

Abstract
Purpose
Aberrant activation of the PI3K pathway has been implicated in resistance to HER2 targeted therapy, but results of clinical trials are confounded by the co-administration of chemotherapy. We investigated the effect of perturbations of this pathway in breast cancers from patients treated with neoadjuvant anti-HER2 targeted therapy without chemotherapy.
Patients and Methods
Baseline tumor samples from patients with HER2 positive breast cancer enrolled in TBCRC006 (NCT00548184), a 12-week neoadjuvant clinical trial with lapatinib plus trastuzumab (plus endocrine therapy for estrogen receptor (ER)-positive tumors), were assessed for PTEN status by immunohistochemistry and PIK3CA mutations by sequencing. Results were correlated with pathologic complete response (pCR).
Results
Of 64 evaluable patients, PTEN immunohistochemistry and PIK3CA mutation analysis were performed for 59 and 46 patients, respectively. PTEN status (dichotomized by H-score median) was correlated with pCR (32% in high PTEN vs. 9% in low PTEN, p=0.04). PIK3CA mutations were identified in 14/46 tumors at baseline (30%) and did not correlate with ER or PTEN status. One patient whose tumor harbored a PIK3CA mutation achieved pCR (p=0.14). When considered together (43 cases), 1/25 cases (4%) with a PIK3CA mutation and/or low PTEN expression levels had a pCR compared to 7/18 cases (39%) with wild-type PI3KCA and high PTEN expression levels (p=0.006).
Conclusion
PI3K pathway activation is associated with resistance to lapatinib and trastuzumab in breast cancers, without chemotherapy. Further studies are warranted to investigate how to use these biomarkers to identify upfront patients who may respond to anti-HER2 alone, without chemotherapy.
Keywords: PIK3CA mutations, PTEN levels, HER2-positive breast cancer, pCR, lapatinib, trastuzumab
INTRODUCTION
About 15–20% of breast cancers have amplification of the human epidermal growth factor receptor 2 gene (ERBB2 or HER2) and overexpression of its protein product HER2 (1). HER2− positive breast cancers (HER2+) have an aggressive clinical behavior although adjuvant chemotherapy markedly improves outcomes (2). The introduction of trastuzumab further improved outcomes when added to chemotherapy (3), however many patients with HER2+ disease have de novo, or develop acquired, resistance to trastuzumab-containing regimens.
HER2 belongs to a membrane tyrosine kinase family (HER1–4). This family works coordinately to activate downstream signaling via, among others, the PI3K/AKT and the mitogen activated protein kinase (MAPK) pathways, which regulate genes involved in cell proliferation, survival, angiogenesis, differentiation, invasion and metastasis (4).
Many potential mechanisms of resistance to trastuzumab have emerged from preclinical studies (5,6). One such mechanism is incomplete blockade of the HER receptor family by trastuzumab (7). Another mechanism is the estrogen receptor (ER) in those tumors that express both ER and HER2 (8–10). Using two different combinations of anti-HER agents, we have shown for the first time that HER2+ xenograft tumors could be eradicated in an in vivo model (10,11). In both cases, simultaneous blockade of ER was required for maximal effects.
These preclinical data led to a neoadjuvant clinical trial (TBCRC 006) in patients with large HER2+ primary breast cancers (median = 6cm), using 12 weeks of lapatinib plus trastuzumab combined with an aromatase inhibitor if tumors were ER-positive. No chemotherapy was given during the neoadjuvant period (12). The pCR was 36% in patients with ER-negative (ER−)/HER2+ breast cancer and 21% in patients with ER-positive (ER+)/HER2+ breast cancer, which is similar to the pCR rates reported upon treatment with a combination of chemotherapy and trastuzumab (12). This and other studies (13,14) strongly argue that a portion of patients does need chemotherapy. Identifying which tumors are likely to be sensitive or resistant can help refine treatment, and possibly spare some patients chemotherapy in the future. Other studies using chemotherapy combined with multiple HER2 inhibitors confirmed the added benefit of more complete blockade of the HER family in inducing pCR (13,15,16).
Despite the use of dual HER2 inhibition, even in combination with chemotherapy, pCR is not observed in all patients. Another mechanism of resistance that could explain this treatment failure is activation of the pathways downstream of HER2 by loss of PTEN, activating mutations in PIK3CA, or CCNE1 (Cyclin E) amplification (17–21). Activation of the PI3K pathway by increased expression of a preexisting mutation or by the expression of a de novo additional mutation has been also shown in acquired lapatinib resistance in experimental models (22). In two prior neoadjuvant trials that included chemotherapy we reported that the presence of PIK3CA mutations or PTEN loss of expression was associated with resistance to trastuzumab but not lapatinib (23). Similar to our findings, Xia et al. reported that lapatinib antitumor activity in HER2+ breast cancer is not dependent upon PTEN status (24). In contrast to our findings, however, recent data from neoadjuvant and adjuvant clinical trials of HER2-targeted therapy with chemotherapy showed no association between PTEN levels and pCR or disease-free survival (25–27). It could be posited that simultaneous use of chemotherapy with HER2 targeted therapy might obscure mechanisms of resistance to the targeted therapy itself. Furthermore, complete loss of PTEN is not required to activate the PI3K pathway suggesting the possibility that this arbitrary cut-off might not be optimal (28).
In the present study we evaluated baseline, pretreatment tumor biopsies from patients with HER2+ breast cancer enrolled in the TBCRC 006 trial that investigated lapatinib and trastuzumab (combined with endocrine therapy if the tumor was also ER+), but did not include chemotherapy. We aimed to determine whether activating PI3KCA mutations or low PTEN expression levels would correlate with de novo resistance (or notably decreased response) to this dual anti-HER2 therapy alone, without chemotherapy.
