

ABSTRACT
Actinobacillus pleuropneumoniae is the causative agent of porcine contagious pleuropneumonia. Overproduction of proinflammatory cytokines, like interleukin-1β (IL-1β), IL-6, tumor necrosis factor alpha, and resistin, in the lung is an important feature of A. pleuropneumoniae infection. These proinflammatory cytokines enhance inflammatory and immunological responses. However, the mechanism that leads to cytokine production remains unclear. As a major virulence factor of A. pleuropneumoniae, lipopolysaccharide (LPS) may act as a potent stimulator of Toll-like receptor 4 (TLR4), triggering a number of intracellular signaling pathways that lead to the synthesis of proinflammatory cytokines. Porcine alveolar macrophages (PAMs) are the first line of defense against pathogenic microbes during pathogen invasion. The results of the present study demonstrate that A. pleuropneumoniae LPS induces PAMs to produce inflammatory cytokines in time- and dose-dependent manners. Moreover, PAMs were activated by A. pleuropneumoniae LPS, resulting in upregulation of signaling molecules, including TLR4, MyD88, TRIF-related adaptor molecule, and NF-κB. In contrast, the activation effects of A. pleuropneumoniae LPS on PAMs could be suppressed by specific inhibitors, like small interfering RNA and Bay11-7082. Taken together, our data indicate that A. pleuropneumoniae LPS can induce PAMs to produce proinflammatory cytokines via the TLR4/NF-κB-mediated pathway. These findings partially reveal the mechanism of the overproduction of proinflammatory cytokines in the lungs of swine with A. pleuropneumoniae infection and may provide targets for the prevention of A. pleuropneumoniae-induced pneumonia. All the data could be used as a reference for the pathogenesis of respiratory infection.
KEYWORDS: Actinobacillus pleuropneumoniae, lipopolysaccharide, porcine alveolar macrophages, Toll-like receptor 4, nuclear factor κB
INTRODUCTION
Porcine pleuropneumonia (PP) is a typical respiratory disease, characterized by hemorrhagic, fibrinous, and necrotic lung lesions (1). This disease affects pigs of all ages and leads to a substantial impact on the economy and ecology of the swine industry and the welfare of animals in the swine industry worldwide (2). Actinobacillus pleuropneumoniae, a Gram-negative bacterium, is the causative agent of PP. A. pleuropneumoniae can be divided into two biotypes (biotype 1 and biotype 2), with biotype 1 having 12 serotypes and biotype 2 having 6 serotypes (3). The virulence of each serotype varies according to the Apx toxins and the surface lipopolysaccharide (LPS) (4). LPS extracted from A. pleuropneumoniae serotype 1 was suggested to be involved in lung infection (5–8). Belanger et al. identified that LPS is the major adhesin of A. pleuropneumoniae (9), and purified LPS was found to be able to induce the excessive production of proinflammatory cytokines like interleukin-1β (IL-1β), IL-6, tumor necrosis factor alpha (TNF-α), and resistin, which contributed to lung lesions similar to those in naturally infected pigs (10, 11).
Porcine alveolar macrophages (PAMs) are strategically situated at the air-surface interface in alveoli and are the predominant macrophages that interact with pathogenic microbes during pathogen invasion (12–17). Similar to other respiratory contagious microbes, A. pleuropneumoniae preferentially binds to the PAMs and endothelial cells of the lower respiratory tract (18). Some studies indicated that A. pleuropneumoniae LPS might induce the overproduction of proinflammatory cytokines by PAMs compared with their level of production in uninfected animals (5, 19).
In this study, we aimed to explore the mechanism that leads to A. pleuropneumoniae LPS-induced cytokine production by PAMs. Toll-like receptor 4 (TLR4) is important to the infective inflammation caused by Gram-negative bacteria (20–23), and the intracellular signaling is achieved via myeloid differentiation primary response protein 88 (MyD88), TRIF-related adaptor molecule (TRAM), and nuclear factor κB (NF-κB) (22, 24–26). Here we found that A. pleuropneumoniae LPS is a TLR4 agonist that mediates proinflammatory activities by interacting with PAMs. We found that A. pleuropneumoniae LPS subsequently activated MyD88, TRAM, and NF-κB through TLR4, and then PAMs secreted large amounts of cytokines. In contrast, the activation effects of A. pleuropneumoniae LPS on PAMs could be suppressed by inhibitors specific for TLR4, MyD88, TRAM, and NF-κB. Taken together, the results in this research indicate that A. pleuropneumoniae LPS can activate PAMs to produce proinflammatory cytokines via the TLR4/NF-κB-mediated pathway.
RESULTS
A. pleuropneumoniae LPS induced PAMs to produce proinflammatory cytokines in time- and dose-dependent manners.
LPS from A. pleuropneumoniae serotype 1 was isolated by the hot phenol-water method (Fig. 1). The relative expression of mRNA for proinflammatory cytokine genes (IL-1β, IL-6, TNF-α, and resistin) in PAMs was measured following exposure to A. pleuropneumoniae LPS. PAMs were incubated with 100 ng/ml A. pleuropneumoniae LPS for 1.5 h, 3 h, 6 h, and 12 h, and the levels of expression of mRNA for the cytokines increased with time (Fig. 2A). PAMs were also exposed to 10, 100, or 1,000 ng/ml A. pleuropneumoniae LPS for 6 h, and the expression of mRNA for the cytokines increased after exposure to 10 and 100 ng/ml but decreased dramatically after exposure to 1,000 ng/ml (Fig. 2C). The levels of the tested cytokines in the supernatant of PAMs following exposure to A. pleuropneumoniae LPS were also measured by ELISA. Consistent with the mRNA expression, the levels of cytokines in the supernatant increased in time- and dose-dependent manners (Fig. 2B and D). Taken together, these results indicate that A. pleuropneumoniae LPS can induce PAMs to produce proinflammatory cytokines in time- and dose-dependent manners.
FIG 1.
