

Abstract
Antitumor vaccines encoding self-antigens generally have low immunogenicity in clinical trials. Several approaches are aimed at improving vaccine immunogenicity, including efforts to alter encoded epitopes. Immunization with epitopes altered for increased affinity for the major histocompatibility complex (MHC) or T-cell receptor (TCR) elicits greater numbers of CD8 T cells but inferior antitumor responses. Our previous results suggested that programmed death 1 (PD-1) and its ligand (PD-L1) increased on antigen-specific CD8 T cells and tumor cells, respectively, after high-affinity vaccination. In this report, we use two murine models to investigate whether the dose, MHC affinity, or TCR affinity of an epitope affected the antitumor response via the PD-1/PD-L1 axis. T cells activated with high-affinity epitopes resulted in prolonged APC:T-cell contact time that led to elevated, persistent PD-1 expression, and expression of other checkpoint molecules, in vitro and in vivo. Immunization with high-affinity epitopes also decreased antitumor efficacy in the absence of PD-1 blockade. Thus, APC:T-cell contact time can be altered by epitope affinity and lead to therapeutically relevant changes in vaccine efficacy mediated by changes in PD-1 expression. These findings have implications for the use of agents targeting PD-1 expression or function whenever high-affinity CD8 T cells are elicited or supplied by means of vaccination or adoptive transfer.
Keywords: Epitope, OT-1, PD-1, affinity, tumor vaccine
Introduction
The goal of anticancer vaccines is to generate a productive pool of tumor-associated antigen (TAA)-specific T cells, including CD8+ cytotoxic T lymphocytes (CTLs). Because most TAA are “self” antigens, the pool of pre-existing CTLs are likely low-affinity, as high-affinity T cells have been centrally deleted (1, 2). These characteristics have hindered vaccines directed at TAA and many approaches that attempt to subvert or avoid self-tolerance have been developed. Initial tumor vaccine efforts were aimed at foreign tumor-associated viral antigens, “non-self” TAA, such as human papilloma virus proteins expressed in cervical cancers (3–5). Other studies have focused on antigens that can be overexpressed in cancers but are not expressed in the thymus and thus may have escaped central tolerance, such as cancer-testis antigens (6, 7) and embryonic or differentiation antigens such as gp100 (8–10), Melan-A (11) and PRAME (12). Several groups also have sought to identify neoepitopes derived from tumor-specific mutations with high MHC affinity to use as tumor-specific vaccine antigens (13–18).
Nonmutated self TAA can also be targeted using modified or xenogeneic epitopes that elicit T cells with cross-reactivity to the native antigen. Specifically, the use of altered peptide ligands (APLs) in which the MHC-I (19) or T-cell receptor (TCR) (20) affinity of the TAA epitope is increased can elicit large numbers of high-affinity T cells with cross-reactivity to the native epitope. However, the resulting increase in T-cell expansion has not consistently produced a greater antitumor therapeutic response (20–22). Similarly, direct adoptive transfer of T cells expressing high-affinity TCR has shown little antitumor efficacy due to the development of tolerance mediated by the tumor microenvironment (23, 24).
We are interested in increasing the immunogenicity of DNA vaccines targeting shared TAA as potential therapies for prostate cancer. We have studied a model antigen, the synovial sarcoma X breakpoint 2 (SSX2) protein. SSX2 is a cancer-testis antigen with two HLA-A2–restricted epitopes that is frequently expressed in metastatic prostate cancer (25, 26). The immunogenicity of a DNA vaccine encoding SSX2 can be enhanced by encoding APLs wherein modifications to anchor residues of MHC class I-restricted epitopes enhance their binding to HLA-A2 (27). Immunization of HLA-A2 transgenic mice with an APL-enhanced DNA vaccine encoding SSX2 elicits higher frequencies of SSX2-specific CD8+ T cells producing Th1 cytokines (27). However, both native and APL-optimized vaccines lead to increased expression of PD-L1 on tumor cells, but antigen-specific CD8+ T cells from mice immunized with the optimized construct expressed higher PD-1 and elicited an inferior antitumor response relative to the native vaccine (28). The antitumor activity of the optimized vaccine can be restored when combined with antibodies blocking PD-1 or PD-L1, or by targeting a tumor line not expressing PD-L1 (28). Thus, TAA-targeted vaccines can be successfully combined with PD-1 blockade as a means of increasing antitumor efficacy. However, these studies also raised questions about the persistence of PD-1 expression after vaccination and whether the increase in PD-1 is common to high-affinity MHC:antigen:TCR combinations, potentially explaining variable antitumor responses observed following other similar approaches.
We sought to investigate here whether PD-1 expression on CD8+ T cells is affected by antigen dose or the affinity of an epitope presented during immunization, and whether this affects the resulting antitumor response. We performed these studies in both an SSX2-expressing tumor model using HLA-A2-restricted epitopes (29) and an OT-1 model using the ovalbumin H2-Kb-restricted epitope, SIINFEKL (30). We found that high antigen dose or high MHC-I or TCR affinity resulted in prolonged antigen-presenting cell (APC):T-cell contact time and elevated, persistent PD-1 expression. Immunization with high-affinity epitopes, or high antigen dose, greatly decreased antitumor efficacy via PD-1-mediated regulation. These results suggest that, with respect to PD-1, optimal APC:T-cell contact times occur when moderate-affinity epitopes are used. For improved efficacy, agents that decrease PD-1 expression or signaling could be combined with antitumor approaches using high-affinity CD8+ T cells, such as vaccines targeting high-affinity APLs (20, 22, 28, 31) or high-affinity neoantigens (13–16), or potentially by T-cell adoptive transfer using high-affinity CD8+ T cells (23, 24).
Materials and Methods
Mice
HLA-A2.01/HLA-DR1-expressing (HHD-II-DR1) mice on a C57BL/6 background were obtained from Charles River Labs courtesy of Dr. François Lemonnier (29). OT-1 (Stock No:003831) and C57BL/6J (B6, Stock No: 000664) were purchased from The Jackson Laboratory (Jax, Bar Harbor, MA). All mice were maintained and treated in microisolator cages under aseptic conditions and all experiments were conducted under an IACUC-approved protocol that conforms to the NIH guide for the care and use of laboratory animals.
Cell Lines
T2 (CRL-1992) and E.G7-OVA (CRL-2113) cells were obtained from ATCC in 2000 and 2015 (Manassas, VA), respectively, and maintained via the ATCC recommended methods for up to 20 passages. RMA-S cells were cultured in RPMI 1640 + l-glutamine, 10% fetal calf serum (FCS), penicillin/streptomycin (200 U/mL), 1% sodium pyruvate (NaPyr), 1% HEPES and 50µM β-MeOH. Cell identity was confirmed by short tandem repeat analysis and testing for mycoplasma was done by DDC Medical (Fairfield, OH). E.G7-OVA cells were lentivirally transduced to express high levels of PD-L1 via the manufacturer’s recommended method (G&P Biosciences, Santa Clara, CA; LTV-mPD-L1), stained for PD-L1 expression (Tonbo, San Diego, CA; 50-1243-U100), sorted using a FACS Aria sorter for live/singlet/PE+ cells, and an E.G7-OVA PD-L1high line was isolated and grown under the same conditions as the parental line (G418 selection).
Peptides and affinity assays
Modified peptides were designed from the amino-acid sequence of SSX2 (GenBank: CAA60111) or chicken ovalbumin to have either reduced or enhanced affinity for HLA-A2 or H-2Kb in the SYFPEITHI and BIMAS prediction algorithms (32, 33). These peptides were synthesized, and the purity and identity of each peptide was confirmed by mass spectrometry and gas chromatography (LifeTein, LLC., Hillsborough, NJ). Peptide binding affinities were assessed with TAP-deficient HLA-A2+ T2 or H-2Kb+ RMA-S cells as described previously (34–36).
Splenocyte preparation
Spleens were collected from OT-1 or HHD mice, processed through a mesh screen, and splenocytes were isolated by centrifugation after red blood cell osmotic lysis with ammonium chloride/potassium chloride lysis buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) (34).