METHODS
Patients and Clinical Samples
The clinical trial TBCRC006 (NCT00548184) was a multicenter single-arm phase II study conducted through the Translational Breast Cancer Research Consortium (12). Institutional review board and scientific committee approval were obtained at the lead site (Baylor College of Medicine) and other participating sites. Written informed consent was obtained from all patients. Patients with HER2-positive invasive breast carcinomas, assessed by immunohistochemistry (IHC) or fluorescence in situ hybridization, according to the 2007 American Society of Clinical Oncology (ASCO)/College of American Pathologists (CAP) guidelines (29), whose tumors were >3 cm by clinical measurement or >2 cm with a palpable ipsilateral axillary lymph node were eligible (Figure 1). Study participants were treated for 12 weeks with lapatinib (Tykerb, supplied by GlaxoSmithKline [London, UK]) 1000 mg orally every day and trastuzumab 4 mg/kg loading dose followed by 2 mg/kg per week. If the tumor was ER+ and/or progesterone receptor (PR)-positive by IHC (according to the 2010 ASCO/CAP guidelines) (30), patients were also treated with letrozole 2.5 mg orally once per day (combined with LHRH agonist of choice in premenopausal women). The rate of pathologic complete response (pCR), defined as disappearance of all invasive carcinoma in the breast, was assessed at the time of surgery according to the guidelines and practice of participating institutions.
Figure 1. Study schema of the neoadjuvant phase II TBCRC 006 clinical trial.
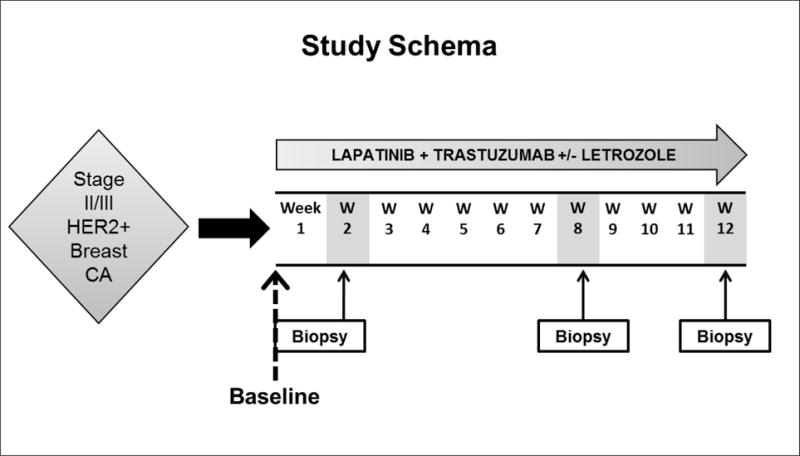
Patients with stage II/III HER2-positive breast cancers were eligible. Patients received lapatinib and trastuzumab with letrozole when estrogen receptor-positive, without chemotherapy, and biopsies were obtained at baseline and weeks 2, 8 and 12 of treatment (at surgery). HER2, human epidermal growth factor 2. Pretreatment baseline biopsies (dashed line) were evaluated for the current study.
Tumor biopsies were collected at baseline (before treatment), and after 2, 8, and 12 weeks of treatment. Collected tissues were placed in formalin for subsequent paraffin embedding [formalin-fixed paraffin embedded tissue (FFPE)] or flash frozen on dry ice. For the present study, only baseline tissue samples were analyzed.
PTEN Immunohistochemical Analysis
PTEN expression was assessed by immunohistochemistry (IHC) on freshly cut 4μm tissue sections of FFPE specimens that were deparaffinized, followed by heat-mediated antigen retrieval using 0.1M Tris-HCL (pH 9.0) buffer. To decrease nonspecific binding, sections were first treated with 3% hydrogen peroxide solution. Tissues were then incubated for 1 hour at room temperature with rabbit polyclonal PTEN (D4.3) antibody (Cell Signaling, Beverly, MA) at a 1:100 dilution in SignalStain antibody diluents (Cell Signaling), followed by incubation with Envision Labelled Polymer-HRP Anti-Rabbit (Dako, Carpenteria, CA), DAB+ solution (Dako), and DAB Sparkle Enhancer (Biocare, Concord, CA) for detection. Immunohistochemical results were evaluated independently by two pathologists (A.C and S.H.) who were blinded to the results of genetic analysis and patient outcome. As a specimen’s internal control, stromal cells were assessed for positive staining; samples devoid of staining in the internal controls were considered uninterpretable. Each section was scored for percent positivity (0–100%) and level of intensity (0–3). The H-score of each sample was calculated by multiplying the percentage and intensity scores.
PIK3CA Mutation Analysis
Eight micron-thick sections from FFPE tumor biopsies were stained with nuclear fast red and microdissected with a needle under a stereomicroscope (Olympus SZ61) to ensure >80% neoplastic cells as previously described (31). Genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen) as previously described (31). For frozen tissues, genomic DNA from tumor biopsies was extracted from frozen tissue using the AllPrep DNA/RNA Mini Kit (Qiagen). PIK3CA mutations were identified either by targeted massively parallel sequencing using the Ion AmpliSeq™ Kit 2.0 and the Ion Ampliseq™ Cancer Hotspot Panel v2 (n=36) (Life Technologies, Carlsbad, CA) according to the manufacturers’ protocols or Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) (n=6) as previously described (31). For Ion AmpliSeq, sequencing alignment, variant calling, and curation were carried out by using the Ion Torrent Suite (TS) software V3.6 and TS Variant Caller Plugin software V3.4.1 (Life Technologies, ThermoFisher Scientific) and as previously described (32), and all PIK3CA mutations identified were manually reviewed using the Integrative Genomics Viewer (IGV) (33). Sequencing analysis of MSK-IMPACT data was performed as previously described (31), and somatic single nucleotide variants (SNVs) were identified using MuTect(34), and small insertions and deletions (indels) using Strelka and VarScan 2 (35,36). For samples with very limited DNA available (n=4), Sanger sequencing of exons 9 and 20 of the PIK3CA gene was performed essentially as previously described (37) (mRNA reference sequence: NM_006218.2). Primers for both exons flank the respective intron/exon junctions. Direct sequence analysis of PCR products was performed in both forward and reverse directions using automated fluorescence dideoxy sequencing methods. Reference sequence NT_005612.16 was used for sequence comparison. The output sequences were analyzed using Mutation Surveyor version 3.97 (Soft Genetics). The mutation nomenclature is based on the convention recommended by the Human Genome Variation Society (38). All mutations identified by targeted massively parallel sequencing were confirmed by Sanger sequencing.