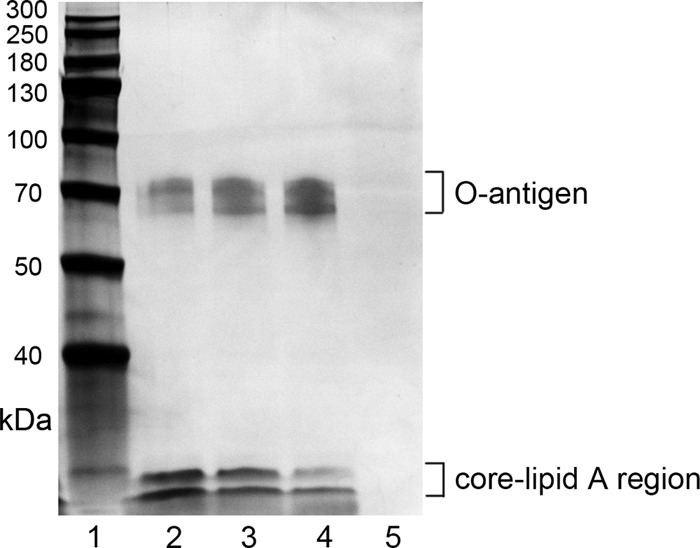
Silver-stained SDS-PAGE profile of purified LPS from A. pleuropneumoniae serotype 1. Lane 1, protein markers; lanes 2 to 4, A. pleuropneumoniae LPS; lane 5, blank control with PBS. This gel showed that purified A. pleuropneumoniae LPS is composed of O antigen, core, and lipid A. Molecular mass markers (in kilodaltons) are indicated on the left. Eight micrograms of A. pleuropneumoniae LPS was loaded in lanes 2 to 4.
FIG 2.

A. pleuropneumoniae LPS-induced proinflammatory cytokine expression is time and dose dependent. PAMs were treated with 100 ng/ml A. pleuropneumoniae LPS for 0, 1.5, 3, 6, and 12 h. (A and B) qRT-PCR was performed to analyze the expression of IL-1β, IL-6, TNF-α, and resistin mRNA (A), and ELISA was used to detect the levels of cytokines in the supernatants (B). PAMs were exposed to 0, 10, 100, or 1,000 ng/ml A. pleuropneumoniae LPS for 6 h. (C and D) qRT-PCR was performed to analyze the expression of IL-1β, IL-6, TNF-α, and resistin mRNA (C), and ELISA was used to detect the levels of cytokines in the supernatants (D). *, P < 0.05 compared with the control group.
The A. pleuropneumoniae LPS-induced response was TLR4 dependent.
TLR4 was abundantly expressed in PAMs, according to the immunocytochemistry assay results (Fig. 3A). We first treated PAMs with increasing amounts of A. pleuropneumoniae LPS. The measurement of TLR4 mRNA showed that A. pleuropneumoniae LPS significantly upregulated the synthesis of TLR4 in a dose-dependent manner (Fig. 3B). To further evaluate the cellular membrane protein required for A. pleuropneumoniae LPS binding, a small interfering RNA (siRNA)-mediated knockdown assay was used. Three different TLR4-specific siRNA sequences (TLR4-1, TLR4-2, and TLR4-3) were transfected to inhibit the expression of the TLR4 gene, and real-time quantitative reverse transcription-PCR (qRT-PCR) results revealed that the expression of TLR4 mRNA decreased by 76%, 67%, and 56%, respectively (Fig. 3C). Thus, the TLR4-1 (siRNA) sequence was chosen for use in the following experiments.
FIG 3.

The A. pleuropneumoniae LPS-induced response is TLR4 dependent. (A) Immunocytochemistry staining of TLR4 in PAMs. PAMs were incubated with 0, 10, 100, or 1,000 ng/ml A. pleuropneumoniae LPS for 6 h. Magnifications, ×400. (B) The measurement of TLR4 mRNA showed that A. pleuropneumoniae LPS significantly upregulated the synthesis of TLR4 in a dose-dependent manner. (C) PAMs were transfected with three different TLR4-specific siRNA sequences for 48 h. qRT-PCR was performed to analyze the efficiency of interference. (D) PAMs were transfected with TLR4-specific siRNA for 48 h, and then the cells were stimulated with 100 ng/ml A. pleuropneumoniae LPS for 6 h. qRT-PCR was performed to analyze the expression of IL-1β, IL-6, TNF-α, and resistin mRNA, and ELISA was used to detect the levels of cytokines in the supernatants. ctrl, blank control (PBS); ctrl-siRNA, a nontargeting siRNA control; LPS, A. pleuropneumoniae LPS. *, P < 0.05 compared with the control group.
The TLR4-specific siRNA was transfected into the cells prior to treatment with A. pleuropneumoniae LPS. The results indicated that the production of cytokines was significantly downregulated in TLR4-knockdown PAMs, whereas the production of IL-1β, IL-6, TNF-α, and resistin was significantly upregulated in PAMs transfected with a nontargeting siRNA sequence compared with that under the unstimulated condition (treatment with phosphate-buffered saline [PBS], used as a blank control) (Fig. 3D). These data indicate that TLR4 in PAMs might play a pivotal role in response to A. pleuropneumoniae LPS.
The A. pleuropneumoniae LPS-induced response was MyD88/TRAM dependent.
LPS-induced TLR4 signaling is classified into MyD88-dependent and TRAM-dependent pathways (27). PAMs were incubated with 100 ng/ml A. pleuropneumoniae LPS for 6 h. The relative quantitative RT-PCR results showed that the MyD88 and TRAM mRNAs were significantly upregulated compared with their levels of regulation under the unstimulated condition (Fig. 4A).
FIG 4.

The A. pleuropneumoniae LPS-induced response is MyD88/TRAM dependent. (A) PAMs were treated with 100 ng/ml A. pleuropneumoniae LPS for 6 h, and the expression of MyD88 and TRAM mRNA was detected by qRT-PCR. (B) PAMs were transfected with three different MyD88-specific siRNA or TRAM-specific siRNA sequences for 48 h. qRT-PCR was performed to detect the efficiency of interference. The results indicated that the expression of MyD88 mRNA decreased by 56%, 73%, and 57%, respectively, and that of TRAM mRNA decreased by 36%, 68%, and 70%, respectively. Accordingly, MyD88-2 (siRNA) and TRAM-3 (siRNA) sequences were chosen for use in the following experiments. PAMs were transfected with MyD88-specific siRNA or TRAM-specific siRNA for 48 h, and then the cells were stimulated with 100 ng/ml A. pleuropneumoniae LPS for 6 h. (C and D) qRT-PCR was performed to analyze the expression of IL-1β, IL-6, TNF-α, and resistin mRNA (C), and ELISA was used to detect the levels of cytokines in the supernatants (D). ctrl, blank control (PBS); ctrl-siRNA, nontargeting siRNA control; LPS, A. pleuropneumoniae LPS. *, P < 0.05 compared with the control group.
