-tyrosine kinase inhibitors (TKIs) (gefitinib) in overall survival (OS) of the glioblastoma multiforme (GBM) patients in the backdrop of mutational status of epidermal growth factor receptor (EGFR) and PTEN genes.
Materials and Methods:
All the patients subjected to resection or biopsies were put on gefitinib, and radiotherapy was delivered as per the hospital protocol. EGFR and PTEN mutational spectrum was performed by single-strand conformation polymorphism followed by DNA sequencing.
Results:
In total, 50% GBM tumors had mutation either in EGFR or PTEN. Median progression-free survival (PFS) and OS observed in patients with EGFR +ve/PTEN −ve were significantly favorable (P < 0.05) which aggregated to 9(7, 11) months and 20 (16, 24) months, respectively, than 6 (4, 8) months and 13 (7, 19) months in patients with PTEN +ve/EGFR −ve. Patients positive for both EGFR/PTEN had lower disease-free survival and OS of 6 and 9 months as compared to 6 (5, 7) and 14 (12, 24) months for those negative for both EGFR/PTEN.
Conclusions:
We conclude that EGFR gene alterations with wild-type PTEN are associated with significantly better PFS and OS in patients treated with anti-TKIs (gefitinib). Combined EGFR and PTEN gene mutation is associated with significantly poor response to gefitinib in terms of median OS.
Keywords: Anti-tyrosine kinases, DNA sequencing, epidermal growth factor receptor, gefitinib, glioblastoma multiforme, mutations, PTEN
Introduction
Glioblastoma multiforme (GBM) presents as frequent and aggressive type of primary brain tumor.[1] Among brain tumors in adults, GBM is the foremost common and found to be implicated more common in western population and among men.[2,3] The incidence of GBM in India is nearly 3/105 population whereas another estimate accounts for 2–5 new cases/105/year.[4] In Kashmir (North India), among the brain tumors, glioma (60%) is most common in which GBM is the most common followed by diffuse and anaplastic astrocytoma.[5] GBM is the foremost deadly sorts of cancers with a median survival of 10–12 months.[6] In contrast to most different kinds of cancer, GBM seldom metastasizes; rather, they induce death through putting resistance to current therapies and invasion into traditional brain tissues.[7]
Commonly known two genetic pathways have been established in GBM development: de novo from glial cells and is mostly common in older patients and secondary GBM develops over months to years from preexisting low-grade astrocytomas and mostly affects younger patients.[8,9] GBM involves a multistep process that goes through a series of potential genetic alterations. Among these, primary GBM tumors exhibit overexpression (>60% of cases) or amplification (>40% of cases) of the epidermal growth factor (EGF) gene[10,11] and prominently include deletion or mutation of the PTEN gene.[9] Owing to failure of treatment for GBM by conventional cytotoxic chemotherapy and radiotherapy, a considerable amount of knowledge for aberrant signaling pathways involved in GBM has elucidated new potential therapeutic targets. These targeted drug therapies may augment better treatment modalities for patients with GBM, which particularly involves EGF receptor (EGFR) inhibitors currently being tested in clinical trials.[11]
Preliminary reports of targeted molecular therapies in GBM have concentrated on the inhibition of tyrosine kinases and growth factor pathways associated with it. In this scenario, gefitinib, a selective small-molecule inhibitor of the EGFR,[12] is generally well tolerated, and patients with GBM in initial clinical trials on this drug produced partial responses after previous radiotherapy.[13] Varied responses have been demonstrated in the backdrop of loss of the PTEN gene and EGFR wherein loss of the former was highly correlated with treatment failure. Evidences show that co-expression of EGFRvIII and PTEN strikingly predicted treatment responses.[14] Now, it seems plausible that PTEN loss could promote resistance to EGFR kinase inhibitors by dissociating EGFR/EGFRvIII inhibition from downstream inhibition of the PI3K signaling pathway.[15]
Thus, in the backdrop of mutational status of EGFR and PTEN genes, the aim of this study conducted first time from Indian subcontinent was to assess the effect of anti-tyrosine kinase inhibitor (TKI) (gefitinib) in combination with surgery on the recurrence and overall survival (OS) of the GBM patients
Materials and Methods
Patients
The present study was carried jointly by the Departments of Neurosurgery, Medical Oncology and Immunology, and Molecular Medicine, Sher-i-Kashmir Institute of Medical Sciences, Srinagar, Jammu and Kashmir, between 2009 and 2012. All consecutive patients with the GBM seen at our institution were considered for the study, and the sample size was calculated as per the hospital records which showed a power of the study >75.