Ex vivo simulation
Splenocytes were cultured at 2 × 106/mL in RPMI 1640 + l-glutamine, 10% FCS, penicillin/streptomycin (200 U/mL), 1% NaPyr, 1% HEPES, 50 µM β-MeOH, and the designated peptide (2 µg/mL). At the time points indicated, cells were stained with the following antibodies: CD3-FITC (BD 555274), CD4-BUV395 (BD 563790), CD8-BUV805 (BD 564920), LAG3-BV711 (BD 563179), PD1-PECF594 (BD 562523), TIM3-APC (eBioscience 17-5871-82), 41BB-PerCPeF710 (eBioscience 46-1371-82), CTLA4-PE (eBioscience 12-1529-42), Live/Dead Ghost dye 780 (Tonbo 13-0865-T100), or corresponding fluorescently labeled IgG controls. Cells were then fixed for 15 min at 4°C in cytofix (BD Biosciences, San Jose, CA; 554655), and frozen in FCS + 10% DMSO. After all time points were collected, cells from all times were thawed, rinsed and resuspended in PBS + 3% FCS + 1 mM EDTA and analyzed by flow cytometry. All antibodies used were at 1:100 dilutions and stained for 30 min at 4°C in a 1:4 dilution of brilliant stain buffer (BD 563794) in PBS + 3% FCS + 1 mM EDTA.
Immunization of HHDII-DR1 mice
6 week-old HHDII-DR1 mice were immunized subcutaneously with 100 µg of an individual SSX2-p103 APL in complete Freund’s adjuvant (Sigma, F5881). Mice were euthanized seven days later and spleens were processed and analyzed via flow cytometry as described above. For these studies, the following antibodies were used: SSX2-p103 HLA-A2 tetramer-APC (NIH Tetramer Core Facility), CD3-FITC (BD 555274), CD4-BUV395 (BD 563790), CD8-BUV805 (BD 564920), PD1-PECF594 (BD 562523), 41BB-PerCPeF710 (eBioscience 46-1371-82), Live/Dead Ghost dye 780 (Tonbo 13-0865-T100) or corresponding fluorescently labeled IgG controls. Examples of flow gating are shown in Supplementary Figs. S1–S6.
Intracellular cytokine staining
Splenocytes were collected from naive OT-1 or immunized HHDII-DR1 mice as described above, cultured with 2 µg/mL (unless otherwise indicated) native SSX2-p103, SIINFEKL APL, a non-specific peptide (negative control), or phorbol 12-myristate 13-acetate (40 ng/mL, PMA, Sigma-Aldrich, St. Louis, MO; P8139) and ionomycin (2.6 µg/mL, Fisher Scientific, Waltham, MA; ICN15507001) as a positive control. After two hours golgistop (0.67µL/mL, BD 554724) was added. Cells were incubated for six additional hours (8 hours total), after which intracellular cytokine staining was performed as per the manufacturer’s protocol (Cytofix/Cytoperm Kit, BD 554714). Antibodies used for cells surface staining were: CD3-FITC (BD 555274), CD4-BUV395 (BD 563790), CD8-BUV805 (BD 564920), LAG3-BV711(BD 563179), PD1-PECF594 (BD 562523), 41BB-PerCPeF710 (eBioscience 46-1371-82). Antibodies used for intracellular staining were: TNFα-PECy7 (BD 557644), IL2-APC (eBioscience 17-7021-82), IFNγ-PE (BD 554412), and Live/Dead Ghost dye 780 (Tonbo 13-0865-T100) or corresponding fluorescently labeled IgG controls. The number of antigen-specific Th1 cells (expressing IL2 and/or TNFα and/or IFNγ) was determined as a percentage of total CD8 T cells via an “OR” Boolean gate (FlowJo software v10.1).
Adoptive transfer and immunization of wild-type C57BL/6 (B6) mice
For adoptive transfer of OT-1 T cells into B6 mice, OT-1 splenocytes were harvested as described above. CD8 T cells were isolated using immunomagnetic negative selection (StemCell, Vancouver, Canada; 19853), rinsed and suspended in PBS, and 2 × 106 cells were adoptively transferred into 6–10 wk old, female, B6 mice via intraperitoneal injection. The day following transfer, mice were immunized subcutaneously with an individual SIINFEKL APL (100 µg) in complete Freund’s adjuvant (Sigma, F5881) or vehicle. Mice were euthanized at the times indicated, spleens were collected, processed as described above, and analyzed via flow cytometry using the following antibodies: SIINFEKL H2Kb tetramer-BV421 (NIH Tetramer Core Facility), CD3-FITC (BD 555274), CD4-BUV395 (BD 563790), CD8-BUV805 (BD 564920), LAG3-BV711 (BD 563179), PD1-PECF594 (BD 562523), TIM3-APC (eBioscience 17-5871-82), 41BB-PerCPeF710 (eBioscience 46-1371-82), CTLA4-PE (eBioscience 12-1529-42), CD44-BV786 (BD 563736) and Live/Dead Ghost dye 780 (Tonbo 13-0865-T100) or corresponding fluorescently labeled IgG controls.
Data collected on different days was normalized using rainbow beads (Spherotech, Lake Forest, IL; RFP-30-5A).
Microscopy
RMA-S cells were loaded with SIINFEKL APL by incubation in full media containing peptide (2 µg/mL) for one hour at 37°C, stained in full media containing 2µM CellTracker Red (Invitrogen, Carlsbad, CA; C-34552) for 30 min at 37°C, rinsed thrice in plain media, and 4×104 cells plated on an ibiTreat coated 8-well μ-slide (Ibidi, Madison, Wisconsin; 80826). Naïve OT-1 CD8 T cells were isolated as described, stained in full media containing 2 µM CellTracker Green (Invitrogen C-2925) for 30 min at 37°C, rinsed thrice in plain media, and 4 × 104 (1:1) cells were added to the wells containing the loaded APC. 20× images in white light and both fluorescent channels were collected every minute for sixty minutes. Imaging was conducted with a Nikon Eclipse Ti-E equipped with a large incubator (encompassing stage, filters and objective lenses) permitting warm air incubation and CO2-control. All images and videos were processed and created in the Fiji package of ImageJ2 (37, 38). To avoid imaging related cell death, cells used for checkpoint expression analysis were set up simultaneously and identically but plated in separate wells and allowed to incubate for 72 hours, then analyzed as described above.
Tumor treatment study
E.G7 PD-L1high cells were rinsed, suspended at 10×106/mL in PBS and 100 µL (1×106 cells) injected subcutaneously into 6–10 wk old, female, B6 mice. Fourteen days following tumor injection, 1×106 naïve OT-1 CD8 T cells were adoptively transferred as described above. The day following adoptive transfer, mice were immunized subcutaneously with 100 µg of an individual SIINFEKL APL in complete Freund’s adjuvant. In the group receiving PD-1 blocking antibodies, antibody (100 µg) was injected intraperitoneally on the day following vaccination. Anti-mouse PD-1 (G4), produced from an Armenian Hamster hybridoma, was a gracious gift from Dr. Lieping Chen (39) and purified using ascites monoclonal antibody production (Envigo, Madison, WI). Tumor volume was measured using calipers and calculated in cubic centimeters according to the following formula: (π/6)*(long axis)*(short axis)2. Tumors obtained at necropsy were digested in media containing collagenase (1 mg/mL) and DNAse I (20 µg/mL, Sigma) for 2 hours at 37°C, and passed through a 100 µm screen to obtain a single-cell suspension. Tumor cells and tumor infiltrating lymphocytes (TILs) were stained and analyzed by flow cytometry using the following antibodies: CD3-FITC (BD 555274), CD4-BUV395 (BD 563790), CD8-BUV805 (BD 564920), LAG3-BV711 (BD 563179), PD1-PECF594 (BD 562523), TIM3-APC (eBioscience 17-5871-82), 41BB-PerCPeF710 (eBioscience 46-1371-82), CTLA4-PE (eBioscience 12-1529-42), CD45-BV510 (BD 563891), PDL1-PE (Tonbo 50-1243-U100) and Live/Dead Ghost dye 780 (Tonbo 13-0865-T100), or corresponding fluorescently labeled IgG controls.
Statistics
Comparison of group means was performed using ANOVA (GraphPad Prism software, v5.01), with Bonferroni’s posttest correction applied for multiple comparisons. Comparison of variables for statistical association or dependency was made using Kendall’s rank correlation coefficient (Mstat software, v6.2.2). For all comparisons, P values equal to or less than 0.05 were considered statistically significant.