To assess the role of intracellular signaling pathways in the A. pleuropneumoniae LPS effects, PAMs were pretreated with inhibitors specific for regulation of the MyD88-dependent pathway (MyD88-specific siRNA) and TRAM-dependent pathway (TRAM-specific siRNA), transfected into the PAMs, and then stimulated with A. pleuropneumoniae LPS (Fig. 4B). Cytokine expression results indicated that the inhibitors of MyD88 and TRAM efficiently inhibited the A. pleuropneumoniae LPS-induced expression of IL-1β, IL-6, TNF-α, and resistin at the mRNA and protein levels by PAMs (Fig. 4C and D). To demonstrate whether A. pleuropneumoniae LPS induced the activation of MyD88/TRAM via the TLR4, we transfected PAMs with siRNA and found that the TLR4-specific siRNA significantly downregulated the expression of MyD88 and TRAM (Fig. 5). These results prove that A. pleuropneumoniae LPS can activate PAMs via MyD88- and TRAM-dependent pathways.
FIG 5.

A. pleuropneumoniae LPS induces activation of MyD88/TRAM via TLR4. PAMs were transfected with TLR4-specific siRNA for 48 h, followed by stimulation with 100 ng/ml A. pleuropneumoniae LPS for 6 h. (A and B) qRT-PCR was performed to analyze the expression of MyD88 and TRAM mRNA (A), and Western blot analysis was used to detect the proteins (B). (C) Image-Pro Plus (version 6.0) software was used to quantify the integrated optical density of MyD88 and TRAM. The results showed that MyD88 and TRAM are significantly downregulated after PAMs are transfected with TLR4-specific siRNA. ctrl, blank control (PBS); ctrl-siRNA, nontargeting siRNA control; LPS, A. pleuropneumoniae LPS; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. *, P < 0.05 compared with the control group.
The A. pleuropneumoniae LPS-induced response was NF-κB dependent.
A. pleuropneumoniae could activate PAMs by NF-κB. Accordingly, we assessed the functional role of NF-κB on the A. pleuropneumoniae LPS-induced proinflammatory responses. The incubation of PAMs with A. pleuropneumoniae LPS resulted in the significant upregulation of p65 and the inhibitor of κB (IκB) at the mRNA level (Fig. 6A). Reductions in the levels of IL-1β, IL-6, TNF-α, and resistin mRNA in the supernatants of A. pleuropneumoniae LPS-stimulated PAMs pretreated with an NF-κB inhibitor (Bay11-7082) were observed (Fig. 6B and C). To demonstrate whether A. pleuropneumoniae LPS induced the activation of NF-κB via TLR4 signaling pathways, we transfected PAMs with siRNA and found that TLR4-specific siRNA significantly downregulated the expression of p65 and IκB and their phosphorylation (Fig. 7). A similar changing pattern was observed when cells were pretreated with MyD88-specific siRNA or TRAM-specific siRNA (Fig. 7). Collectively, these results support the suggestion that A. pleuropneumoniae LPS induces IL-1β, IL-6, TNF-α, and resistin production via TLR4-MyD88-dependent/TRAM-dependent signaling pathway-mediated activation of NF-κB.
FIG 6.

The A. pleuropneumoniae LPS-induced response is NF-κB dependent. (A) PAMs were treated with 100 ng/ml A. pleuropneumoniae LPS for 6 h, and the expression of p65 and IκB mRNA was detected by qRT-PCR. PAMs were pretreated with 5 mM Bay11-7082 for 2 h before 100-ng/ml A. pleuropneumoniae LPS treatment. (B) qRT-PCR was performed to analyze IL-1β, IL-6, TNF-α, and resistin mRNA expression. (C) ELISA was used to detect the levels of cytokines in supernatants. PBS was used as a blank control. LPS, A. pleuropneumoniae LPS. *, P < 0.05 compared with the control group.
FIG 7.

A. pleuropneumoniae LPS induces the activation of NF-κB via the TLR4-MyD88-dependent/TRAM-dependent pathways. PAMs were transfected with TLR4-specific siRNA, MyD88-specific siRNA, or TRAM-specific siRNA for 48 h, followed by stimulation with 100 ng/ml A. pleuropneumoniae LPS for 6 h. (A and B) The expression of p65 and IκB mRNA was analyzed by qRT-PCR (A), and the proteins were detected by Western blot analysis (B). (B) The level of p65 and IκB phosphorylation was also tested by Western blot analysis. (C) Image-Pro Plus (version 6.0) software was used to quantify the integrated optical density of p65 and IκB and their phosphorylation. The results showed that p65 and IκB and their phosphorylation were significantly downregulated after PAMs were transfected with TLR4-specific siRNA, MyD88-specific siRNA, or TRAM-specific siRNA. Dimethyl sulfoxide (DMSO)-LPS was used as a treatment sample. PBS was used as a blank control. p-P65, phosphorylated p65; p-IκB, phosphorylated IκB; ctrl-siRNA, nontargeting siRNA control; LPS, A. pleuropneumoniae LPS. *, P < 0.05 compared with the control group.
DISCUSSION
The results in our present research confirm that A. pleuropneumoniae LPS can induce PAMs to produce proinflammatory cytokines via the TLR4/NF-κB-mediated pathway, thus illuminating a possible mechanism that leads to cytokine production in lungs with A. pleuropneumoniae infection. PAMs are the first line of defense against pathogenic microbe infections in the lung. We found that the production of proinflammatory cytokines in A. pleuropneumoniae LPS-stimulated cells was triggered through TLR4. In these situations, the effects of A. pleuropneumoniae LPS could be suppressed by specific inhibitors, like siRNA and Bay11-7082. These findings are in agreement with previous reports, in which LPS was a positive regulator of inflammatory effects triggered through TLR4 (22, 27, 28).