Patients were included in the study after written informed consent. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards, and ethical approval was obtained from Institutional Ethical Committee.
The surgically resected tissue samples taken through stereotactic/open biopsy of GBM tumors, were collected directly into sterile vials containing chilled phosphate-buffered saline (pH = 7.2), and frozen at 70°C for molecular investigations. The normal brain tissue was a 2 mm × 2 mm × 1 mm block procured while performing corticectomy for the same lesion.
After entry into the study, patients were evaluated for detailed history, physical and systemic examination. All the patients were subjected to radiological examinations such as X-ray chest, contrast-enhanced computed tomography (CECT) brain, and contrast-enhanced magnetic resonance imaging (MRI) brain.
All the patients were subjected to gross-total resection, subtotal resection, or biopsy depending on the patient's status and tumor location. Once the pathology was confirmed, all the patients were put on gefitinib at an initial oral dose of 250 mg/day[16] and radiotherapy was delivered as per the hospital protocol. Patients were treated with concurrent chemoradiotherapy that included temozolomide. Radiotherapy was delivered as 60 Grays in 30 fractions at 2 Gray per fraction, 5 days a week for a period of 6 weeks. The gross tumor volume (GTV) was determined by pre- and post-operative MRI imaging using enhanced T1 and fluid-attenuated inversion recovery/T2. The GTV was expanded by 2–3 cm to generate clinical target volume, to account for subdiagnostic tumor infiltration. Radiation fields were reduced after 46 Grays to prescribe boost radiation to gross disease. Patients received oral temozolomide 75 mg/m2/day for the duration of radiotherapy. Four weeks after the completion of concurrent chemoradiotherapy, patients received 3-weekly six cycles (175 mg/m2 orally daily 5 days) of temozolomide. All patients received oral premedication during treatment. Patients who received dexamethasone and/or enzyme-inducing antiepileptic drugs without toxicities after 2 weeks of receiving gefitinib had the gefitinib dose escalated to 500 mg/day. Therapy was continued until disease progression, significant clinical decline, unacceptable toxicity, or patient decision. Toxicity was graded using the National Cancer Institute Common Toxicity Criteria, version 2.0.[17]
For Grade 2 skin rashes and diarrhea that were unacceptable to the patient for symptomatic reasons, gefitinib was temporarily withheld until resolution and subsequently restarted at the same dose. If symptomatic Grade 2 diarrhea and skin rash recurred after reinstituting gefitinib at the same dose, treatment was held until resolution to Grade 1 or less, and gefitinib was reinstituted at a lower dose. If a patient dose was lowered, no increase was undertaken.
All the patients were monitored initially biweekly, thereafter monthly for complete blood count, liver function tests, kidney function test, and X-ray chest. All the patients were subjected to follow-up MRI/CECT head at the 1st week and every 3 months thereafter. Survival time of the patients was deduced which was taken as the length of time from either the date of diagnosis or the start of treatment for GBM that patients diagnosed with the disease are still alive. Survival time was calculated from the date of first surgery to the date of death or date of last contact if lost to follow-up evaluation. Follow-up ranged from 6 months to a maximum of 25 months. Progression-free survival (PFS) was defined as the time from first surgery to first evidence of tumor progression on CT or MRI or to death whereas tumor progression was defined as the appearance of new lesions, an increase in tumor extension by 25% on CT or MRI, a worsening in the clinical/neurological condition, or an increased need for corticosteroids.[18]
Histopathologically confirmed GBM tissues and corresponding normal tissues were used for mutational analysis of PTEN and EGFR gene.
Detection of mutations in epidermal growth factor receptor and PTEN genes
DNA was extracted from the tissues by phenol-chloroform method and by Qiagen DNA extraction kit (Zymo Research Corporation, USA).
EGFR and PTEN genes were amplified using previously described specific primers [Table 1]. PCR amplification was carried out in a 50 μL volume container with 50 ng of genomic DNA, 1xPCR buffer containing 15 mM MgCl2, 100 mM each of dATP, dGTP, dTTP, dCTP, and 1.5 U of Taq DNA polymerase (Biotools; Madrid, Spain), and 1 μM of forward and reverse primers (Genscript; Piscataway, NJ, USA).