Results
Immunization with high-affinity epitopes elicits CD8+ T cells with higher PD-1 expression
We have previously reported that HLA-A2 transgenic mice vaccinated with a DNA vaccine encoding SSX2 APL with increased MHC binding (pTVG-SSX2RF) developed a greater number of systemic Th1-biased, multifunctional, and SSX2-specific CD8 T cells than animals immunized with the native construct (27). However, PD-1 expression was also increased on SSX2-specific CD8 T cells. When animals implanted with SSX2-expressing tumors were immunized, the tumors in animals receiving the APL vaccine developed much faster when compared to those from mice immunized with the native construct (28). This inferior antitumor response was abrogated by eliminating PD-L1 expression from the tumor or by the addition of PD-1 or PD-L1 blocking antibodies at the time of immunization (28). We consequently sought to investigate the persistence of PD-1 expression following vaccination and whether the increase in PD-1 is common to high-affinity MHC:antigen:TCR combinations or unique to this SSX2 antigen model. Using two epitope prediction algorithms, we generated a large panel of APLs derived from the dominant HLA-A2-restricted SSX2 epitope (p103–111) (Table 1) (32, 33). Affinity was assessed directly by measuring the peptide’s ability to stabilize HLA-A2 on TAP-deficient T2 cells (Fig. 1A). To assess the effect of altering MHC-I affinity on checkpoint receptor expression following immunization, HHD-II (HLA-A2 transgenic) mice were immunized with each of five peptides spanning the widest range of affinities (green bars, Fig. 1A). As was observed previously, immunization with the high-affinity peptides resulted in a greater number of SSX2-specific T cells (Fig. 1B) that expressed Th1 cytokines (Fig. 1C) (27, 28). PD-1 expression on epitope-specific CD8 T cells increased (Fig. 1D), with strong association to the peptide affinity for HLA-A2 (P = 0.01, two-sided Kendall's rank correlation).
Table 1. Amino acid sequence of peptides used with observed and predicted MHC-I affinities.
Observed relative binding represents stabilization (normalized mean fluorescence intensity) of HLA-A2 or H-2Kb expression on TAP-deficient T2 or RMA-S cells by test peptide relative to a non-binding peptide. Predicted binding by SYFPEITHI or BIMAS algorithms is also shown. Peptides in gray were those used for subsequent analyses.
| Protein | Peptide name |
AA Sequence |
Observed relative binding |
SYFPEITHI prediction |
Bimas prediction |
|---|---|---|---|---|---|
| SSX2 | IP | RLQGISPKP | 1.07 | 15 | 0.02 |
| LA | RAQGISPKI | 1.31 | 17 | 0.15 | |
| LI | RIQGISPKI | 2.34 | 21 | 1.44 | |
| Nat | RLQGISPKI | 3.61 | 23 | 10.43 | |
| IA | RLQGISPKA | 3.79 | 19 | 4.97 | |
| IL | RLQGISPKL | 3.89 | 25 | 21.36 | |
| IV | RLQGISPKV | 3.88 | 25 | 69.55 | |
| RF-IV | FLQGISPKV | 4.25 | 26 | 319.94 | |
| RF-IA | FLQGISPKA | 4.78 | 20 | 22.85 | |
| RF | FLQGISPKI | 4.97 | 24 | 47.99 | |
| OVA - MHC affinity panel | IP-FT | SIPNTEKL | 0.92 | 15 | 10.45 |
| LY | SIINFEKY | 0.98 | 15 | 0.52 | |
| FL | SIINLEKL | 1.25 | 15 | 1.74 | |
| LP | SIINFEKP | 1.72 | 15 | 0.17 | |
| FT | SIINTEKL | 3.34 | 15 | 3.49 | |
| LI | SIINFEKI | 5.91 | 21 | 5.23 | |
| OVA | SIINFEKL | 6.46 | 25 | 17.42 | |
| LV | SIINFEKV | 6.70 | 21 | 1.72 | |
| OVA - TCR affinity panel | SE | EIINFEKL | 6.52 | 24 | 15.84 |
| NG | SIIGFEKL | 6.75 | 25 | 17.42 | |
| IY | SIYNFEKL | 6.66 | 30 | 87.12 | |
| NQ | SIIQFEKL | 6.94 | 25 | 17.42 |
Figure 1. Increasing the MHC I affinity of SSX2-p103 resulted in increased activation, expansion, and PD-1 expression.
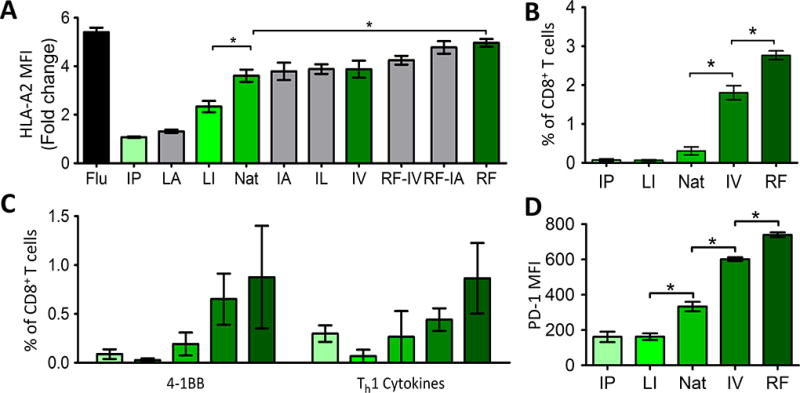
A) T2 cells were incubated with the indicated peptides for six hours, stained for HLA-A2, and analyzed by flow cytometry. HLA-A2 MFI is displayed as the fold change over cells that were incubated with a non-specific peptide B) Mice were immunized with each peptide and after one week the spleens were collected. The percentage of CD8 T cells that were positive for SSX2-p103-tetramer staining was determined. C) Splenocytes from immunized mice were stimulated with the native SSX2-p103 peptide, and 4-1BB expression and intracellular cytokine staining for one or more Th1 (IFNγ, TNFα, IL2) cytokines, was assessed by flow cytometry. D) PD-1 amounts on the tetramer+ cells from B. APL abbreviations are shown in Table 1. Letters along the X axis indicate AA changes in the SSX2-p103 sequence as indicated in Table 1. Nat = SSX2-p103 native sequence, Flu = HLA-A2 restricted flu epitope positive control. Results shown are representative of two independent experiments with N=6 replicates each, *= P < 0.05, two-way ANOVA with Bonferroni’s posttest. Representative flow cytometry gating strategy shown in Supplementary Fig. S1.
Stimulation with high-affinity APL resulted in an increase in checkpoint receptor expression
Gallegos and colleagues reported TCR downregulation and increased CTLA-4 expression on a high-affinity T-cell clone, when compared to the same clone expressing a low-affinity TCR (40). In addition, a low-affinity subset of melanoma-specific T cells cannot express PD-1 (41). Therefore, one explanation for our findings could be that immunization with different APLs led to expansion of different populations of T cells that inherently expressed PD-1 differently. To remove the potential variable of expanding different T-cell populations, we used the ovalbumin model in which CD8+ T cells from transgenic OT-1 mice express a monoclonal TCR specific for the dominant ovalbumin 257–264 epitope (SIINFEKL [OVA]) in the context of H-2Kb (30). We first designed APLs with different predicted affinity for H-2Kb by altering anchor residue modifications to affect MHC class I binding (Table 1). Previously identified OVA-derived peptides with altered affinity for the OT-1 TCR were also used (20, 40). Using these APLs, and different concentrations of OVA peptide, we evaluated their MHC-I affinity based on their ability to stabilize H-2Kb expression on TAP-deficient RMA-S cells (Fig. 2A). As expected, peptides with modifications to anchor residues demonstrated decreased affinity for MHC-I at equivalent concentrations. Changes to the TCR-binding portion of the peptides did not affect H-2Kb affinity (Fig. 2A). The functional avidity of these peptides and concentrations was assessed by stimulating OT-I mouse splenocytes with each peptide for eight hours and assessing Th1 cytokine production as a measure of T-cell activation (Fig. 2B). As expected, stimulation of OT-1 cells using APL with decreased MHC affinity, reduced TCR affinity, or reduced antigen dose led to reduced Th1 cytokine production (Fig. 2B).
Figure 2. Amino acid substitutions in the SIINFEKL (OVA) peptide affect MHC-I or TCR affinity.