Statistical Analysis
Data were summarized by descriptive analysis. Chi-square tests (not continuity corrected; used when expected values > 5) or Fisher’s Exact tests were performed to assess the associations between pCR and PTEN expression and/or PIK3CA mutation status, and between PTEN expression, PIK3CA mutation status with ER status and with each other. Odds ratios and 95% conference intervals were calculated to measure the strength of the associations.
RESULTS
Patient demographic and clinical characteristics are summarized in Table 1. Sixty-six patients with HER2+ breast cancer were enrolled in the neoadjuvant TBCRC 006 trial of whom 64 patients were evaluable for response. Seventeen patients had a pCR (27%) following lapatinib and trastuzumab treatment (plus endocrine treatment in patients with ER+/HER2+ disease). The rate of pCR was 21% in the ER-positive subgroup and 36% in the ER-negative subgroup.
Table 1.
Patient Demographic and Clinical Characteristics
| Characteristic | Evaluable* Patients (n=64) | PTEN IHC Available (n=59) | PIK3CA Mutation Available (n=46) | PTEN and PIK3CA Available (n=43) |
|---|---|---|---|---|
| Age, years | ||||
| Median | 50 | 50 | 50.5 | 50 |
| Range | 31–74 | 31–74 | 31–70 | 31–70 |
|
| ||||
| Tumor size, cm | ||||
| Median | 6 | 6 | 6 | 6 |
| Range | 1.5–30 | 2–30 | 1.5–30 | 2–30 |
|
| ||||
| Biomarkers† | ||||
| ER positive | 31 (51%) | 31(52%) | 22 (49%) | 22 (51%) |
| PR positive | 22 (36%) | 22 (37%) | 17 (38%) | 17 (40%) |
|
| ||||
| Pathologic complete response | ||||
| Yes | 17 (27%) | 14 (24%) | 10 (22%) | 8 (18%) |
| No | 47 (73%) | 45 (76%) | 36 (78%) | 35 (81%) |
Abbreviations: ER, estrogen receptor; PR, progesterone receptor.
64 patients in the study were evaluable for efficacy
ER, PR were from central review IHC, 61 out of 64 patients’ data were available
Tissue from baseline biopsies was available from 59 evaluable patients for PTEN IHC studies and 46 had sufficient tissue available for DNA extraction and PIK3CA mutation analysis (Figure 2). Fourteen out of the 59 patients whose primary tumors at baseline were subjected to PTEN IHC analysis had a pCR (24%) and 10 out of the 46 patients whose baseline primary tumors were subjected to PIK3CA analysis had a pCR (22%). In the 43 patients, where both PTEN and PIK3CA analysis were performed, eight had a pCR (19%)
Figure 2.
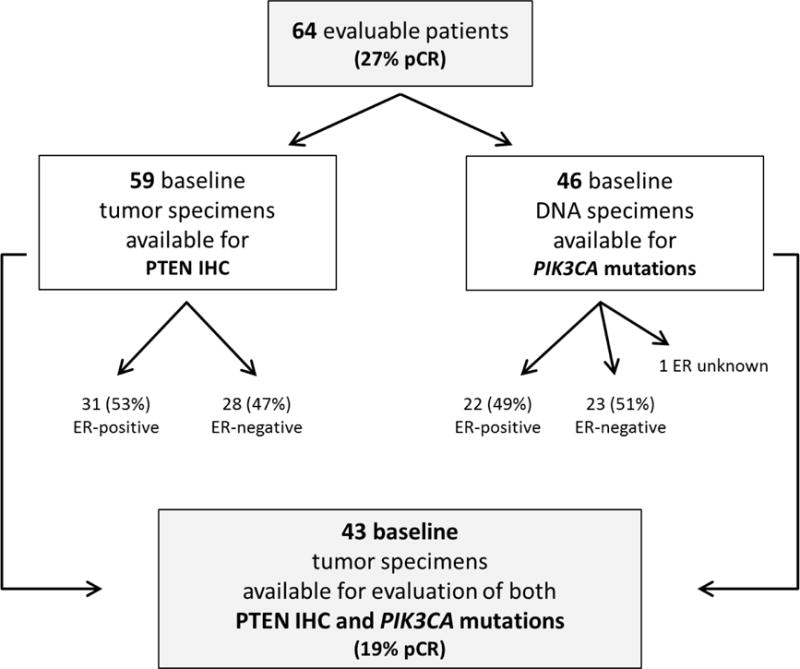
Evaluable material for baseline biomarker analysis and associated pathologic complete response (pCR) rates
PTEN Immunohistochemistry
Given that reduced expression levels of PTEN can contribute to downstream activation of the PI3K pathway, we measured PTEN protein expression by IHC and correlated its expression level with pCR. PTEN IHC was successfully performed and scored on all 59 available baseline biopsy tumors, which revealed a PTEN median H-score of 100 (range 0–300). Figure 3 shows examples of results of staining demonstrating negative, weak, and strong staining.
Figure 3. Immunohistochemical staining and associated H scores for PTEN in baseline specimens.
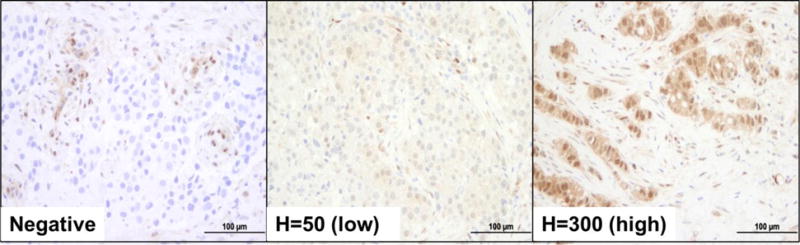
Representative micrographs of breast cancer biopsies showing negative (left), low (middle), and high (right) PTEN expression levels. PTEN status was dichotomized by H-score median. Please note the stromal cells, which were used as internal positive controls. Scale bars, 100μm.