Table 1.
Clinico-pathological characteristics of GBM patients
Results
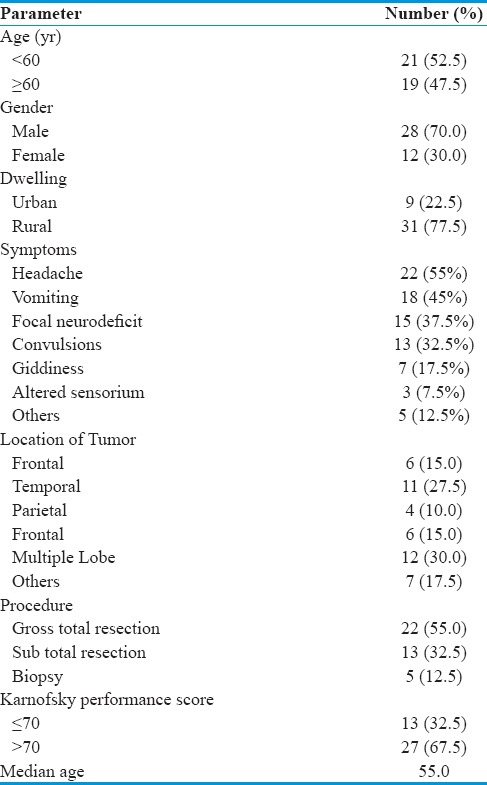
The present study comprised forty histologically confirmed cases of GBM. Majority of the patients, i.e., 28 (70.0%) were male. The common presenting symptoms included headache (55%), vomiting (45%), and convulsions (37.5%), and other symptoms included altered sensorium, amnesia, slurred speech, and forgetfulness and their details are provided in Table 1.
Tumor location on the left side was seen in 19 (47.5%) as against 17 (42.5%) on the right side whereas midline tumors were observed in only 4 (10%) of patients. Majority of the tumors, i.e., 12 (30%) involved more than one lobe whereas single-lobe involvement was most common (11:27.5%) in the temporal lobe. Other sites involved were frontal, parietal, midline, occipital, and thalamic [Table 1]. Majority of the patients, i.e. 22 (55%) were subjected to gross-total resection whereas 13 (32.5%) were subjected to subtotal resection, and biopsy only was performed in 5 (12.5%) patients. Twenty-seven (67.5%) patients had Karnofsky performance score (KPS) of >70 and 13 (32.5%) had ≤70 with mean KPS as 78.2 ± 9.8 [Table 1].
Overall mutations of EGFR and PTEN genes identified in this study have been previously published.[19] The overall frequencies of EGFR and PTEN mutations in GBM patients are shown in Table 2. In total, 50% (20 of 40) GBM tumors studied had mutation of either in EGFR or PTEN gene.
Table 2.
Frequency and distribution of EGFR and PTEN mutations in GBM patients
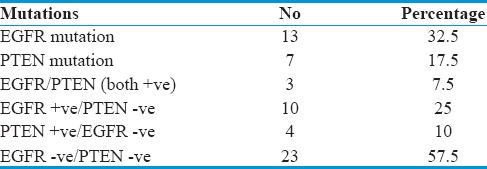
EGFR mutation was present in 13 (32.5%) and PTEN gene mutations in 7 (17.5%) patients. Both EGFR and PTEN mutations were found in 3 samples (7.5%). The samples which showed EGFR mutations and negative for PTEN were detected in 10 (25%) patients (EGFR +ve/PTEN ve). The samples which showed PTEN mutations but absent in EGFR (PTEN +ve/EGFR − ve) were present in 4 (10%) patients. No mutations were seen in both the genes (EGFR/PTEN both − ve) in 23 patients (57.5%) [Table 2].
All the patients were put on gefitinib 250–500 mg/day. Most common side effects noted were rash (26%) and diarrhea (22%). Rash and diarrhea occurred mostly at dose of 500 mg/day. Other toxicities that were encountered included aspartate transaminase and alanine transaminase elevation, conjunctivitis, anorexia, and weight loss. To assess the impact of TKIs, patients put on gefitinib were prospectively followed for their OS and disease-free survival (DFS).