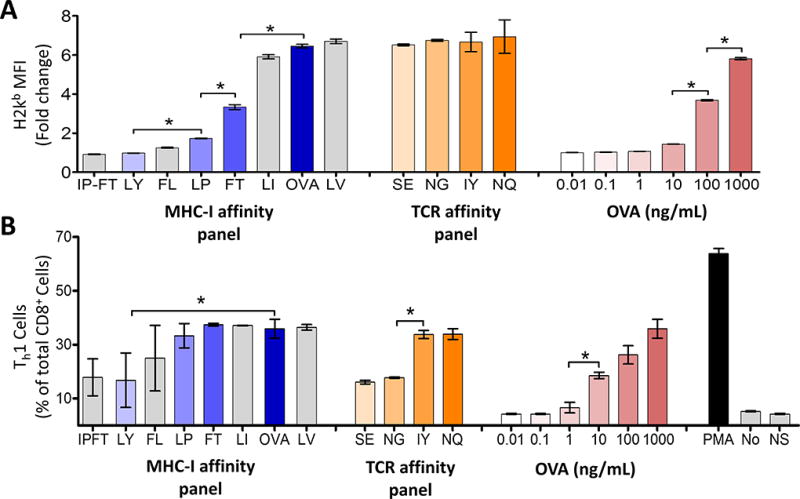
A) TAP-deficient RMA-S cells were incubated with the indicated peptides (Table 1) for six hours, at which time the cells were stained for MHC-I and analyzed by flow cytometry. MHC-I MFI is displayed as the fold change over cells that were incubated with a non-specific peptide. B) OT-1 mouse splenocytes were incubated with the indicated peptides and cytokine expression was assessed by intracellular cytokine staining. The number of cells expressing IL2 and/or TNFα and/or IFNγ is shown as the percent of total CD8 T cells. Highlighted peptides were selected for further analysis. The letters along the x axis indicate AA changes in the SIINFEKL sequence as indicated in Table 1, ex. IP-FT = SIPNTEKL. No = vehicle control, NS = non-specific peptide control, SSX2 p103–111 (RLQGISPKI), PMA = PMA and ionomycin positive control. Results shown are representative of two independent experiments with N=6 replicates each, *= P < 0.05, two-way ANOVA followed by Bonferroni’s posttest correction. Representative flow cytometry gating strategy shown in Supplementary Fig. S2.
To assess how altering the antigen affinity for H-2Kb, the TCR, or antigen dose affected the expression of regulatory checkpoint receptors, OT-1 splenocytes were cultured with each of the different peptides for four days and activation (4-1BB expression), TCR expression, and the expression of different immune checkpoint receptors (PD-1, LAG3, TIM-3, and CTLA-4) were assessed (Fig. 3). APLs with higher MHC or TCR affinity led to greater activation of T cells with greater expression of 4-1BB and decreased expression of CD3. Activation using APLs with higher affinity or peptide dose elicited increased expression of T-cell checkpoint molecules PD-1, LAG3, CTLA-4, and TIM3, with expression peaking 24–48 hours after initial encounter. High doses of the APLs with higher MHC or TCR affinity led to prolonged expression of PD-1 and TIM-3 throughout the four days observed in vitro, whereas expression of 4-1BB and regulatory markers LAG3 and CTLA-4 tended to decrease over time (Fig. 3).
Figure 3. Varying the MHC-I affinity, TCR affinity or antigen dose alters the phenotype of CD8 T cells ex vivo.
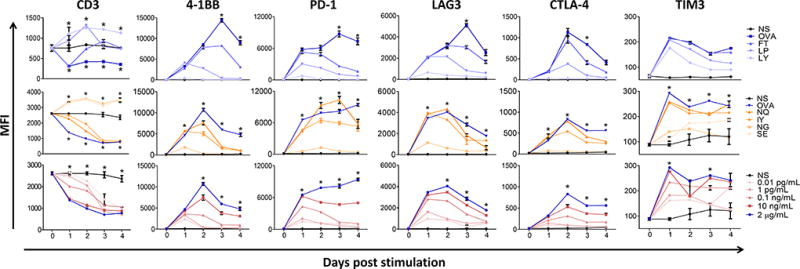
Mouse splenocytes (OT-1) were collected and immediately stimulated with SIINFEKL (OVA) or one of the variants that alters the MHC-I affinity (top), TCR affinity (middle) or different concentrations of SIINFEKL (bottom). The CD3 (TCR) levels, activation, and checkpoint receptor expression of CD8 T cells was monitored every 24 hours for 96 hours. Results are representative of two independent experiments with N=3 mice each. NS = non-specific peptide, SSX2 p103–111 (RLQGISPKI). Statistical analyses compare OVA to FT (top), OVA to NG (middle), and 2µg/mL to 0.1ng/mL OVA (bottom). For CD3 levels, OVA, FT, NG and 0.1ng/mL OVA were compared to NS. *= P < 0.05, two-way ANOVA followed by Bonferroni correction. Representative flow cytometry gating strategy shown in Supplementary Fig. S3.
The persistent expression of PD-1 and TIM3 after stimulation with high-affinity APLs in vitro was similar to what we observed in vivo with the HLA-A2 model (Fig. 1) (28). However, in that system immunization with epitopes with higher affinities elicited higher numbers of CD8 T cells. Hence, to evaluate changes in vivo on the quality of the response in the same CD8 T-cell population, without changes to the magnitude of CD8 response, OT-1 T cells were adoptively transferred into wild-type (WT) C57BL/6 mice. Mice were immunized the next day with peptides of high- or moderate-affinity for H-2Kb, the TCR, and with lower dose of the high-affinity OVA peptide. Spleens were collected on the indicated days and OVA-tetramer+ CD8+ T cells were evaluated for markers of activation and expression of T-cell checkpoint molecules. Although OT-1 T cells were activated with each APL condition, as demonstrated by increases in 4-1BB expression, the expression of regulatory checkpoint molecules, and PD-1 in particular, on these cells was highest following immunization with the high-affinity epitope (Fig. 4A). Given that immunization with Freund’s adjuvant can lead to persistence of the antigen and collection of T cells at the site of immunization (42), we conducted concurrent studies using a single immunization with peptide in PBS, without oil adjuvant. Although this immunization was less effective in activating T cells (lower expression of 4-1BB), PD-1 expression was similarly persistently elevated on adoptively transferred OT-1 cells at least 6 days following immunization, suggesting that the persistence of PD-1 expression was not due to persistent antigen stimulation (Fig. 4A). Finally, PD-1 and LAG3 expression, albeit lower than following initial activation, remained significantly elevated at least 28 days after vaccination (Fig. 4B) demonstrating not only increased, but persistent, expression of these checkpoints as a result of a single vaccination with a high-affinity epitope.
Figure 4. Vaccination with high-affinity peptides led to persistent PD-1 and LAG3 expression in vivo.
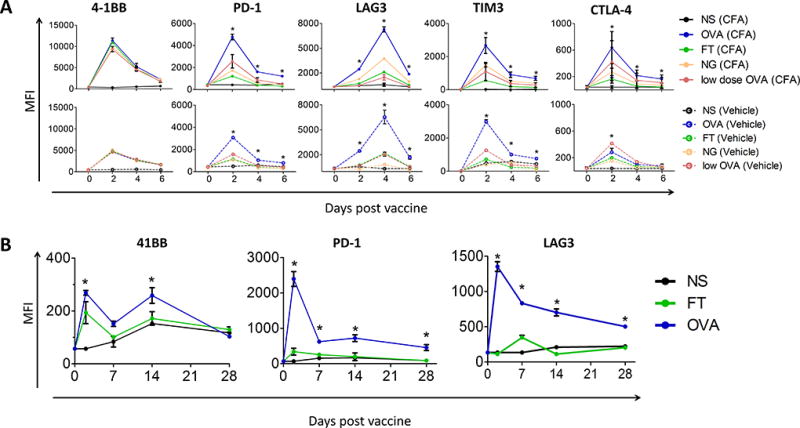
A) C57BL/6 mice received 2×106 OT-1 cells by adoptive transfer and the next day were immunized with 100µg OVA, 100µg of one of the variants, or 0.1 µg (low dose) of OVA with Freund’s adjuvant (top, CFA) or alone in PBS (bottom, vehicle). Spleens were harvested from individual groups every two days for six days. The MFI of indicated receptors was assessed on SIINFEKL-tetramer+ CD45+ CD8+ T cells directly ex vivo. B) C57BL/6 mice were immunized as in panel A with 100µg FT, OVA, or NS control in CFA. Expression of 4-1BB (activation), PD-1, and LAG3 were assessed on splenocytes on days 2, 7, 14 and 28. Results shown are representative of two independent experiments with N=5 mice/group. Statistical comparisons are made between groups receiving OVA and FT. *= P < 0.05, two-way ANOVA with Bonferroni correction. (NS = non-specific peptide, SSX2 p103–111 (RLQGISPKI)). Representative flow cytometry gating strategy shown in Supplementary Fig. S4.