Since PI3K activation may occur with low levels of PTEN but does not require complete loss of the protein (28,39), PTEN status was dichotomized by the median H-score of 100 as low (< 100) or high (≥100). In this series, 37 HER2+ breast cancers at baseline showed high PTEN expression levels (H-score ≥100) and 22 baseline HER2+ breast cancers low PTEN expression levels (H-score <100) (Figure 3).
When correlated with treatment response, we found a statistically significant difference between PTEN expression levels and pCR (Table 2). pCR was observed in 12 out of 37 patients (32%) with high PTEN expression levels and only 2 out of 22 patients (9%) with low PTEN expression levels (p=0.04, Fisher’s exact test) (Table 2). Complete PTEN loss by IHC was found in only 6 (10%) patients. One of these (17%) had a pCR, while there were 13 pCRs (25%) out of 53 patients with any PTEN expression (p=1, Fisher’s exact test). Thus, the median value proved a better discriminator.
Table 2.
Activation of PI3K Pathway by Low PTEN expression levels and/or PIK3CA Mutations as Pretreatment Predictive Markers of Response to the Trastuzumab + Lapatinib regimen
| Marker | pCR | No pCR | OR | 95% CI | p* | ||
|---|---|---|---|---|---|---|---|
|
| |||||||
| n | % | n | % | ||||
| PTEN (n=59) | 0.04 | ||||||
| High (≥100) | 12 | 32 | 25 | 68 | 4.80 | 0.96–23.97 | |
| Low (< 100) | 2 | 9 | 20 | 91 | 1 | – | |
|
| |||||||
| PIK3CA (n=46) | 0.14 | ||||||
| WT | 9 | 28 | 23 | 72 | 5.09 | 0.58–44.78 | |
| Mutant | 1 | 7 | 13 | 93 | 1 | – | |
|
| |||||||
| PTEN low or PIK3CA mutation (n=43) | |||||||
| No | 7 | 39 | 11 | 61 | 15.27 | 1.67–139.68 | 0.006 |
| Yes | 1 | 4 | 24 | 96 | 1 | – | |
Abbreviations: PI3K, phosphoinositol-3 kinase; pCR, pathologic complete response; OR, odds ratio; PTEN, phosphatase and tensin homolog; WT, wild type.
Chi-square or Fisher’s exact test, whichever is appropriate.
PIK3CA Mutation Analysis
In addition to loss of PTEN expression, PIK3CA mutations may result in activation of the PI3K pathway downstream from HER2, abrogating the impact of inhibition of the receptor layer and resulting in treatment resistance.
In the 46 patient samples with sufficient tissue for DNA extraction and assessment of PIK3CA mutations, 14 (30%) harbored activating mutations (Table 3), including mutations at residues R88 (n=1), E542 (n=2), E545 (n=1), H1047 (n=9) and G1049 (n=1). Nine out of 32 patients with wild-type PIK3CA achieved pCR (28%). None of the patients with the classical PIK3CA hotspot mutations (i.e. E542K, E545K or H1047R) evolved to pCR. However, one patient with an activating R88 PIK3CA mutation (1/14), however, (1/14) achieved a pCR (7%, p=0.14).
Table 3.
Baseline PIK3CA Mutation Frequency
| Mutation | p110α domain | n | % |
|---|---|---|---|
| p.R88Q | p85-binding | 1 | 2.2 |
| p.E542K | Helical | 2 | 4.4 |
| p.E545K | Helical | 1 | 2.2 |
| p.G1049R | Catalytic | 1 | 2.2 |
| p.H1047L | Catalytic | 1 | 2.2 |
| p.H1047R | Catalytic | 8 | 17.4 |
| Wild-type | 32 | 69.6 |
Association of PIK3CA mutations and PTEN status with pCR
Given that both PIK3CA mutations and PTEN expression levels are surrogates for PI3K pathway activation, we next analyzed the series of patients for which both PIK3CA mutational status and PTEN expression level were available (n=43, Table 2).
Out of the 18 patients whose HER2+ breast cancers expressed high PTEN expression levels and had no PIK3CA mutation, 7 patients had a pCR (39%). In contrast, one patient (1/25) achieved pCR whose tumor harbored a PIK3CA mutation or had low PTEN expression (p=0.006) (Table 2).
Moreover, PIK3CA mutation status and PTEN status were independent of ER status (p= 0.92 and 0.76, respectively) and of each other (p=0.89, Chi-Squared test), suggesting these are independent variables or that a larger sample is required to detect a correlation.
These findings support the hypothesis that deregulation of the PI3K pathway is associated with resistance to anti-HER2 therapy.
DISCUSSION
Potent inhibition of HER2 with combination therapy targeting HER receptors has shown a clinically meaningful response rate and rate of pCR in the neoadjuvant setting without chemotherapy (12–14). In our TBCRC006 trial the combination of lapatinib and trastuzumab (with the addition of endocrine therapy in patients with ER-positive tumors) for 12 weeks resulted in a pCR rate of 27%. Recently, the study by Prat et al. reported an overall pCR rate of 30% with the same dual anti-HER2 regimen administered for 18 weeks (14). The NeoSphere trial, showed a similar result in the arm with trastuzumab plus pertuzumab and no chemotherapy, with pCR rate of 17% (13). The potential of treating a subset of patients with targeted therapy alone and without chemotherapy is attractive, as it would spare patients the toxicity and cost of chemotherapy.