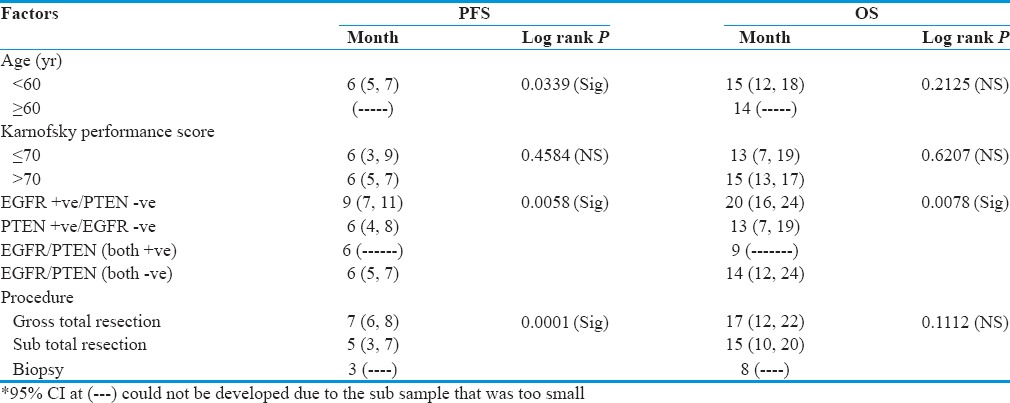
Kaplan–Meier (KM) curves were constructed to assess OS, and differences among groups were analyzed by the log-rank test. Kaplan–Meier survival analysis was performed to evaluate any possible association between mutational spectrum of EGFR/PTEN genes and its impact on treatment with anti-TKIs (gefitinib) and OS of patients. Median PFS was 6 (5, 7) and median OS was 15 (12, 18) months in patients who were <60 years of age compared to 14 months in patients ≥60 years of age [Figure 1a]. There was no difference in the median OS between the two genders in our study. As far as the overall mutations are concerned, they were almost equally present in both the genders [Table 3].
Figure 1.

(a) Kaplan–Meier curve of overall survival and progression-free survival as a function age. (b) Kaplan–Meier curve of overall survival and progression-free survival as a function gender. (c) Kaplan–Meir curve to evaluate effect of mutational spectrum of epidermal growth factor receptor/PTEN as a component of overall survival. (d) Kaplan–Meir curve to evaluate effect of mutational spectrum of epidermal growth factor receptor/PTEN in relation to progression-free survival
Table 3.
Pattern of mutations and survival across age and gender

KP analysis showed that median OS was better, 15 months in patients with KPS >70 compared to 13 (7, 19) months in patients with KPS ≤70 but was not statistically significant. In our study, median PFS was better 7 (6, 8) months in patients with gross-total resection compared to patients who were subjected to subtotal resection 5 (3, 7) months or biopsy only 3(--) months (P > 0.05). Median OS was also better 17 (12, 22) months in patients who were subjected to gross-total resection compared to the patients subjected to subtotal resection 15 (10, 20) months or biopsy only 8 (--) months, respectively [Figure 1b].
Survival score (OS and DFS) was deduced from the GBM patients put on gefitinib by KM curves to evaluate effect of mutational spectrum of EGFR/PTEN [Figure 1c and d]. A better median PFS and median OS were observed in patients who were EGFR + ve/PTEN –ve which aggregated to 9 (7, 11) months and 20 (16, 24) months, respectively, as compared to patients with EGFR − ve/PTEN +ve where survival was 6 (4, 8) months and 13 (7, 19) months, respectively. Patients positive for both EGFR/PTEN (+) had lower DFS and OS of 6 (--) and 9 (--) months whereas patients negative for both EGFR/PTEN (-) had 6 (5, 7) months and 14 (12, 24) months, respectively. The finding was observed to be significantly associated with the response to gefitinib in patients with EGFR +ve/PTEN −ve in terms of survival [Table 4].
Table 4.
Different characters affecting progression free survival and overall survival
Discussion
The association of genetic alterations with brain tumor behavior has aroused multiple investigations into the prognostic value of various genetic markers.[20] The main focus of investigations has lead more deep into GBM pathogenesis, and in this relation, elucidation of the PTEN and EGFR oncogenes has been the primary target of analysis. Recent evidence shows that about 10%–20% of unselected GBM patients showed significant tumor regression in response to EGFR kinase inhibitors.[14,21] A definite connection has been established between a response and resistance to EGFR kinase inhibitors in GBM patients that is mediated by loss of the PTEN gene derangement.[15] To understand the molecular basis for drug response in the backdrop of their mutation spectrum in EGFR and PTEN gene, we conducted a prospective analysis of tumor tissues from patients who responded and/or did not respond to EGFR kinase inhibitor therapy in combination with surgery and radiotherapy to analyze PFS and OS of the GBM patients.