High-affinity antigens resulted in significantly increased APC:T-cell contact time
Increasing the duration of the MHC:peptide:TCR complex increases T-cell activation in an OVA-expressing S. typhimurium model in which the SIINFEKL sequence was altered to change H-2Kb affinity (43). In addition, regulatory receptor expression is associated with increased TCR signaling (44). Therefore, we hypothesized that increasing the duration of APC:T-cell contact time, leading to increased TCR signaling, could be responsible for the increase in regulatory receptor expression observed above. As such, we recorded the contact time between APCs incubated with select peptides and naive OT-1 T cells using time-lapse microscopy. As antigen affinity for H-2Kb, the TCR, or antigen dose increased, so did the APC:T-cell contact time (Fig. 5, Supplementary Videos 1–3, P = 0.0001, two-way ANOVA). Furthermore, OT-1 T cells cultured with peptide-loaded APCs for three days showed increased 4-1BB, PD-1 and LAG3 expression that were all significantly correlated to the average APC:T-cell contact times for each peptide (P < 0.05, two-sided Kendall’s rank correlation; Fig. 5, bottom). Together with the information from Fig. 3, these findings demonstrated that increasing antigen affinity for H-2Kb, TCR, or increasing antigen dose led to stabilization of the interaction between APCs and T cells, which in turn led to increased T-cell activation and prolonged expression of one or more T-cell checkpoint molecules, notably PD-1.
Figure 5. APC:T-cell contact time corresponds to antigen affinity or dose.
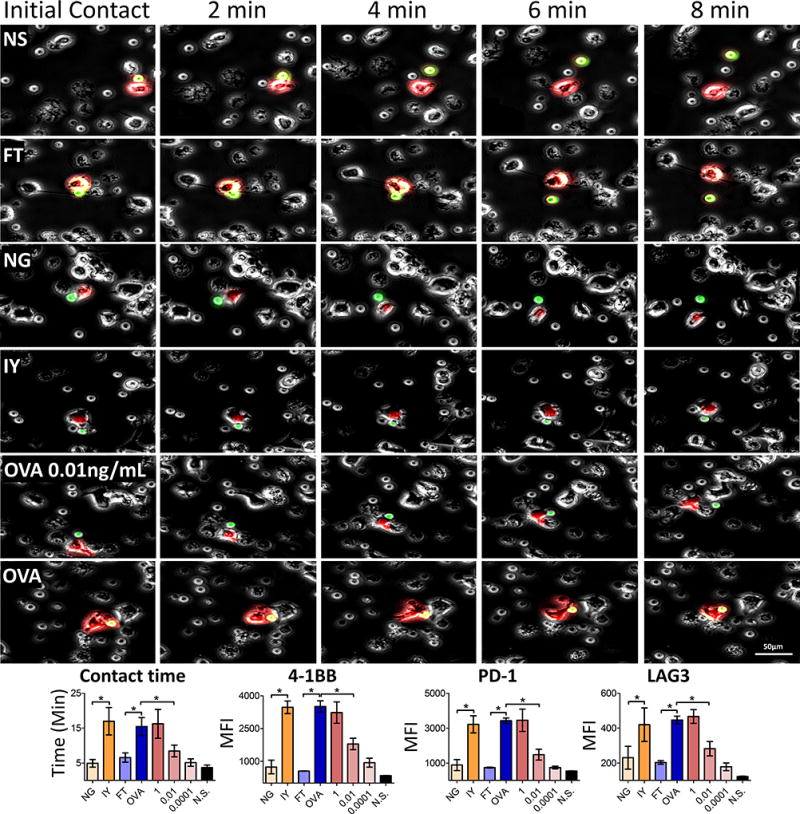
Top) APCs were stained with CellTracker red (CMTPX), incubated with the indicated peptides or various concentrations of OVA (ng/mL), and allowed to incubate for one hour. Isolated OT-1 T cells stained with CellTracker green (CMFDA) were added to the peptide loaded APCs. Upon addition of the T cells, 20× time-lapse microscopy was conducted for one hour at one minute intervals to monitor APC:T-cell contact times. Images are representative of cells interacting for the mean time observed with the respective peptide. Color was removed from the surrounding cells for ease of tracking. Bottom) Contact time for each peptide was quantified. Prior to imaging, cells were removed, allowed to incubate together for 72 hours and analyzed by flow cytometry. Association between contact time and peptide affinity was determined via two-sided Kendall’s rank correlation. NS = non-specific peptide, SSX2 p103–111 (RLQGISPKI). * = P < 0.05, two-way ANOVA followed by Bonferroni correction. For contact time calculations, N=30 APC:T-cell interactions were evaluated for each experimental condition. For MFI of 4-1BB, PD-1 and LAG3 expression in the bottom panels, data shown is for 6-well replicates. Representative flow cytometry gating strategy shown in Supplementary Fig. S5.
Moderate-affinity peptides led to decreased PD-1 expression and more antitumor activity
To determine if the changes in PD-1 expression after different APL-activating conditions lead to differences in antitumor efficacy, we evaluated the in vivo effect of OT-1 T cells activated with different peptides on ovalbumin-expressing tumors. C57BL/6 mice were implanted with PD-L1+ ovalbumin-expressing E.G7 cells and left for 14 days until tumors were palpable (~0.05 cm3). OT-1 T cells (2 ×106) were then adoptively transferred into the mice. The following day mice were immunized once with either OVA, an equivalent dose of APL with lower affinity for MHC class I (SIINTEKL), an equivalent dose of APL with lower affinity for the TCR (SIIGFEKL), lower dose OVA, high dose OVA with anti-PD-1 antibody administered 24 hours after immunization, or PD-1 blocking antibody alone. Activation of OT-1 cells with OVA peptide elicited no significant antitumor response, and this was mitigated using a lower dose of the peptide or by blocking PD-1 after immunization (Fig. 6A,). APLs with moderate-affinity for either MHC class I or TCR resulted in greater antitumor response and, in the case of the low MHC-I affinity FT peptide, significantly increased the number of CD8 TILs when compared to high-affinity vaccination (Fig. 6B). As previously observed, a corresponding increase in PD-L1 was detected on tumors from animals immunized with the high-affinity epitope (Fig. 6C). Activation and regulatory receptor expression was assessed on CD8 TILs (CD45+CD3+CD8+CD4−) (Fig. 6D). As expected from in vitro studies, TILs from the group immunized with the high-affinity OVA epitope expressed the highest 4-1BB, consistent with recruitment of activated CD8+ T cells, but these cells also expressed higher PD-1, as well as higher LAG3 and TIM3 (Fig. 6D). TILs from groups that were immunized with moderate-affinity APL, or lower dose of the high-affinity epitope, had lower expression of PD-1, as well as LAG3 and TIM3. Therefore, the increased and prolonged PD-1 expression (and potentially other regulatory molecules) following activation of CD8 T cells with a high-affinity epitope can lead to inferior antitumor immunity.
Figure 6. Moderate affinity vaccine antigens elicited greatest antitumor response.