However, to consider the use of targeted therapy alone in the future, it is essential to accurately distinguish patients who would benefit from this approach from those requiring chemotherapy. Results from the ALTTO trial showed a small and non-significant benefit for adding lapatinib to standard of care chemotherapy and trastuzumab (40). Similar, albeit statistically significant, results were observed in APHINITY when pertuzumab was added to trastuzumab (41). This highlights the importance of identifying patients who may benefit from a certain treatment as well as those in whom a de-escalation strategy might be pursued. Paclitaxel plus trastuzumab adjuvant therapy in patients with small, node-negative HER2-positive tumors is one de-escalation strategy that reported excellent outcomes (42). Identification of patients with resistant tumors and clarification of mechanisms of resistance, might identify drugs to block these escape pathways and overcome resistance. The PI3K pathway has been implicated in resistance to anti-HER2 therapy and it is plausible to speculate that patients with PI3K pathway activation on the basis of PTEN down-regulation or PIK3CA mutations might benefit from the addition of inhibitors of this pathway (17–19,21).
Our study is unique in that it allowed us to understand the influence of aberrant activation of this pathway via decreased levels of PTEN and/or the presence of activating PIK3CA mutations in patients treated exclusively with targeted therapy only and without the confounding effects of chemotherapy. Here, we report results of our molecular studies, on baseline tumor biopsies obtained from the TBCRC006 trial, investigating the potential role of PTEN expression and activating mutations in PIK3CA in resistance to lapatinib combined with trastuzumab. Our data show that low PTEN expression is associated with significantly lower response to anti-HER2 therapy with a low pCR rate of 9% vs. 32% with high PTEN. Having a significant difference in pCR when data were dichotomized by the median argues that complete loss of PTEN is not necessary, and that a partial loss may be enough to release its regulatory effect on PI3K signaling resulting in its activation (28,39), and subsequently treatment resistance. These results are consistent with other reports suggesting that moderate reductions in PTEN are sufficient to activate the pathway (28).
Several studies in the neoadjuvant, the adjuvant, and the metastatic settings investigating the role of PTEN levels in response to HER2-targeted therapies have yielded conflicting results (19,21,43,44). In a recent analysis of the neoadjuvant GeparQuattro study, higher PTEN levels, evaluated by a quantitative immunofluorescent assay, were associated with pCR after anti-HER2 treatment in combination with chemotherapy (45). In contrast, the expression of PTEN protein by IHC did not predict response to trastuzumab and lapatinib-based therapies in the NeoALTTO trial (25). Retrospective analysis of two large adjuvant trials of chemotherapy plus trastuzumab also failed to demonstrate any association between PTEN protein levels and resistance to trastuzumab (26,27). In addition to the use of chemotherapy, which could obscure the effect of PTEN loss on resistance to trastuzumab, these studies used a different primary antibody or required complete loss of PTEN by IHC to dichotomize between low and high. Our findings may reconcile the discrepancies between these studies, in that our data suggest that low but not necessarily absent PTEN as assessed by IHC staining might suffice to confer resistance to dual anti-HER2 inhibition.
One possible explanation for our findings is that dichotomization into positive and negative subsets using a cut-point of absent PTEN staining imparts less effect because of dilution of the “positive-PTEN” group of tumors with the group of “low-PTEN” tumors that have activated the PI3K pathway. In support of this idea are results from a recent study using a Pten hypermorphic mouse model (39). This study shows that subtle downregulation of PTEN levels results in pleiotropic biological consequences in the breast tissue, including the activation of PI3K downstream signaling, induction of gene expression signatures associated with cell cycle and tissue hyper-proliferation, and development of breast tumors at the highest frequency among a spectrum of other tumors (39). Similarly, in a prior study using an inducible siRNA we showed that moderate reductions in PTEN activated the PI3K pathway in breast cancer cells. The fact that PTEN is infrequently inactivated at the genomic level in breast cancer, especially within the HER2 and the luminal subtypes (46), further emphasizes the importance of subtle reductions in its expression for the activation of the PI3K pathway and resistance to HER2 targeted therapy. A clinically useful assay to measure PTEN with appropriate cutoff values remains a challenge and warrants further study.
Our data also show that only one tumor with a non-hotpsot PIK3CA activating mutations had a pCR (7% vs. 28% with wild-type PIK3CA, p=0.14) highlighting the potential role of these activating mutations in resistance to anti-HER2 treatment by downstream activation of the pathway, thereby circumventing potent inhibition of the receptor layer and rendering the treatment ineffective. Similar to our findings, recent data from the large phase III NeoALTTO trial also suggested that tumors with PI3KCA mutations are less likely to achieve a pCR from HER2 targeted therapy combined with chemotherapy (47). Reduced pCR rates were most pronounced in the arm of dual HER2-targeted therapy with lapatinib plus trastuzumab. The lower but still substantial pCR rates observed in tumors with PI3KCA mutations in NeoALTTO is likely due to the use of chemotherapy for all patients. Our results are also concordant with recent reports from the Neosphere (48) and the GeparSixto and GeparQuinto studies (49). Of note, the two Gepar studies also suggested that the largest differences in pCR rate between PIK3CA mutant and wild-type was found in patients receiving dual HER2 blockade. Although chemotherapy might partially overcome the resistance to lapatinib plus trastuzumab caused by PIK3CA mutations, the favorable effect of HER2 inhibition on chemosensitivity would be lost in such tumors. Overcoming resistance to HER2 inhibition caused by PIK3CA mutations would likely also improve the cytotoxic effects of the chemotherapy itself by effectively blocking the HER2/PI3K/AKT cell survival pathway. The role of PIK3CA is different in ER+/HER2− tumors, where PIK3CA mutations are associated with luminal A tumors with a better prognosis and improved response to endocrine therapy (50).
Here, only one patient out of 24 with either low PTEN or a PIK3CA activating mutation (in the 43 cohort of patients for which both were measured) had a pCR (4% vs. 39% with wild-type PIK3CA/high PTEN, p=0.006). Equally important, PTEN and PIK3CA status were not correlated with each other or with ER expression arguing they are independent variables that can lead to deregulation of the PI3K pathway.