GBM incidence occurs mostly between the sixth and seventh decade, but these tumors appear to show its increasing trend in young adults.[22,23] Incidence in men is approximately 40% more than in women, and accordingly, we observed that in our study, 28 (70%) were male and 12 (30%) were female with a male:female ratio of 2.3:1. All the patients were put on gefitinib 250–500 mg/day. Among the most common side effects, noted rash (26%) and diarrhea (22%) occurred mostly at dose of 500 mg/day, which are in agreement with the previous studies.[24,25,26]
Fukuoka et al. (2003) and Ranson et al. (2002),[26,27] in two large randomized phase II trials, analyzed the effectiveness and toxicity of gefitinib (250 mg or 500 mg/day) wherein the former conducted trial of gefitinib for nonsmall cell lung cancer patients. The study observed no difference between rate of response and survivals following the two completely different dose schedules whereas the adverse event rates were higher in 500 mg/day. In phase I studies, gefitinib at doses lower than 250 mg/day showed better responses and disease stabilizations, and gefitinib at 500 mg/day in intermittent schedules has been shown to be effective.[28,29]
In our series of GBM patients, both PTEN and EGFR gene mutations aggregated to 50% (20 of 40). Although a major proportion of the samples were exclusive for mutations in PTEN and EGFR, three mutations were commonly found in same samples in both genes and were thus observed to be overlapping in 15% of the GBM cases. This shows that a good proportion of GBM cases harbor both mutations implicating EGFR and PTEN as mutually inclusive genetic events. Our study thus is in agreement with Smith et al. who also observed the same frequency of genetic alterations of EGFR and PTEN in GBM patients.[30]
Although in our study, we did not find a major difference in PFS and OS between two age groups, OS was better in patients <60 years of age compared to those with age ≥60 years (P > 0.05). Median PFS could not be calculated in patients with age ≥60 due to the subsample that was too small. Most of the previous studies concluded that patient's age had the greatest effect on survival. In a study conducted by Donato et al.,[31] it was observed that patients under 61 years of age had a significantly prolonged survival. Filippini et al. (2008)[32] observed a strong evidence for the effectiveness of surgery even for elderly patients, provided that they had an adequate performance status.[33]
We observed that median OS was better 15 months in patients with KPS >70 compared to 13 (7, 19) months in patients with KPS ≤70 (P > 0.05). Most of the previous studies have also concluded that median OS is better in patients with good preoperative KPS.[31]
Whether the extent of resection is a factor significantly associated with the survival advantage is much debated, but some reports found that more extensive resection was associated with longer survival[33,34] whereas others showed no relation.[35,36] Our results also support a significant increase in PFS and a nonsignificant increase in OS associated with extensive surgical resection compared with partial resection/biopsy although precise evaluation of PFS after surgery by postoperative imaging was not available in all patients. In our study, median PFS was better 7 (6, 8) months in patients with gross-total resection compared to patients who were subjected to subtotal resection 5 (3, 7) or biopsy only 3 (--) months, (P < 0.05). Median OS was also better 17 (12, 22) months in patients who were subjected to gross-total resection compared to the patients subjected to subtotal resection 15 (10, 20) months or biopsy only 8 (--) months, respectively.