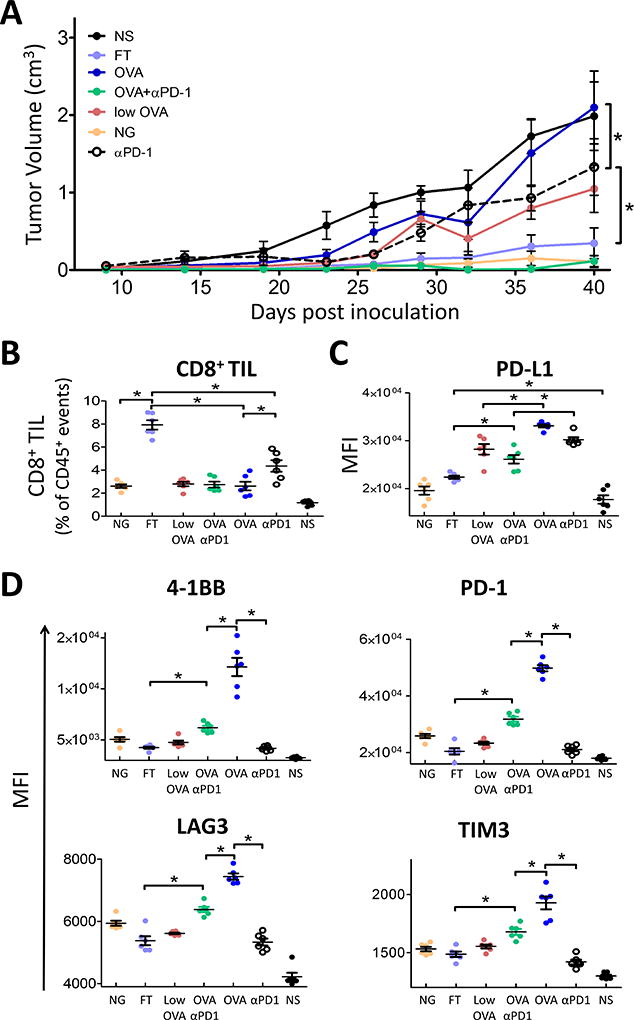
Wild-type C57BL/6 (n=5) mice were injected with PD-L1 transduced E.G7-OVA tumor cells. When tumors were palpable (day 14), 2 × 106 OT-1 T cells were adoptively transferred into the mice. The following day mice were immunized with the indicated peptides or 0.1 µg (low dose) of OVA in CFA; PD-1 blocking antibodies were given the day following vaccination. A) Tumor growth was monitored over time. Complete tumor responses were observed in 2/5 animals receiving FT, 1/5 animals receiving NG, and 2/5 animals receiving OVA+αPD1. On day 40, tumors were removed and analyzed by flow cytometry. B) The number CD8+ TIL was calculated as a percent of CD45+ cells. C) PD-L1 was assessed on the CD45− population. D) Activation and immune checkpoint expression was assessed on CD45+CD3+CD4−CD8+ TIL. NS = non-specific peptide, SSX2 p103–111 (RLQGISPKI). Results are representative of four independent experiments, N=5 mice each. * = P < 0.05 two-way ANOVA with Bonferroni’s posttest correction. Representative flow cytometry gating strategy shown in Supplementary Fig. S6.
Discussion
In this report we found that modifications to a CD8+ T-cell epitope that permitted longer APC:T-cell contact time (effectively prolonging TCR signaling) led to persistent expression of T-cell checkpoint molecules, notably PD-1. This occurred in vitro as a T-cell intrinsic response to activation, and in vivo following immunization. Prolonged expression led to decreased antitumor response when used as a vaccine in vivo in the absence of PD-1 blockade. Given similar findings in both an HLA-A2-restricted model and in the OVA H-2Kb model, we propose that this is generalizable and that increases in the magnitude and duration of T-cell expression of PD-1 occur after activation with high affinity antigens. We do not yet know the molecular mechanism by which prolonged T-cell activation may lead to prolonged or persistent expression of PD-1 or other inhibitory receptors. Early gene changes in activated CD8+ T cells that associate with changes in persistence of PD-1 expression will be evaluated in future studies. Notwithstanding, this phenomenon may help explain prior findings and provide a general framework for improving the efficacy of antitumor vaccines. These findings are significant to the field of tumor immunology for the following reasons: 1) they help explain why high epitope doses and high-affinity epitopes have not uniformly demonstrated superior antitumor efficacy when used as antitumor vaccines (20, 22, 28, 31); 2) they potentially explain similar findings of poor antitumor efficacy of adoptive transfer therapies using high-affinity T cells (23, 24); 3) they anticipate likely problems that will arise with the use of high-affinity neoepitope vaccines (13–16); 4) they help explain how PD-1/PD-L1 blockade as a monotherapy works, in particular for tumors with a high likelihood of having CD8+ T cells specific for high-affinity tumor-specific mutant neoepitopes (45–50) (e.g. those with high mutation burden); and 5) these findings suggest a major point of intervention, namely blockade of checkpoint molecule expression or function at the time of T-cell activation, as a strategy to improve antitumor vaccine efficacy.
We have previously reported that a DNA vaccine, modified to encode epitopes with greater affinity for MHC class I, elicited a higher frequency of epitope-specific CD8 T cells that expressed multiple Th1 cytokines (28). However, this vaccine demonstrated inferior antitumor efficacy compared to an unmodified vaccine. We found that this MHC class I modified vaccine elicited CD8 T cells with higher PD-1 expression as the basis for this inferior antitumor response. In the current report, we found that epitopes with high MHC affinity, or which lead to prolonged TCR signaling via changes to the MHC:peptide:TCR complex, lead to prolonged PD-1 expression (and likely LAG3 and TIM3 expression) on CD8 T cells following activation (Fig. 4B). Although others have similarly found that immunization with high affinity epitopes can lead to inferior antitumor immunity, our findings here implicate the expression of immune checkpoints as the cause. For example, Gross and colleagues reported that the CTL repertoire towards high-affinity epitopes was partially dysfunctional and unable to protect mice from a lethal challenge with a murine telomerase reverse transcriptase (mTERT) expressing EL4-HHD tumor cell line (22). In contrast, mice developing CTL responses against low-affinity mTERT epitopes exhibited potent antitumor immunity (22). Likewise, Gaiger et al. showed that immunization with high-affinity MHC class I restricted peptides induced peptide-specific CTL that specifically lysed TRAMP-C cells in vitro but peptide immunization did not show any effect on TRAMP-C tumor growth in vivo (51). Andersen et al., demonstrated that mice immunized with intermediate MHC-I affinity peptides more frequently generated a CTL response (36). Unfortunately, none of these studies evaluated the role of T-cell checkpoint expression as a possible cause.
Other groups have similarly demonstrated inferior antitumor immunity following epitope changes that affect TCR affinity or antigen dose. Slansky and colleagues found that increasing the affinity of the peptide-MHC-I (pMHC) complex for the TCR correlated with increased proliferation and IFNγ production from a T-cell clone in vitro, but again resulted in decreased CTL function in vivo (20). They suggested that these high-affinity APL may functionally inactivate the relevant responding T cells by expanding cells with defective cytokine profiles, or that T cells expanded with the high-affinity APL were more susceptible to peripheral deletion (20). Similarly, following peptide-pulsed dendritic cells (DC) vaccination, and altering the antigen density by pulsing with higher concentrations of peptide or by changing the pMHC-binding affinity, Bullock and colleagues showed that the most effective peptide density was a moderate one; immunization with higher density or affinity resulted in an attenuated CD8 T-cell response (8, 52). Finally, Corse and colleagues have studied the effects of TCR/pMHC binding characteristics on in vivo T-cell immunity by tracking T-cell activation, effector, and memory responses to immunization with peptides exhibiting a range of TCR/pMHC half-lives and in vitro T-cell activation potencies (21). Contrary to predictions from in vitro studies, and similar to our results, they found that optimal in vivo T-cell responses occurred using ligands with intermediate TCR/pMHC half-lives. The diminished in vivo response to the ligand exhibiting the longest TCR/pMHC half-life was associated with attenuation of intracellular signaling, expansion, and function over a broad range of time points. Again, the expression of CD8 T-cell checkpoint molecules was not assessed in these studies.
Similar findings of decreased antitumor efficacy using adoptive T-cell therapy with high-affinity T cells have also been reported. For example, Janicki et al showed that high-affinity tumor-specific T cells were more susceptible to loss of CTL function after tumor infiltration when compared to low affinity T cells in a murine renal carcinoma model (23). Our findings would suggest that this might have been mediated by increased PD-1 expression following high-affinity MHC:peptide:TCR T-cell activation. Likewise, Zhu and colleagues reported that, in contrast to low-affinity T cells, high-affinity T cells initially delayed subcutaneous B16 melanoma tumor growth, however persistence in the tumor microenvironment resulted in reduced IFNγ production and CD107a mobilization. Indeed, blockade of PD-1 prevented T-cell tolerization and restored tumor immunity (24), suggesting that activation of these cells similarly produced prolonged PD-1 expression. Supporting this hypothesis, Simon et al. reported that PD-1 expression identified antigen-specific tumor-infiltrating T-cell clonotypes of high-affinity, but with less antitumor efficacy (41).
Taken together, these results show that antitumor therapies involving either immunization with high-affinity epitopes, or by adoptive transfer of high-affinity tumor-specific CTL populations, may be generally less effective due to persistent expression of PD-1 and/or other T-cell checkpoint molecules. Although this can potentially be reversed with T-cell checkpoint blockade, it implies that PD-1 blockade, or other therapies that interfere with PD-1 expression or function, may be particularly advantageous during priming, and notably for vaccines designed to encode or target high-affinity epitopes. This is particularly relevant for personalized vaccine approaches aimed at high-affinity neoepitopes generated by tumor-specific mutations. That approach may lead to the generation of high-affinity, tumor-specific CD8 T cells, but they will likely have higher PD-1 expression and thus have lower antitumor efficacy in the absence of agents that decrease PD-1 expression or function. Likewise, adoptive T-cell therapies using high-affinity TCR may best be combined with PD-1 blockade.