Our study has limitations; our cohort of patients is relatively small, and we were not able to obtain PIK3CA mutational status on all tumors. The difference in pCR between PIK3CA mutant and wild-type tumors did not achieve statistical significance on its own, albeit it did when combined with PTEN. Our results need validation in a large cohort. Still, we observed differences in response in patients with low PTEN and PIK3CA mutations that are statistically significant and may be clinically meaningful. If validated these in larger prospective cohorts, our findsings will enable the design of prospective clinical trials testing de-escalation strategies (e.g., no or less chemotherapy) in carefully selected HER2-positive breast cancer patients. This work in ongoing.
In the age of precision medicine and increasing awareness of the burden of healthcare costs, the current approach of simply adding new agents to existing regimens for a small benefit and without proper patient selection is scientifically unsound and economically unsustainable. More individualized approaches in HER2-positive breast cancer that are based on patient and tumor characteristics are warranted.
Acknowledgments
Research support: This work was supported in part by NCI grants P50CA58183 and P50CA186784 (SPORE), P30CA125123, P30CA008748, and R01CA72038, as well as grants from the Komen Foundation for the Cure, the Avon Foundation, the Breast Cancer Research Foundation, the Cancer Prevention & Research Institute of Texas CPRIT RP 140102, the Conquer Cancer Foundation - Gianni Bonadonna Breast Cancer Research Fellowship and the Translational Breast Cancer Research Consortium.
None of the funding agencies had any role in the design, analysis, or reporting of analyses.
Footnotes
Authors’ Contributions:
Mothaffar Rimawi: Study design, data collection, data analysis, data interpretation, and writing
Carmine De Angelis: Study design, data collection, data analysis, data interpretation, and writing
Alejandro Contreras: Study design, data collection, data analysis, data interpretation, and writing
Fresia Pareja: Study design, data analysis, data interpretation, and writing
Felipe C. Geyer: Study design, data analysis, data interpretation, and writing
Kathleen A. Burke: Study design, data analysis, data interpretation, and writing
Sabrina Herrera: Study design, data analysis, data interpretation, and writing
Tao Wang: Study design, data analysis, data interpretation, and writing
Ingrid A Mayer: Study design, data collection, data interpretation, and writing
Andres Forero: Study design, data collection, data interpretation, and writing
Rita Nanda: Study design, data collection, data interpretation, and writing
Matthew P. Goetz: Study design, data collection, data interpretation, and writing
Jenny C. Chang: Study design, data collection, data interpretation, and writing
Ian E. Krop: Study design, data interpretation, and writing
Antonio C. Wolff: Study design, data interpretation, and writing
Anne C. Pavlick: Study design, data collection, data interpretation, and writing
Suzanne A. W. Fuqua: Study design, data collection, data interpretation, and writing
Carolina Gutierrez: Study design, data collection, data interpretation, and writing
Susan G. Hilsenbeck: Study design, data analysis, data interpretation, and writing
Marilyn M. Li: Study design, data analysis, data interpretation, and writing
Britta Weigelt: Study design, data collection, data analysis, data interpretation, and writing
Jorge S. Reis-Filho: Study design, data collection, data analysis, data interpretation, and writing
C. Kent Osborne: Study design, data collection, data analysis, data interpretation, and writing
Rachel Schiff: Study design, data collection, data analysis, data interpretation, and writing.
Conflicts of Interest:
Mothaffar F. Rimawi: Research grant from GlaxoSmithKline (to Institution), Consulting with Genentech.
Carmine De Angelis: Nothing to disclose
Alejandro Contreras: Nothing to disclose
Fresia Pareja: Nothing to disclose
Felipe C. Geyer: Nothing to disclose
Kathleen A. Burke: Nothing to disclose
Sabrina Herrera: Nothing to disclose
Tao Wang: Nothing to disclose
Ingrid A Mayer: Nothing to disclose
Andres Forero: Research grants from GlaxoSmithKline and Genentech (to Institution)
Rita Nanda: Nothing to disclose
Matthew P. Goetz: Nothing to disclose
Jenny C. Chang: Nothing to disclose
Ian E. Krop: Consulting: Genentech/Roche. Research grant from Genentech/Roche (to Institution)
Antonio C. Wolff: Research grant from Genentech (to Institution)
Anne C. Pavlick: Nothing to disclose
Suzanne A. W. Fuqua: Nothing to disclose
Carolina Gutierrez: Nothing to disclose
Susan G. Hilsenbeck: Nothing to disclose
Marilyn M. Li: Nothing to disclose
Britta Weigelt: Nothing to disclose
Jorge S. Reis-Filho: Nothing to disclose
C. Kent Osborne: Nothing to disclose
Rachel Schiff: Nothing to disclose
References
- 1.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244(4905):707–12. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 2.Thor AD, Berry DA, Budman DR, Muss HB, Kute T, Henderson IC, et al. erbB-2, p53, and efficacy of adjuvant therapy in lymph node-positive breast cancer. J Natl Cancer Inst. 1998;90(18):1346–60. doi: 10.1093/jnci/90.18.1346. [DOI] [PubMed] [Google Scholar]
- 3.Zardavas D, Fouad TM, Piccart M. Optimal adjuvant treatment for patients with HER2-positive breast cancer in 2015. Breast. 2015;24(Suppl 2):S143–8. doi: 10.1016/j.breast.2015.07.034. [DOI] [PubMed] [Google Scholar]
- 4.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7(7):505–16. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 5.Rimawi MF, Schiff R, Osborne CK. Targeting HER2 for the treatment of breast cancer. Annu Rev Med. 2015;66:111–28. doi: 10.1146/annurev-med-042513-015127. [DOI] [PubMed] [Google Scholar]