Although gefitinib is generally well tolerated, patients with GBM in initial clinical trials with gefitinib had minimal tumor response and no improvement in OS.[37] In one phase II study, response to gefitinib showed that 13% patients remained progression free for a minimum of 6 months.[37] In the following phase I/II study conducted by the North American Brain Tumor Consortium, a partial response was shown after previous RT in around 13% patients with GBM.[25] In our study, we observed that median PFS and median OS was better 9 (7, 11) and 20 (16, 24) months, respectively, in patients who were EGFR +ve/PTEN –ve as compared to patients with PTEN +ve/EGFR –ve 6 (4, 8) and 13 (7, 19) months, EGFR/PTEN (both +ve) 6 (--) and 9 (--) months, and EGFR/PTEN (both −ve) 6 (5, 7) and 14 (12, 24) months, and this difference was found statistically significant (P < 0.05). Hence, EGFR kinase inhibitor (gefitinib) showed significant response in terms of PFS and OS in our patients who had alterations in EGFR gene and intact PTEN gene. Data till date analyzed from various clinical trials and preclinical models[14,38] report that treatment with EGFR kinase inhibitors is irresponsive in PTEN deficient tumors even if they derive activating EGFR mutations. This potentially causes upfront resistance to EGFR kinase inhibitors in highly PTEN-deficient tumors or when resistance is acquired in molecularly heterogeneous tumors wherein PTEN deficient cells develop a selective growth advantage during chemotherapy.
Median PFS and median OS in patients who were PTEN +ve/EGFR −ve were 6 (4, 8) months and 13 (7, 19) months, respectively, and in patients who were PTEN/EGFR (both +ve) were 6 (--) and 9 (--) months, respectively. Hence, patients with mutation in PTEN gene showed poor response to gefitinib in terms of median PFS and median OS than patients without alterations in PTEN. The fact that EGFR gene derangements with intact wild-type PTEN are associated with significantly better PFS and OS in patients treated with EGFR inhibitors (gefitinib). These observations are in agreement with the previous studies.[39,40,41] The PTEN gene is lost or mutated in ~40% of GBM,[1] and retention of PTEN protein expression has been linked with responses to EGFR TKIs in GBM patients[21] suggesting that the detection of functional PTEN may inform the successful deployment of targeted therapeutics in this currently intractable disease.
Conclusions
We conclude that EGFR gene alterations with wild-type PTEN are associated with significantly better PFS and OS in patients treated with EGFR inhibitors (gefitinib). Combined EGFR and PTEN gene mutation is associated with significantly poor response to gefitinib in terms of median OS. Molecularly targeted therapies can potentially provide novel cancer therapies by selectively inhibiting these aberrant pathways, but this needs to be evaluated in further studies in GBM patients of the ethnic Kashmiri population.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
The authors gratefully acknowledge the financial support provided by the Sher-i-Kashmir Institute of Medical Sciences, Kashmir, for this work. Our thanks are also due to the Technical Staff of the Department of Neurosurgery, Sher-i-Kashmir Institute of Medical Sciences, who helped us in procurement of samples. The study was partially funded by the Sher-i-Kashmir Institute of Medical Sciences Hospital.
References
- 1.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 2.Grossman SA, Batara JF. Current management of glioblastoma multiforme. Semin Oncol. 2004;31:635–44. doi: 10.1053/j.seminoncol.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Uddin S, Jarmi T, Hariharan S. Glioblastoma Multiforme. [Last accessed on 2005 Mar 15]. Available from: http://www.emedicine.com/NEURO/topic147.htm .
- 4.Buckner JC. Factors influencing survival in high-grade gliomas. Semin Oncol. 2003;30(6 Suppl 19):10–4. doi: 10.1053/j.seminoncol.2003.11.031. [DOI] [PubMed] [Google Scholar]
- 5.Bhat AR, Altaf K, Tariq HR, Wani MA, Ramzan AU, Iqbal SJ, et al. Analysis of surgical outcome of brain tumors in Kashmir-A 26 year experience at Skims. J Med Sci. 2008;11:152–8. [Google Scholar]