A. pleuropneumoniae LPS, an important virulence factor, exerts cytotoxic effects on various cell types. In in vivo experiments, A. pleuropneumoniae LPS could stimulate alveolar and intravascular macrophages to release toxic oxygen metabolites, proteolytic enzymes, and inflammatory cytokines (2, 4, 29). A. pleuropneumoniae LPS could also activate factor XII to initiate the coagulation, fibrinolysis, and kinin systems, resulting in endothelial cell injury (3, 10, 30). In this study, the morphological changes showed that a high dose (1,000 ng/ml) of A. pleuropneumoniae LPS could lead to cytopathic effects (CPE) on PAMs (Fig. 8D) and decreases in the levels of cytokine mRNA expression (Fig. 2C).
FIG 8.

Morphological changes of PAMs induced with A. pleuropneumoniae LPS. PAMs were treated with A. pleuropneumoniae LPS and cultured for 6 h. (A) Control PAMs; (B) PAMs treated with 10 ng/ml A. pleuropneumoniae LPS; (C) PAMs treated with 100 ng/ml A. pleuropneumoniae LPS; (D) PAMs treated with 1,000 ng/ml A. pleuropneumoniae LPS. Note that the high dose (1,000 ng/ml) of A. pleuropneumoniae LPS could lead to a cytopathic effect (CPE) on the PAMs. Magnifications, ×200.
Alveolar macrophages are resident phagocytes of the alveolar space (31). Recent studies with animals with acute lung injury have indicated that an uncontrolled inflammatory response in PAMs is important in the pathogenesis of pleural pneumonia (2, 4, 5). Moreover, much research on macrophages has focused on the secretion of cytokines (32–35). IL-1β, IL-6, and TNF-α are cytokines mainly secreted by macrophages and have multiple functions in regulating the immune response and the acute-phase reaction (APR) (36, 37). IL-6 mediates the APR and modulates the overactivity of inflammation (38). Resistin has been described to be a protein found in the inflammatory zone (FIZZ) (39, 40). Resistin levels were suggested to show a strong correlation to those of other inflammatory markers, like C-reactive protein (41), TNF-α (42), IL-6 (43), and soluble vascular adhesion molecules (44). In this study, the expression of resistin was significantly increased in PAMs following exposure to A. pleuropneumoniae LPS, similar to the resistin expression in diseased lungs (45, 46).
Toll-like receptors (TLRs) are the primary pattern recognition receptors that act as the sensors of invading pathogens in macrophages (8, 23, 47). At present, although research has reported that A. pleuropneumoniae could adhere to cells such as SJPL cells and stimulate the NF-κB induction through the TLR pathways (8), the specific molecular mechanism remains unclear. Among the TLRs, TLR4 plays a pivotal role in the host response to LPS (22, 27, 28). Here, we found that the expression of TLR4 was significantly increased in PAMs following exposure to A. pleuropneumoniae LPS and this effect could be inhibited by TLR4-specific siRNA. TLR4-mediated signaling pathways could induce the expression of inflammatory cytokines by MyD88-dependent and TRAM-dependent pathways (25, 48). MyD88 is essential for responses against microbial components in the early phase of NF-κB activation, while TRAM is involved in the late phase of NF-κB activation and has an essential role in the MyD88-independent cascade of TLR4-induced signals (26). Interestingly, we observed that both MyD88 and TRAM were involved in the production of cytokines induced by A. pleuropneumoniae LPS. To further disclose the link between TLR4 and MyD88/TRAM, the expression of MyD88 and TRAM mRNA was tested following the transfection of TLR4-specific siRNA into PAMs. We found that MyD88/TRAM mRNA was significantly decreased, suggesting that the A. pleuropneumoniae LPS-induced activation of MyD88/TRAM is dependent on TLR4. However, there might be other receptors involved in A. pleuropneumoniae LPS-induced cytokine production.
NF-κB, a key transcription factor of macrophages, consists of the p50 and p65 subunits (49–51). In unstimulated cells, NF-κB is sequestered by the inhibitor of κB (IκB) in the cytosol (52). Upon stimulation, IκB is phosphorylated by the activated cellular IκB kinase (IKK) complex, triggering its degradation and resulting in the translocation of NF-κB into the nucleus to promote the transcription of target genes (53). In this study, the macrophage immunostimulatory properties of A. pleuropneumoniae LPS were observed by measuring the activation of NF-κB, and we found that proteins and/or mRNA coding for p65 and IκB were significantly upregulated. In contrast, after being transfected with TLR4-specific siRNA, MyD88-specific siRNA, and TRAM-specific siRNA, the expression of p65 and IκB and their phosphorylation were suppressed. Therefore, it was inferred that A. pleuropneumoniae LPS might induce the activation of NF-κB via TLR4-dependent pathways.
In conclusion, this study partially revealed the mechanism that leads to cytokine production in the lungs of swine with A. pleuropneumoniae infection (Fig. 9). Our results show that A. pleuropneumoniae LPS contributes to the production of IL-1β, IL-6, TNF-α, and resistin and to the activation of PAMs via MyD88, TRAM, and NF-κB by interacting with TLR4. These observations elucidate a possible mechanism of lung injury induced by A. pleuropneumoniae and help to provide targets for the prevention and control of PP. All the data can be used as a reference for analysis of the pathogenesis of respiratory infection.
FIG 9.