- 6.Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. 2012;17(1):1–16. doi: 10.1615/critrevoncog.v17.i1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghosh R, Narasanna A, Wang SE, Liu S, Chakrabarty A, Balko JM, et al. Trastuzumab has preferential activity against breast cancers driven by HER2 homodimers. Cancer Res. 2011;71(5):1871–82. doi: 10.1158/0008-5472.CAN-10-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang YC, Morrison G, Gillihan R, Guo J, Ward RM, Fu X, et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers–role of estrogen receptor and HER2 reactivation. Breast cancer research: BCR. 2011;13(6):R121. doi: 10.1186/bcr3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Konecny G, Pauletti G, Pegram M, Untch M, Dandekar S, Aguilar Z, et al. Quantitative association between HER-2/neu and steroid hormone receptors in hormone receptor-positive primary breast cancer. J Natl Cancer Inst. 2003;95(2):142–53. doi: 10.1093/jnci/95.2.142. [DOI] [PubMed] [Google Scholar]
- 10.Arpino G, Gutierrez C, Weiss H, Rimawi M, Massarweh S, Bharwani L, et al. Treatment of human epidermal growth factor receptor 2-overexpressing breast cancer xenografts with multiagent HER-targeted therapy. J Natl Cancer Inst. 2007;99(9):694–705. doi: 10.1093/jnci/djk151. [DOI] [PubMed] [Google Scholar]
- 11.Rimawi MF, Wiechmann LS, Wang YC, Huang C, Migliaccio I, Wu MF, et al. Reduced dose and intermittent treatment with lapatinib and trastuzumab for potent blockade of the HER pathway in HER2/neu-overexpressing breast tumor xenografts. Clin Cancer Res. 2011;17(6):1351–61. doi: 10.1158/1078-0432.CCR-10-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rimawi MF, Mayer IA, Forero A, Nanda R, Goetz MP, Rodriguez AA, et al. Multicenter phase II study of neoadjuvant lapatinib and trastuzumab with hormonal therapy and without chemotherapy in patients with human epidermal growth factor receptor 2-overexpressing breast cancer: TBCRC 006. J Clin Oncol. 2013;31(14):1726–31. doi: 10.1200/JCO.2012.44.8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gianni L, Pienkowski T, Im YH, Roman L, Tseng LM, Liu MC, et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. The lancet oncology. 2012;13(1):25–32. doi: 10.1016/S1470-2045(11)70336-9. [DOI] [PubMed] [Google Scholar]
- 14.Llombart-Cussac A, Cortes J, Pare L, Galvan P, Bermejo B, Martinez N, et al. HER2-enriched subtype as a predictor of pathological complete response following trastuzumab and lapatinib without chemotherapy in early-stage HER2-positive breast cancer (PAMELA): an open-label, single-group, multicentre, phase 2 trial. Lancet Oncol. 2017;18(4):545–54. doi: 10.1016/S1470-2045(17)30021-9. [DOI] [PubMed] [Google Scholar]
- 15.Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2012;379(9816):633–40. doi: 10.1016/S0140-6736(11)61847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneeweiss A, Chia S, Hickish T, Harvey V, Eniu A, Hegg R, et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA) Annals of oncology: official journal of the European Society for Medical Oncology. 2013;24(9):2278–84. doi: 10.1093/annonc/mdt182. [DOI] [PubMed] [Google Scholar]
- 17.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. doi: 10.1016/j.ccr.2007.08.030. doi S1535-6108(07)00262-0 [pii] 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 18.Fujita T, Doihara H, Kawasaki K, Takabatake D, Takahashi H, Washio K, et al. PTEN activity could be a predictive marker of trastuzumab efficacy in the treatment of ErbB2-overexpressing breast cancer. British journal of cancer. 2006;94(2):247–52. doi: 10.1038/sj.bjc.6602926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esteva FJ, Guo H, Zhang S, Santa-Maria C, Stone S, Lanchbury JS, et al. PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. The American journal of pathology. 2010;177(4):1647–56. doi: 10.2353/ajpath.2010.090885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scaltriti M, Eichhorn PJ, Cortes J, Prudkin L, Aura C, Jimenez J, et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(9):3761–6. doi: 10.1073/pnas.1014835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallardo A, Lerma E, Escuin D, Tibau A, Munoz J, Ojeda B, et al. Increased signalling of EGFR and IGF1R, and deregulation of PTEN/PI3K/Akt pathway are related with trastuzumab resistance in HER2 breast carcinomas. British journal of cancer. 2012;106(8):1367–73. doi: 10.1038/bjc.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brady SW, Zhang J, Seok D, Wang H, Yu D. Enhanced PI3K p110alpha signaling confers acquired lapatinib resistance that can be effectively reversed by a p110alpha-selective PI3K inhibitor. Mol Cancer Ther. 2014;13(1):60–70. doi: 10.1158/1535-7163.MCT-13-0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dave B, Migliaccio I, Gutierrez MC, Wu MF, Chamness GC, Wong H, et al. Loss of phosphatase and tensin homolog or phosphoinositol-3 kinase activation and response to trastuzumab or lapatinib in human epidermal growth factor receptor 2-overexpressing locally advanced breast cancers. J Clin Oncol. 2011;29(2):166–73. doi: 10.1200/JCO.2009.27.7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia W, Husain I, Liu L, Bacus S, Saini S, Spohn J, et al. Lapatinib antitumor activity is not dependent upon phosphatase and tensin homologue deleted on chromosome 10 in ErbB2-overexpressing breast cancers. Cancer research. 2007;67(3):1170–5. doi: 10.1158/0008-5472.CAN-06-2101. [DOI] [PubMed] [Google Scholar]