Our proposed model may also explain why anti-PD-1 or anti-PD-L1 therapy works as a single agent therapy for some tumor types but not others. In general, it has been recognized that these therapies are more effective against cancer types with high mutation burdens, such as melanoma (45), lung cancer (46, 47), bladder cancer, microsatellite instable (MSI) gastroesophageal cancers, and MSI- or polymerase epsilon-ultramutated endometrial (POLE) cancers (46, 49). An increased rate of mutation is more likely to alter existing antigen sequences to result in high-affinity neo-antigens that are recognized as immunogens and result in expansion of tumor-specific CD8 T cells. We anticipate that these resulting CD8+ T cells have increased and persistent PD-1 expression, leaving them more susceptible to regulation in a PD-L1-expressing tumor microenvironment, but more responsive to PD-1/PD-L1 blockade. Indeed, a recent report examining POLE and MSI endometrial cancers found higher numbers of CD8+ TILs, but also overexpression PD-1 on those TILs when compared with microsatellite-stable tumors (50). PD-L1 expression was also more frequent in POLE and MSI tumors (50). Similarly, Lizotte et al. reported immunologically “hot clusters” of T cells that were found in lung tumors with higher mutation burden which consisted of abundant CD8+ T cells expressing high levels of PD-1 and TIM-3. These “hot clusters” were not found in tumors with lower mutation burden (46).
In this report, we found PD-1 and other checkpoint molecule expression can be persistently expressed on T cells following activation with epitopes of high MHC class I or TCR affinity or with increased antigen dose. Expression of these molecules can increase transiently following T-cell activation, and here we show that persistent expression depends on the strength of T-cell activation and that this activation-induced increased expression subsequently impacts antitumor activity. Our studies did not assess the signaling pathways or gene expression differences responsible for the differences in PD-1 expression. Future studies will explore the exact mechanisms by which longer APC:T-cell interactions lead to persistent checkpoint molecule expression. We hypothesize this is mediated by signaling downstream of the TCR, or recruitment/rearrangement of signaling molecules to the immunological synapse with prolonged contact time. High throughput methods are being used to examine the immunological synapse via imaging flow cytometry (53). Future experiments will use a similar method or more traditional microscopy to assess changes in the localization of molecules within the immunological synapse, which could be altered following prolonged interaction. Alternatively, intracellular signaling could be changed in response to longer TCR signaling, as such other studies will seek to determine changes in the signaling pathways that lead to persistent PD-1 expression.
In summary, our findings demonstrate that persistent expression of checkpoint molecules can negatively impact the antitumor efficacy of activated T cells against tumors expressing their ligands following activation with high affinity epitopes in two different mouse models. With additional confirmation, this has implications for the design of vaccines with the goal of eliciting high-affinity T cells, such as those for eliciting CD8 T cells specific for high-affinity tumor-specific neoepitopes, and for adoptive T-cell therapies using high-affinity T cells. In addition, these findings also suggest that blockade of the expression or function of different T-cell checkpoint molecules might be optimally timed concurrent with T-cell activation strategies that increase the magnitude of expression of these molecules on tumor-specific T cells.
Supplementary Material
Acknowledgments
We thank Mark E. Burkard, MD, PhD for the use of the Nikon microscope, Dr. François Lemonnier for providing the HHDII-DR1 mice, and Dr. Lieping Chen for the anti-PD-1 hybridoma.
Grant Support: This work is supported for CDZ by 5T32 CA157322-05 (NIH), and for all authors by Department of Defense Prostate Cancer Research Program W81XWH-15-1-0492 and NIH P30 CA014520.
DGM has ownership interest, has received research support, and serves as a consultant to Madison Vaccines, Inc. which has licensed technologies related to those described in this report.
Footnotes
Conflicts of Interest: None of the other authors have relevant potential conflicts of interest.
Author contributions: CDZ designed and conducted all experiments, data analysis, statistical tests and wrote and edited the manuscript; VTC assisted with experimental design and technique, data analysis and edited the manuscript; DGM oversaw the experimental design and study conduct, and edited the manuscript.
References
- 1.Oh J, Shin JS. The Role of Dendritic Cells in Central Tolerance. Immune Netw. 2015;15:111–20. doi: 10.4110/in.2015.15.3.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see) Nat Rev Immunol. 2014;14:377–91. doi: 10.1038/nri3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trimble CL, Frazer IH. Development of therapeutic HPV vaccines. Lancet Oncol. 2009;10:975–80. doi: 10.1016/S1470-2045(09)70227-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim MS, Sin J-I. Both antigen optimization and lysosomal targeting are required for enhanced anti-tumour protective immunity in a human papillomavirus E7-expressing animal tumour model. Immunology. 2005;116:255–66. doi: 10.1111/j.1365-2567.2005.02219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mattarollo SR, Frazer IH, Leggatt GR. Regulation of immune responses to HPVinfection and during HPV- directed immunotherapy. 2011;239:85–98. doi: 10.1111/j.1600-065X.2010.00966.x. [DOI] [PubMed] [Google Scholar]
- 6.Kronig H, Hofer K, Conrad H, Guilaume P, Muller J, Schiemann M, et al. Allorestricted T lymphocytes with a high avidity T-cell receptor towards NY-ESO-1 have potent anti-tumor activity. Int J Cancer. 2009;125:649–55. doi: 10.1002/ijc.24414. [DOI] [PubMed] [Google Scholar]
- 7.Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5:615–25. doi: 10.1038/nrc1669. [DOI] [PubMed] [Google Scholar]
- 8.Bullock TNJ, Colella TA, Engelhard VH. The Density of Peptides Displayed by Dendritic Cells Affects Immune Responses to Human Tyrosinase and gp100 in HLA-A2 Transgenic Mice. The Journal of Immunology. 2000;164:2354–61. doi: 10.4049/jimmunol.164.5.2354. [DOI] [PubMed] [Google Scholar]
- 9.Hawkins WG, Gold JS, Dyall R, Wolchok JD, Hoos A, Bowne WB, et al. Immunization with DNA coding for gp100 results in CD4 T-cell independent antitumor immunity. Surgery. 2000;128:273–80. doi: 10.1067/msy.2000.107421. [DOI] [PubMed] [Google Scholar]
- 10.Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of "self"-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. The Journal of experimental medicine. 1998;188:277–86. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Appay V, Jandus C, Voelter V, Reynard S, Coupland SE, Rimoldi D, et al. New generation vaccine induces effective melanoma-specific CD8+ T cells in the circulation but not in the tumor site. J Immunol. 2006;177:1670–8. doi: 10.4049/jimmunol.177.3.1670. [DOI] [PubMed] [Google Scholar]