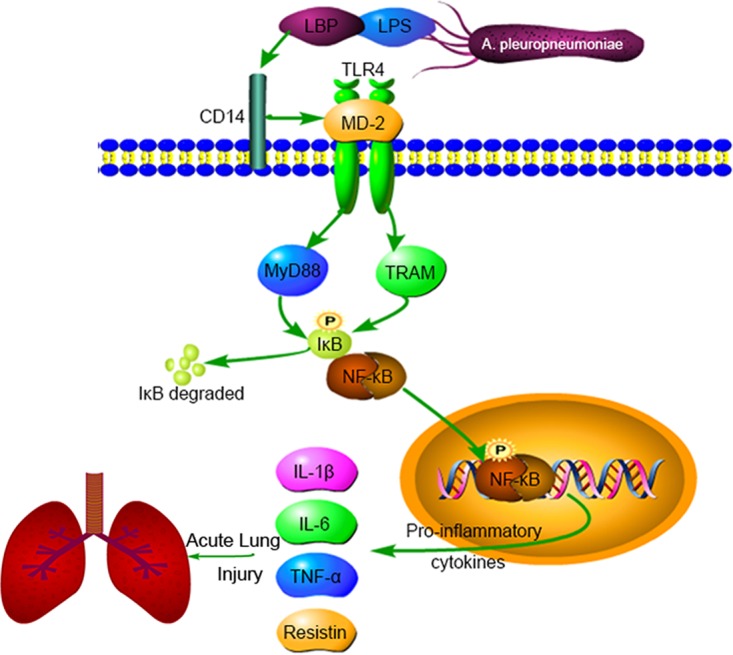
A. pleuropneumoniae LPS induces the production of IL-1β, IL-6, TNF-α, and resistin via the TLR4-MyD88-dependent/TRAM-dependent and NF-κB-mediated pathways in PAMs cells. These observations may partly explain the mechanism of pleural pneumonia induced by A. pleuropneumoniae. LBP, lipopolysaccharide binding protein.
MATERIALS AND METHODS
Cell line.
PAMs (3D4/21) were bought from the American Type Culture Collection (ATCC CRL-2843) and were cultured in Dulbecco modified Eagle medium (DMEM; Gibco, Life Technologies, Carlsbad, CA, USA) with 10% fetal bovine serum (Gibco, Life Technologies). Cells were cultivated in a humidified incubator with 5% CO2 at 37°C. Before A. pleuropneumoniae LPS treatment, PAMs were seeded into 6-well plates with serum-free DMEM for 24 h.
Bacterial strains and preparation of LPS.
A. pleuropneumoniae serotype 1 was obtained from the Veterinary College of Sichuan Agricultural University. The bacterial strain was cultured in brain heart infusion (BHI) broth or agar (Oxoid Ltd., Basingstoke, UK) supplemented with 10% fetal calf serum and 15 mg/ml NAD and incubated at 37°C in 5% CO2. A. pleuropneumoniae LPS was prepared by the hot phenol-water method. The purified LPS preparations were free from proteins and nucleic acids. Fractions were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with silver staining.
Immunocytochemistry.
PAMs were seeded on coverslips in 6-well plates at 2 × 105 cells/well. For immunofluorescence staining of TLR4, PAMs were fixed with 4% paraformaldehyde for 30 min at 37°C, permeabilized with 0.1% Triton X-100 for 10 min, and blocked with 3% bovine serum albumin (BSA) for 1 h. Then the PAMs were incubated with rabbit anti-porcine TLR4 antibody (1:400) (Abgent) at 4°C overnight. After washing, the samples were incubated with fluorescein isothiocyanate-conjugated goat anti-rabbit IgG (1:400) for 10 min. The slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) to stain the cell nuclei and analyzed by confocal laser scanning microscopy.
ELISA.
The levels of porcine IL-1β, IL-6, TNF-α, and resistin cytokines were measured by a sandwich enzyme-linked immunosorbent assay (ELISA) using pair-matched antibodies from R&D Systems (Minneapolis, MN, USA). Briefly, PAMs were seeded at a density of 2 × 105 cells per well and cultured for 1 h before stimulation with A. pleuropneumoniae LPS. For the siRNA-treated cells, cells were cultured for 1 h with treatment with A. pleuropneumoniae LPS. After 1 h, the cells were replaced with DMEM, followed by stimulation with A. pleuropneumoniae LPS. Supernatants in 6-well plates were collected and analyzed by ELISA according to the instruction manual. Twofold dilutions of recombinant porcine IL-1β, IL-6, TNF-α, and resistin (R&D Systems) were used to generate standard curves.
qRT-PCR.
PAMs were collected and placed in TRIzol reagent, and the RNA in the cells was extracted according to the instruction manual (TaKaRa Biomedical Technology, Beijing, China). Purified mRNA was resuspended in 30 μl RNase-free water. Total RNA from the samples was stored at −80°C. Quantification of RNA was analyzed by use of a NanoDrop spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE, USA). Reverse transcriptions were performed with a TaKaRa RT reagent kit with a gDNA Eraser. Real-time quantitative reverse transcription-PCR (qRT-PCR) was then performed using a QuantStudio real-time PCR system (Applied Biosystems). A two-step PCR protocol was used to perform the qRT-PCR in the present study. The two-step PCR protocol was as follows: step 1, denaturation at 95°C for 5 s with detection off, and step 2, combined annealing/extension at 60°C for 40 s with detection on. Water was used to replace cDNA in each run as a negative control in the PCR amplification. The relative gene expression was determined by the ΔΔCT threshold cycle (CT) method. The sequences of the primers used for PCR are listed in Table 1.
TABLE 1.
Sequences of primers used for qRT-PCR
| Primer name | Sequence (5′–3′) |
|
|---|---|---|
| Forward primer | Reverse primer | |
| IL-1β | GGCCCCAAAGAGATGAAGTG | CTGGGAGGAGGGATTCATCG |
| IL-6 | TTCAGTCCAGTCGCCTTCTC | TGGCATCACCTTTGGCATCT |
| TNF-α | GCATGATCCGAGACGTGGAG | AACCTCGAAGTGCAGTAGGC |
| Resistin | GCTCTCTCCCTCCTCTTCCT | GCGACATCCCGGATCTTCT |
| TLR4 | GCGTGCAGGTGGTTCCTA | AGCACCTGCAGTTCTGGAAA |
| MyD88 | ATGGCTGCAGGAGGCT | TTTAGGAAGAGCGACAGGCG |
| TRAM | GCCACGGTGTGGATACAAGT | ACATGCAACATCTTCGCCAC |
| p65 | CATGAGCTCGTGGGGAAAGA | CACACTGGATCCCCAGGTTC |
| IκB | TGCAGGCCACCAACTACAAT | TCAACAAGAGCGACACCAGG |
| GAPDH | CCACTGGTGTCTTCACGACC | GGTTCACGCCCATCACAAAC |
Western blot analysis.