- 6.Legler JM, Ries LA, Smith MA, Warren JL, Heineman EF, Kaplan RS, et al. Cancer surveillance series [corrected]: Brain and other central nervous system cancers: Recent trends in incidence and mortality. J Natl Cancer Inst. 1999;91:1382–90. doi: 10.1093/jnci/91.16.1382. [DOI] [PubMed] [Google Scholar]
- 7.Giese A, Bjerkvig R, Berens ME, Westphal M. Cost of migration: Invasion of malignant gliomas and implications for treatment. J Clin Oncol. 2003;21:1624–36. doi: 10.1200/JCO.2003.05.063. [DOI] [PubMed] [Google Scholar]
- 8.Kleihues P, Ohgaki H. Primary and secondary glioblastomas: From concept to clinical diagnosis. Neuro Oncol. 1999;1:44–51. doi: 10.1093/neuonc/1.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tysnes BB, Mahesparan R. Biological mechanisms of glioma invasion and potential therapeutic targets. J Neurooncol. 2001;53:129–47. doi: 10.1023/a:1012249216117. [DOI] [PubMed] [Google Scholar]
- 10.Benjamin R, Capparella J, Brown A. Classification of glioblastoma multiforme in adults by molecular genetics. Cancer J. 2003;9:82–90. doi: 10.1097/00130404-200303000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Kesari S, Ramakrishna N, Sauvageot C, Stiles CD, Wen PY. Targeted molecular therapy of malignant gliomas. Curr Neurol Neurosci Rep. 2005;5:186–97. doi: 10.1007/s11910-005-0046-8. [DOI] [PubMed] [Google Scholar]
- 12.Jendrossek V, Belka C, Bamberg M. Novel chemotherapeutic agents for the treatment of glioblastoma multiforme. Expert Opin Investig Drugs. 2003;12:1899–924. doi: 10.1517/13543784.12.12.1899. [DOI] [PubMed] [Google Scholar]
- 13.Lieberman FS, Cloughesy T, Fine H, Kuhn J, Lamborn K, Malkin M, et al. NABTC phase I/II trial of ZD-1839 for recurrent malignant gliomas and unresectable meningiomas. J Clin Oncol. 2004;22:1510. [Google Scholar]
- 14.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–24. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 15.Mellinghoff IK, Cloughesy TF, Mischel PS. PTEN-mediated resistance to epidermal growth factor receptor kinase inhibitors. Clin Cancer Res. 2007;13(2 Pt 1):378–81. doi: 10.1158/1078-0432.CCR-06-1992. [DOI] [PubMed] [Google Scholar]
- 16.Franceschi E, Cavallo G, Lonardi S, Magrini E, Tosoni A, Grosso D, et al. Gefitinib in patients with progressive high-grade gliomas: A multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO) Br J Cancer. 2007;96:1047–51. doi: 10.1038/sj.bjc.6603669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 18.Macdonald DR, Cascino TL, Schold SC Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8:1277–80. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 19.Sajad HA, Arshad AP, Abdul RB, Altaf UR, Nayil KM, Sarabjit SC, et al. EGFR and PTEN gene mutation status in glioblastoma patients and their prognostic impact on patient's survival. J Carcinog Mutagen. 2015;6:2. [Google Scholar]
- 20.Smith JS, Jenkins RB. Genetic alterations in adult diffuse glioma: Occurrence, significance, and prognostic implications. Front Biosci. 2000;5:D213–31. doi: 10.2741/smith. [DOI] [PubMed] [Google Scholar]
- 21.Haas-Kogan DA, Prados MD, Tihan T, Eberhard DA, Jelluma N, Arvold ND, et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005;97:880–7. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- 22.Radhakrishnan K, Mokri B, Parisi JE, O’Fallon WM, Sunku J, Kurland LT. The trends in incidence of primary brain tumors in the population of Rochester, Minnesota. Ann Neurol. 1995;37:67–73. doi: 10.1002/ana.410370113. [DOI] [PubMed] [Google Scholar]
- 23.Bhat AR, Muhammed AW, Kirmani AR, Raina TH, Ramzan AU, Shafiq A, et al. Malignant brain tumors (brain cancer) in orchard farmers of Kashmir linked to pesticides. Curr Neurobiol. 2010;1:137–50. [Google Scholar]
- 24.Baselga J, Rischin D, Ranson M, Calvert H, Raymond E, Kieback DG, et al. Phase I safety, pharmacokinetic, and pharmacodynamic trial of ZD1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with five selected solid tumor types. J Clin Oncol. 2002;20:4292–302. doi: 10.1200/JCO.2002.03.100. [DOI] [PubMed] [Google Scholar]