- 12.Yan M, Himoudi N, Basu BP, Wallace R, Poon E, Adams S, et al. Increased PRAME antigen-specific killing of malignant cell lines by low avidity CTL clones, following treatment with 5-Aza-2'-Deoxycytidine. Cancer Immunol Immunother. 2011;60:1243–55. doi: 10.1007/s00262-011-1024-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 14.Gallou C, Rougeot A, Graff-Dubois S, Kosmatopoulos K, Menez-Jamet J. A general strategy to optimize immunogenicity of HLA-B*0702 restricted cryptic peptides from tumor associated antigens: Design of universal neo-antigen like tumor vaccines for HLA-B*0702 positive patients. Oncotarget. 2016 doi: 10.18632/oncotarget.11086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Buuren MM, Calis JJ, Schumacher TN. High sensitivity of cancer exome-based CD8 T cell neo-antigen identification. Oncoimmunology. 2014;3:e28836. doi: 10.4161/onci.28836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572–6. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 17.Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72:1081–91. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 18.Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–6. doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engels B, Engelhard VH, Sidney J, Sette A, Binder DC, Liu RB, et al. Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer cell. 2013;23:516–26. doi: 10.1016/j.ccr.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McMahan RH, McWilliams JA, Jordan KR, Dow SW, Wilson DB, Slansky JE. Relating TCR-peptide-MHC affinity to immunogenicity for the design of tumor vaccines. The Journal of clinical investigation. 2006;116:2543–51. doi: 10.1172/JCI26936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corse E, Gottschalk RA, Krogsgaard M, Allison JP. Attenuated T cell responses to a high-potency ligand in vivo. PLoS biology. 2010;8:e1000481-e. doi: 10.1371/journal.pbio.1000481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gross D-A, Graff-Dubois S, Opolon P, Cornet S, Alves P, Bennaceur-Griscelli A, et al. High vaccination efficiency of low-affinity epitopes in antitumor immunotherapy. The Journal of clinical investigation. 2004;113:425–33. doi: 10.1172/JCI19418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janicki CN, Jenkinson SR, Williams NA, Morgan DJ. Loss of CTL function among high-avidity tumor-specific CD8+ T cells following tumor infiltration. Cancer Res. 2008;68:2993–3000. doi: 10.1158/0008-5472.CAN-07-5008. [DOI] [PubMed] [Google Scholar]
- 24.Zhu Z, Singh V, Watkins SK, Bronte V, Shoe JL, Feigenbaum L, et al. High-Avidity T Cells Are Preferentially Tolerized in the Tumor Microenvironment. Cancer Research. 2012;73:595–604. doi: 10.1158/0008-5472.CAN-12-1123. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Smith HA, McNeel DG. The SSX Family of Cancer-Testis Antigens as Target Proteins for Tumor Therapy. Clinical and Developmental Immunology. 2010;2010:1–18. doi: 10.1155/2010/150591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith HA, Cronk RJ, Lang JM, McNeel DG. Expression and immunotherapeutic targeting of the SSX family of cancer-testis antigens in prostate cancer. Cancer research. 2011;71:6785–95. doi: 10.1158/0008-5472.CAN-11-2127. [DOI] [PubMed] [Google Scholar]
- 27.Smith Ha, Rekoske BT, McNeel DG. DNA vaccines encoding altered peptide ligands for SSX2 enhance epitope-specific CD8+ T-cell immune responses. Vaccine. 2014;32:1707–15. doi: 10.1016/j.vaccine.2014.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rekoske BT, Smith HA, Olson BM, Maricque BB, McNeel DG. PD-1 or PD-L1 Blockade Restores Antitumor Efficacy Following SSX2 Epitope-Modified DNA Vaccine Immunization. Cancer immunology research. 2015;3:946–55. doi: 10.1158/2326-6066.CIR-14-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pajot A, Michel ML, Fazilleau N, Pancre V, Auriault C, Ojcius DM, et al. A mouse model of human adaptive immune functions: HLA-A2.1-/HLA-DR1-transgenic H-2 class I-/class II-knockout mice. Eur J Immunol. 2004;34:3060–9. doi: 10.1002/eji.200425463. [DOI] [PubMed] [Google Scholar]
- 30.Clarke SR, Barnden M, Kurts C, Carbone FR, Miller JF, Heath WR. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol Cell Biol. 2000;78:110–7. doi: 10.1046/j.1440-1711.2000.00889.x. [DOI] [PubMed] [Google Scholar]
- 31.Corse E, Gottschalk Ra, Allison JP. Strength of TCR-peptide/MHC interactions and in vivo T cell responses. Journal of immunology (Baltimore, Md : 1950) 2011;186:5039–45. doi: 10.4049/jimmunol.1003650. [DOI] [PubMed] [Google Scholar]
- 32.Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. Journal of immunology (Baltimore, Md : 1950) 1994;152:163–75. [PubMed] [Google Scholar]
- 33.Rammensee H-G, Friede T, Stevanović S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 34.Smith HA, McNeel DG. Vaccines targeting the cancer-testis antigen SSX-2 elicit HLA-A2 epitope-specific cytolytic T cells. Journal of immunotherapy (Hagerstown, Md : 1997) 2011;34:569–80. doi: 10.1097/CJI.0b013e31822b5b1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olson BM, Frye TP, Johnson LE, Fong L, Knutson KL, Disis ML, et al. HLA-A2-restricted T-cell epitopes specific for prostatic acid phosphatase. Cancer immunology, immunotherapy: CII. 2010;59:943–53. doi: 10.1007/s00262-010-0820-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andersen ML, Ruhwald M, Nissen MH, Buus S, Claesson MH. Self-peptides with intermediate capacity to bind and stabilize MHC class I molecules may be immunogenic. Scand J Immunol. 2003;57:21–7. doi: 10.1046/j.1365-3083.2003.01182.x. [DOI] [PubMed] [Google Scholar]
- 37.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nature methods. 2012;9:676–82. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schindelin J, Rueden CT, Hiner MC, Eliceiri KW. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol Reprod Dev. 2015;82:518–29. doi: 10.1002/mrd.22489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–96. [PubMed] [Google Scholar]
- 40.Gallegos AM, Xiong H, Leiner IM, Sušac B, Glickman MS, Pamer EG, et al. Control of T cell antigen reactivity via programmed TCR downregulation. Nature Immunology. 2016 doi: 10.1038/ni.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simon S, Vignard V, Florenceau L, Dreno B, Khammari A, Lang F, et al. PD-1 expression conditions T cell avidity within an antigen-specific repertoire. Oncoimmunology. 2015;5:e1104448-e. doi: 10.1080/2162402X.2015.1104448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hailemichael Y, Overwijk WW. Peptide-based anticancer vaccines: The making and unmaking of a T-cell graveyard. Oncoimmunology. 2013;2:e24743. doi: 10.4161/onci.24743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carreño LJ, Bueno SM, Bull P, Nathenson SG, Kalergis AM. The half-life of the T-cell receptor/peptide-major histocompatibility complex interaction can modulate T-cell activation in response to bacterial challenge. Immunology. 2007;121:227–37. doi: 10.1111/j.1365-2567.2007.02561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith TRF, Verdeil G, Marquardt K, Sherman La. Contribution of TCR signaling strength to CD8+ T cell peripheral tolerance mechanisms. Journal of immunology (Baltimore, Md : 1950) 2014;193:3409–16. doi: 10.4049/jimmunol.1401194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jazirehi AR, Lim A, Dinh T. PD-1 inhibition and treatment of advanced melanoma-role of pembrolizumab. Am J Cancer Res. 2016;6:2117–28. [PMC free article] [PubMed] [Google Scholar]
- 46.Lizotte PH, Ivanova EV, Awad MM, Jones RE, Keogh L, Liu H, et al. Multiparametric profiling of non-small-cell lung cancers reveals distinct immunophenotypes. JCI Insight. 2016;1:e89014. doi: 10.1172/jci.insight.89014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen YM. Immune checkpoint inhibitors for nonsmall cell lung cancer treatment. J Chin Med Assoc. 2016 doi: 10.1016/j.jcma.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 48.Zhou TC, Sankin AI, Porcelli SA, Perlin DS, Schoenberg MP, Zang X. A review of the PD-1/PD-L1 checkpoint in bladder cancer: From mediator of immune escape to target for treatment. Urol Oncol. 2016 doi: 10.1016/j.urolonc.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 49.Gargiulo P, Della Pepa C, Berardi S, Califano D, Scala S, Buonaguro L, et al. Tumor genotype and immune microenvironment in POLE-ultramutated and MSI-hypermutated Endometrial Cancers: New candidates for checkpoint blockade immunotherapy? Cancer Treat Rev. 2016;48:61–8. doi: 10.1016/j.ctrv.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 50.Howitt BE, Shukla SA, Sholl LM, Ritterhouse LL, Watkins JC, Rodig S, et al. Association of Polymerase e-Mutated and Microsatellite-Instable Endometrial Cancers With Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol. 2015;1:1319–23. doi: 10.1001/jamaoncol.2015.2151. [DOI] [PubMed] [Google Scholar]
- 51.Gaiger A, Reese V, Disis ML, Cheever MA. Immunity to WT1 in the animal model and in patients with acute myeloid leukemia. Blood. 2000;96:1480–9. [PubMed] [Google Scholar]
- 52.Bullock TNJ, Mullins DW, Engelhard VH. Antigen Density Presented By Dendritic Cells In Vivo Differentially Affects the Number and Avidity of Primary, Memory, and Recall CD8+ T Cells. The Journal of Immunology. 2003;170:1822–9. doi: 10.4049/jimmunol.170.4.1822. [DOI] [PubMed] [Google Scholar]
- 53.Markey KA, Gartlan KH, Kuns RD, MacDonald KP, Hill GR. Imaging the immunological synapse between dendritic cells and T cells. J Immunol Methods. 2015;423:40–4. doi: 10.1016/j.jim.2015.04.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.