PAMs were lysed with radioimmunoprecipitation assay lysis buffer, and the lysate was transferred into a 1.5-ml Eppendorf tube and centrifuged at 13,000 × g for 20 min at 4°C to collect the supernatant. The protein concentration was determined with a bicinchoninic acid assay kit (Beyotime Biotechnology). Proteins (15 μg) were loaded and separated on 12% ExpressPlus PAGE gels (GenScript Corporation, Piscataway, NJ, USA), followed by analysis with a one-step complete Western blotting kit (GenScript Corporation, Piscataway, NJ, USA) according to the instruction manual. Briefly, membranes were incubated with pretreat solution on a shaker for 5 min at room temperature and then incubated at 4°C for 12 h with primary antibodies against TLR4, MyD88, TRAM, p65, and IκB (Abgent) and against phosphorylated p65 (Ser536) and phosphorylated IκB (Cell Signaling Technology). After the gels were washed 3 times for 10 min each time, the bands were displayed on the membranes after addition of tetramethylbenzidine buffer for 5 min. Image-Pro Plus (version 6.0) software (Media Cybernetics, Inc.) was used to quantify the integrated optical density of the bands. Three repeated experiments were carried out to study protein expression.
siRNA transfection.
Small-interfering RNAs (siRNAs) that target porcine TLR4, MyD88, and TRAM were designed and synthesized by Invitrogen (Life Technologies). siRNA or control siRNA (150 nM) was transfected into the PAMs in each well by using the Lipofectamine 3000 reagent (Life Technologies), according to the instruction manual. Downregulation of genes was evaluated by qRT-PCR. The sequences of the primers used for siRNA are listed in Table 2.
TABLE 2.
Sequences of primers used for siRNA
| Primer name | Sequence (5′–3′) |
|
|---|---|---|
| Sense primers | Antisense primers | |
| TLR4-1 | GGUGCUGGAUUUAUCCAGAUGUGAA | UUCACAUCUGGAUAAAUCCAGCACC |
| TLR4-2 | GAGCUUAAUGUGGCUCACAAUCAUA | UAUGAUUGUGAGCCACAUUAAGCUC |
| TLR4-3 | CAACCUGGAGCAUUUGGAUCUUUCU | AGAAAGAUCCAAAUGCUCCAGGUUG |
| Control TLR4 | GGUGUAGUUAUCUACAGGUUCGGAA | UUCCGAACCUGUAGAUAACUACACC |
| MyD88-1 | CCUCUUUCAAUAGCCGGGUAUGAUA | UAUCAUACCCGGCUAUUGAAAGAGG |
| MyD88-2 | GAGAAAGGAGGUGAAGGCCUGGAAA | UUUCCAGGCCUUCACCUCCUUUCUC |
| MyD88-3 | CACCAGUCUCCUGAGUUCUUAUAUG | CAUAUAAGAACUCAGGAGACUGGUG |
| Control MyD88 | CCUUUACUAGACCGGUGUAGUCAUA | UAUGACUACACCGGUCUAGUAAAGG |
| TRAM-1 | AAGCCACGGUGUGGAUACAAGUCAA | UUGACUUGUAUCCACACCGUGGCUU |
| TRAM-2 | CCUCACGAGACAGAUUGCACGAUAU | AUAUCGUGCAAUCUGUCUCGUGAGG |
| TRAM-3 | UGCCGUGUGGCAGACAGCAUUUACA | UGUAAAUGCUGUCUGCCACACGGCA |
| Control TRAM | AAGACGGUGUGGAUACAAGUCCCAA | UUGGGACUUGUAUCCACACCGUCUU |
Statistical analysis.
All data were analyzed statistically using GraphPad Prism software (version 4.03; GraphPad Software Inc., San Diego, CA, USA) by application of the Mann-Whitney U test or one-way analysis of variance with Bartlett's test. Differences were considered significant at a P value of <0.05.
ACKNOWLEDGMENT
This study was supported by a Sichuan Agricultural University Science and Technology Department support project (no. 2013NZ0032).
REFERENCES
- 1.Bossé JT, Li Y, Rogers J, Crespo RF, Li Y, Chaudhuri RR, Holden MTG, Maskell DJ, Tucker AW, Wren BW. 2017. Whole genome sequencing for surveillance of antimicrobial resistance in Actinobacillus pleuropneumoniae. Front Microbiol 8:311. doi: 10.3389/fmicb.2017.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiers K, Waele TD, Pasmans F, Ducatelle R, Haesebrouck F. 2010. Virulence factors of Actinobacillus pleuropneumoniae involved in colonization, persistence and induction of lesions in its porcine host. Vet Res 41:65. doi: 10.1051/vetres/2010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bossé JT, Janson H, Sheehan BJ, Beddek AJ, Rycroft AN, Kroll JS, Langford PR. 2002. Actinobacillus pleuropneumoniae: pathobiology and pathogenesis of infection. Microbes Infect 4:225–235. doi: 10.1016/S1286-4579(01)01534-9. [DOI] [PubMed] [Google Scholar]
- 4.Sassu EL, Frömbling J, Duvigneau JC, Miller I, Müllebner A, Gutiérrez AM, Grunert T, Patzl M, Saalmüller A, Altrock AV. 2017. Host-pathogen interplay at primary infection sites in pigs challenged with Actinobacillus pleuropneumoniae. BMC Vet Res 13:64. doi: 10.1186/s12917-017-0979-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramjeet M, Deslandes V, St Michael F, Cox AD, Kobisch M, Gottschalk M, Jacques M. 2005. Truncation of the lipopolysaccharide outer core affects susceptibility to antimicrobial peptides and virulence of Actinobacillus pleuropneumoniae serotype 1. J Biol Chem 280:39104–39114. doi: 10.1074/jbc.M502852200. [DOI] [PubMed] [Google Scholar]
- 6.Bélanger M, Dubreuil D, Harel J, Girard C, Jacques M. 1990. Role of lipopolysaccharides in adherence of Actinobacillus pleuropneumoniae to porcine tracheal rings. Infect Immun 58:3523–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paradis SE, Dubreuil D, Rioux S, Gottschalk M, Jacques M. 1994. High-molecular-mass lipopolysaccharides are involved in Actinobacillus pleuropneumoniae adherence to porcine respiratory tract cells. Infect Immun 62:3311–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Auger E, Deslandes VM. 2009. Host-pathogen interactions of Actinobacillus pleuropneumoniae with porcine lung and tracheal epithelial cells. Infect Immun 77:1426–1441. doi: 10.1128/IAI.00297-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belanger M, Begin C, Jacques M. 1995. Lipopolysaccharides of Actinobacillus pleuropneumoniae bind pig hemoglobin. Infect Immun 63:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Udeze FA, Latimer KS, Kadis S. 1987. Role of Haemophilus pleuropneumoniae lipopolysaccharide endotoxin in the pathogenesis of porcine Haemophilus pleuropneumonia. Am J Vet Res 48:768–773. [PubMed] [Google Scholar]