- 25.Herbst RS, Maddox AM, Rothenberg ML, Small EJ, Rubin EH, Baselga J, et al. Selective oral epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 is generally well-tolerated and has activity in non-small-cell lung cancer and other solid tumors: Results of a phase I trial. J Clin Oncol. 2002;20:3815–25. doi: 10.1200/JCO.2002.03.038. [DOI] [PubMed] [Google Scholar]
- 26.Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected] J Clin Oncol. 2003;21:2237–46. doi: 10.1200/JCO.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 27.Ranson M, Hammond LA, Ferry D, Kris M, Tullo A, Murray PI, et al. ZD1839, a selective oral epidermal growth factor receptor-tyrosine kinase inhibitor, is well tolerated and active in patients with solid, malignant tumors: Results of a phase I trial. J Clin Oncol. 2002;20:2240–50. doi: 10.1200/JCO.2002.10.112. [DOI] [PubMed] [Google Scholar]
- 28.Kris MG, Natale RB, Herbst RS, Lynch TJ, Jr, Prager D, Belani CP, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: A randomized trial. JAMA. 2003;290:2149–58. doi: 10.1001/jama.290.16.2149. [DOI] [PubMed] [Google Scholar]
- 29.Nakagawa K, Tamura T, Negoro S, Kudoh S, Yamamoto N, Yamamoto N, et al. Phase I pharmacokinetic trial of the selective oral epidermal growth factor receptor tyrosine kinase inhibitor gefitinib (’Iressa’, ZD1839) in Japanese patients with solid malignant tumors. Ann Oncol. 2003;14:922–30. doi: 10.1093/annonc/mdg250. [DOI] [PubMed] [Google Scholar]
- 30.Smith JS, Tachibana I, Passe SM, Huntley BK, Borell TJ, Iturria N, et al. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst. 2001;93:1246–56. doi: 10.1093/jnci/93.16.1246. [DOI] [PubMed] [Google Scholar]
- 31.Donato V, Papaleo A, Castrichino A, Banelli E, Giangaspero F, Salvati M, et al. Prognostic implication of clinical and pathologic features in patients with glioblastoma multiforme treated with concomitant radiation plus temozolomide. Tumori. 2007;93:248–56. doi: 10.1177/030089160709300304. [DOI] [PubMed] [Google Scholar]
- 32.Filippini G, Falcone C, Boiardi A, Broggi G, Bruzzone MG, Caldiroli D, et al. Prognostic factors for survival in 676 consecutive patients with newly diagnosed primary glioblastoma. Neuro Oncol. 2008;10:79–87. doi: 10.1215/15228517-2007-038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lacroix M, Abi-Said D, Fourney DR, Gokaslan ZL, Shi W, DeMonte F, et al. A multivariate analysis of 416 patients with glioblastoma multiforme: Prognosis, extent of resection, and survival. J Neurosurg. 2001;95:190–8. doi: 10.3171/jns.2001.95.2.0190. [DOI] [PubMed] [Google Scholar]
- 34.Ammirati M, Vick N, Liao YL, Ciric I, Mikhael M. Effect of the extent of surgical resection on survival and quality of life in patients with supratentorial glioblastomas and anaplastic astrocytomas. Neurosurgery. 1987;21:201–6. doi: 10.1227/00006123-198708000-00012. [DOI] [PubMed] [Google Scholar]
- 35.Franklin CI. Does the extent of surgery make a difference in high grade malignant astrocytoma? Australas Radiol. 1992;36:44–7. doi: 10.1111/j.1440-1673.1992.tb03073.x. [DOI] [PubMed] [Google Scholar]
- 36.Gamburg ES, Regine WF, Patchell RA, Strottmann JM, Mohiuddin M, Giese A, et al. Cost of migration: Invasion of malignant gliomas and implications for treatment. J Clin Oncol. 2003;21:1624–36. doi: 10.1200/JCO.2003.05.063. [DOI] [PubMed] [Google Scholar]
- 37.Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL, et al. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22:133–42. doi: 10.1200/JCO.2004.08.110. [DOI] [PubMed] [Google Scholar]
- 38.Bianco R, Shin I, Ritter CA, Yakes FM, Basso A, Rosen N, et al. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene. 2003;22:2812–22. doi: 10.1038/sj.onc.1206388. [DOI] [PubMed] [Google Scholar]
- 39.She QB, Solit DB, Ye Q, O’Reilly KE, Lobo J, Rosen N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell. 2005;8:287–97. doi: 10.1016/j.ccr.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–7. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 41.Li B, Yuan M, Kim IA, Chang CM, Bernhard EJ, Shu HK. Mutant epidermal growth factor receptor displays increased signaling through the phosphatidylinositol-3 kinase/AKT pathway and promotes radioresistance in cells of astrocytic origin. Oncogene. 2004;23:4594–602. doi: 10.1038/sj.onc.1207602. [DOI] [PubMed] [Google Scholar]