- 25.Nuciforo PG, Aura C, Holmes E, Prudkin L, Jimenez J, Martinez P, et al. Benefit to neoadjuvant anti-human epidermal growth factor receptor 2 (HER2)-targeted therapies in HER2-positive primary breast cancer is independent of phosphatase and tensin homolog deleted from chromosome 10 (PTEN) status. Annals of oncology: official journal of the European Society for Medical Oncology. 2015;26(7):1494–500. doi: 10.1093/annonc/mdv175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perez EA, Dueck AC, McCullough AE, Chen B, Geiger XJ, Jenkins RB, et al. Impact of PTEN protein expression on benefit from adjuvant trastuzumab in early-stage human epidermal growth factor receptor 2-positive breast cancer in the North Central Cancer Treatment Group N9831 trial. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31(17):2115–22. doi: 10.1200/JCO.2012.42.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stern HM, Gardner H, Burzykowski T, Elatre W, O’Brien C, Lackner MR, et al. PTEN Loss Is Associated with Worse Outcome in HER2-Amplified Breast Cancer Patients but Is Not Associated with Trastuzumab Resistance. Clin Cancer Res. 2015;21(9):2065–74. doi: 10.1158/1078-0432.CCR-14-2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu X, Creighton CJ, Biswal NC, Kumar V, Shea M, Herrera S, et al. Overcoming endocrine resistance due to reduced PTEN levels in estrogen receptor-positive breast cancer by co-targeting mammalian target of rapamycin, protein kinase B, or mitogen-activated protein kinase kinase. Breast Cancer Res. 2014;16(5):430. doi: 10.1186/s13058-014-0430-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2007;25(1):118–45. doi: 10.1200/JCO.2006.09.2775. [DOI] [PubMed] [Google Scholar]
- 30.Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. Arch Pathol Lab Med. 2010;134(6):907–22. doi: 10.1043/1543-2165-134.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piscuoglio S, Ng CK, Murray M, Burke KA, Edelweiss M, Geyer FC, et al. Massively parallel sequencing of phyllodes tumours of the breast reveals actionable mutations, and TERT promoter hotspot mutations and TERT gene amplification as likely drivers of progression. J Pathol. 2016;238(4):508–18. doi: 10.1002/path.4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang F, Li MM. Clinical application of amplicon-based next-generation sequencing in cancer. Cancer genetics. 2013;206(12):413–9. doi: 10.1016/j.cancergen.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 33.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–76. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28(14):1811–7. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 37.Eberle CA, Piscuoglio S, Rakha EA, Ng CK, Geyer FC, Edelweiss M, et al. Infiltrating epitheliosis of the breast: characterization of histological features, immunophenotype and genomic profile. Histopathology. 2016;68(7):1030–9. doi: 10.1111/his.12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Society HGV. Sequence Variant Nomenclature. 2016;15:11. < http://varnomen.hgvs.org/>. [Google Scholar]
- 39.Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, et al. Subtle variations in Pten dose determine cancer susceptibility. Nature genetics. 2010;42(5):454–8. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piccart-Gebhart M, Holmes E, Baselga J, de Azambuja E, Dueck AC, Viale G, et al. Adjuvant Lapatinib and Trastuzumab for Early Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer: Results From the Randomized Phase III Adjuvant Lapatinib and/or Trastuzumab Treatment Optimization Trial. J Clin Oncol. 2016;34(10):1034–42. doi: 10.1200/JCO.2015.62.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.von Minckwitz G, Procter M, de Azambuja E, Zardavas D, Benyunes M, Viale G, et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N Engl J Med. 2017 doi: 10.1056/NEJMoa1703643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tolaney SM, Barry WT, Dang CT, Yardley DA, Moy B, Marcom PK, et al. Adjuvant paclitaxel and trastuzumab for node-negative, HER2-positive breast cancer. N Engl J Med. 2015;372(2):134–41. doi: 10.1056/NEJMoa1406281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–27. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 44.Loi S, Michiels S, Lambrechts D, Salgado R, Sirtaine N, Fumagalli D, et al. Tumor PIK3CA mutations, lymphocyte infiltration, and recurrence-free survival (RFS) in early breast cancer (BC): results from the FinHER trial. J Clin Oncol. 2012;30(Suppl) Abstr. 507. [Google Scholar]
- 45.Loibl S, Darb-Esfahani S, Huober J, Klimowicz A, Furlanetto J, Lederer B, et al. Integrated analysis of PTEN and p4EBP1 protein expression as predictors for pCR in HER2-positive breast cancer. Clin Cancer Res. 2016;22(11):2675–83. doi: 10.1158/1078-0432.CCR-15-0965. [DOI] [PubMed] [Google Scholar]
- 46.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Majewski IJ, Nuciforo P, Mittempergher L, Bosma AJ, Eidtmann H, Holmes E, et al. PIK3CA Mutations Are Associated With Decreased Benefit to Neoadjuvant Human Epidermal Growth Factor Receptor 2-Targeted Therapies in Breast Cancer. J Clin Oncol. 2015 doi: 10.1200/JCO.2014.55.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bianchini G, Kiermaier A, Bianchi GV, Im YH, Pienkowski T, Liu MC, et al. Biomarker analysis of the NeoSphere study: pertuzumab, trastuzumab, and docetaxel versus trastuzumab plus docetaxel, pertuzumab plus trastuzumab, or pertuzumab plus docetaxel for the neoadjuvant treatment of HER2-positive breast cancer. Breast cancer research: BCR. 2017;19(1):16. doi: 10.1186/s13058-017-0806-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loibl S, von Minckwitz G, Schneeweiss A, Paepke S, Lehmann A, Rezai M, et al. PIK3CA Mutations Are Associated With Lower Rates of Pathologic Complete Response to Anti-Human Epidermal Growth Factor Receptor 2 (HER2) Therapy in Primary HER2-Overexpressing Breast Cancer. J Clin Oncol. 2014 doi: 10.1200/JCO.2014.55.7876. [DOI] [PubMed] [Google Scholar]
- 50.Guarneri V, Generali DG, Frassoldati A, Artioli F, Boni C, Cavanna L, et al. Double-blind, placebo-controlled, multicenter, randomized, phase IIb neoadjuvant study of letrozole-lapatinib in postmenopausal hormone receptor-positive, human epidermal growth factor receptor 2-negative, operable breast cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32(10):1050–7. doi: 10.1200/JCO.2013.51.4737. [DOI] [PubMed] [Google Scholar]