- 11.Fenwick BW, Osburn BI, Olander HJ. 1986. Isolation and biological characterization of two lipopolysaccharides and a capsular-enriched polysaccharide preparation from Haemophilus pleuropneumoniae. Am J Vet Res 47:1433–1441. [PubMed] [Google Scholar]
- 12.Chen C, Yang S, Zhang M, Zhang Z, Zhang SB, Wu B, Hong J, Zhang W, Lin J, Okunieff P, Zhang L. 2017. Triptolide mitigates radiation-induced pneumonitis via inhibition of alveolar macrophages and related inflammatory molecules. Oncotarget 8:45133–45142. doi: 10.18632/oncotarget.16456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, Qin W, Zhang J, Bao C, Zhang H, Che Y, Sun C, Gu J, Feng X, Du C, Han W, Richard PL, Lei L. 2016. Adh enhances Actinobacillus pleuropneumoniae pathogenicity by binding to OR5M11 and activating p38 which induces apoptosis of PAMs and IL-8 release. Sci Rep 6:24058. doi: 10.1038/srep24058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lambrecht BN. 2006. Alveolar macrophage in the driver's seat. Immunity 24:366–368. doi: 10.1016/j.immuni.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 15.Hathroubi S, Beaudry F, Provost C, Martelet L, Segura M, Gagnon CA, Jacques M. 2016. Impact of Actinobacillus pleuropneumoniae biofilm mode of growth on the lipid A structures and stimulation of immune cells. Innate Immun 22:1–10. doi: 10.1177/1753425916649676. [DOI] [PubMed] [Google Scholar]
- 16.Baarsch MJ, Foss DL, Murtaugh MP. 2000. Pathophysiologic correlates of acute porcine pleuropneumonia. Am J Vet Res 61:684–690. doi: 10.2460/ajvr.2000.61.684. [DOI] [PubMed] [Google Scholar]
- 17.Meng A, Wang B, Zhang X, Qi N, Liu D, Wu J. 2015. Additive suppression of LPS-induced IL-10 and TNF-α by pre-treatment of dexamethasone and SB203580 in a murine alveolar macrophage cell line (MH-S). Inflammation 38:1260–1266. doi: 10.1007/s10753-014-0093-x. [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Qin W, Ruidong Z, Liu S, Zhang H, Sun C, Feng X, Gu J, Du C, Han W. 2015. Differential gene expression profiling of Actinobacillus pleuropneumoniae during induction of primary alveolar macrophage apoptosis in piglets. Microb Pathog 78:74–86. doi: 10.1016/j.micpath.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 19.Paradis SE, Dubreuil JD, Gottschalk M, Archambault M, Jacques M.. 1999. Inhibition of adherence of Actinobacillus pleuropneumoniae to porcine respiratory tract cells by monoclonal antibodies directed against LPS and partial characterization of the LPS receptors. Curr Microbiol 39:313–320. doi: 10.1007/s002849900465. [DOI] [PubMed] [Google Scholar]
- 20.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162:3749–3752. [PubMed] [Google Scholar]
- 21.Xu C, Chen G, Yang W, Xu Y, Xu Y, Huang X, Liu J, Feng Y, Xu Y, Liu B. 2015. Hyaluronan ameliorates LPS-induced acute lung injury in mice via Toll-like receptor (TLR) 4-dependent signaling pathways. Int Immunopharmacol 28:1050–1058. doi: 10.1016/j.intimp.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 22.Kawai T, Akira S. 2007. Signaling to NF-κB by Toll-like receptors. Trends Mol Med 13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Akira S. 2001. Toll-like receptors and innate immunity. Adv Immunol 78:1–56. [DOI] [PubMed] [Google Scholar]
- 24.Takeda K, Akira S. 2004. TLR signaling pathways. Semin Immunol 16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S. 2003. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol 4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 26.Lu YC, Yeh WC, Ohashi PS. 2008. LPS/TLR4 signal transduction pathway. Cytokine 42:145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 27.Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ. 2007. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist eritoran. Cell 130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Ramjeet M, Cox AD, Hancock MA, Mourez M, Labrie J, Gottschalk M, Jacques M. 2008. Mutation in the LPS outer core biosynthesis gene, galU, affects LPS interaction with the RTX toxins ApxI and ApxII and cytolytic activity of Actinobacillus pleuropneumoniae serotype 1. Mol Microbiol 70:221–235. doi: 10.1111/j.1365-2958.2008.06409.x. [DOI] [PubMed] [Google Scholar]
- 30.Serebrin S, Rosendal S, Valdivieso-Garcia A, Little PB. 1991. Endothelial cytotoxicity of Actinobacillus pleuropneumoniae. Res Vet Sci 50:18–22. doi: 10.1016/0034-5288(91)90047-R. [DOI] [PubMed] [Google Scholar]
- 31.Chelen CJ, Fang Y, Freeman GJ, Secrist H, Marshall JD, Hwang PT, Frankel LR, Dekruyff RH, Umetsu DT. 1995. Human alveolar macrophages present antigen ineffectively due to defective expression of B7 costimulatory cell surface molecules. J Clin Invest 95:1415–1421. doi: 10.1172/JCI117796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao J, Tang J, Chen Q, Tang D, Liu M, Luo M, Wang Y, Wang J, Zhao Z, Tang C. 2015. miR-429 regulates alveolar macrophage inflammatory cytokine production and is involved in LPS-induced acute lung injury. Biochem J 471:281–291. doi: 10.1042/BJ20131510. [DOI] [PubMed] [Google Scholar]
- 33.Kavanová L, Prodělalová J, Nedbalcová K, Matiašovic J, Volf J, Faldyna M, Salát J. 2015. Immune response of porcine alveolar macrophages to a concurrent infection with porcine reproductive and respiratory syndrome virus and Haemophilus parasuis in vitro. Vet Microbiol 180:28–35. doi: 10.1016/j.vetmic.2015.08.026. [DOI] [PubMed] [Google Scholar]
- 34.Dai H, Pan L, Lin F, Ge W, Li W, He S. 2015. Mechanical ventilation modulates Toll-like receptors 2, 4, and 9 on alveolar macrophages in a ventilator-induced lung injury model. J Thorac Dis 7:616–624. doi: 10.3978/j.issn.2072-1439.2015.02.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sijan Z, Antkiewicz DS, Heo J, Kado NY, Schauer JJ, Sioutas C, Shafer MM. 2015. An in vitro alveolar macrophage assay for the assessment of inflammatory cytokine expression induced by atmospheric particulate matter. Environ Toxicol 30:836–851. doi: 10.1002/tox.21961. [DOI] [PubMed] [Google Scholar]
- 36.Talreja J, Talwar H, Ahmad N, Rastogi R, Samavati L. 2016. Dual inhibition of Rip2 and IRAK1/4 regulates IL-1β and IL-6 in sarcoidosis alveolar macrophages and peripheral blood mononuclear cells. J Immunol 197:1368–1378. doi: 10.4049/jimmunol.1600258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ziltener P, Reinheckel T, Oxenius A. 2016. Neutrophil and alveolar macrophage-mediated innate immune control of Legionella pneumophila lung infection via TNF and ROS. PLoS Pathog 12:e1005591. doi: 10.1371/journal.ppat.1005591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gabay C. 2006. Interleukin-6 and chronic inflammation. Arthritis Res Ther 8(Suppl 2):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hannan FA, Culligan KG. 2015. Human resistin and the RELM of inflammation in diabesity. Diabetol Metab Syndr 7:54. doi: 10.1186/s13098-015-0050-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Molica F, Morel S, Kwak BR, Rohnerjeanrenaud F, Steffens S. 2015. Adipokines at the crossroad between obesity and cardiovascular disease. Thromb Haemostasis 113:553–566. doi: 10.1160/TH14-06-0513. [DOI] [PubMed] [Google Scholar]
- 41.Al-Daghri N, Chetty R, McTernan PG, Al-Rubean K, Al-Attas O, Jones AF, Kumar S. 2005. Serum resistin is associated with C-reactive protein and LDL-cholesterol in type 2 diabetes and coronary artery disease in a Saudi population. Cardiovasc Diabetol 4:10. doi: 10.1186/1475-2840-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ayeser T, Basak M, Arslan K, Sayan I. 2016. Investigating the correlation of the number of diagnostic criteria to serum adiponectin, leptin, resistin, TNF-alpha, EGFR levels and abdominal adipose tissue. Diabetes Metab Syndr 10:S165–S169. doi: 10.1016/j.dsx.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 43.Deshmukh SK, Srivastava SK, Bhardwaj A, Singh AP, Tyagi N, Marimuthu S, Dyess DL, Zotto VD, Carter JE, Singh S. 2015. Resistin and interleukin-6 exhibit racially-disparate expression in breast cancer patients, display molecular association and promote growth and aggressiveness of tumor cells through STAT3 activation. Oncotarget 6:11231–11241. doi: 10.18632/oncotarget.3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lozano-Nuevo JJ, Estrada-Garcia T, Vargas-Robles H, Escalante-Acosta BA, Rubio-Guerra AF. 2011. Correlation between circulating adhesion molecules and resistin levels in hypertensive type-2 diabetic patients. Inflamm Allergy Drug Targets 10:27–31. doi: 10.2174/187152811794352024. [DOI] [PubMed] [Google Scholar]
- 45.Forrest O, Chopyk D, Ingersoll S, Yoo D, Brown M, Tangpricha V, Tirouvanziam R, Rada B. 2015. Human resistin, a neutrophil-derived metabolic and inflammatory mediator correlates with decreased lung function and NETosis in cystic fibrosis airway disease (HUM1P.261). J Immunol 194(1 Suppl):52.10. [Google Scholar]
- 46.Ballantyne D, Scott H, Macdonald-Wicks L, Gibson PG, Wood LG. 2016. Resistin is a predictor of asthma risk and resistin:adiponectin ratio is a negative predictor of lung function in asthma. Clin Exp Allergy 46:1–10. doi: 10.1111/cea.12614. [DOI] [PubMed] [Google Scholar]
- 47.Takeda K, Akira S. 2007. Toll-like receptors. Curr Protoc Immunol Chapter 14:Unit 14.12. doi: 10.1002/0471142735.im1412s77. [DOI] [PubMed] [Google Scholar]
- 48.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. 2003. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science 301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 49.Pålsson-McDermott EM, O'Neill LA. 2004. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 113:153–162. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hayden MS, Ghosh S. 2008. Shared principles in NF-κB signaling. Cell 132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 51.Hoffmann A, Levchenko A, Scott ML, Baltimore D. 2002. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science 298:1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 52.Park J, Min JS, Kim B, Chae UB, Yun JW, Choi MS, Kong IK, Chang KT, Lee DS. 2015. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-κB pathways. Neurosci Lett 584:191–196. doi: 10.1016/j.neulet.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 53.Petr V, Legi S, Vojtech N. 2013. Andrographolide protects against LPS-induced acute lung injury by inactivation of NF-κB. PLoS One 8:e56407. doi: 10.1371/journal.pone.0056407. [DOI] [PMC free article] [PubMed] [Google Scholar]