

Abstract
We evaluated the impact of early embryonic deletion of huntingtin (htt) from pyramidal neurons on cortical development, cortical neuron survival and motor behavior, using a cre-loxP strategy to inactivate the mouse htt gene (Hdh) in emx1-expressing cell lineages. Western blot confirmed substantial htt reduction in cerebral cortex of these Emx-httKO mice, with residual cortical htt in all likelihood restricted to cortical interneurons of the subpallial lineage and/or vascular endothelial cells. Despite the loss of htt early in development, cortical lamination was normal, as revealed by layer-specific markers. Cortical volume and neuron abundance were, however, significantly less than normal, and cortical neurons showed reduced brain-derived neurotrophic factor (BDNF) expression and reduced activation of BDNF signaling pathways. Nonetheless, cortical volume and neuron abundance did not show progressive age-related decline in Emx-httKO mice out to 24 months. Although striatal neurochemistry was normal, reductions in striatal volume and neuron abundance were seen in Emx-httKO mice, which were again not progressive. Weight maintenance was normal in Emx-httKO mice, but a slight rotarod deficit and persistent hyperactivity were observed throughout the lifespan. Our results show that embryonic deletion of htt from developing pallium does not substantially alter migration of cortical neurons to their correct laminar destinations, but does yield reduced cortical and striatal size and neuron numbers. The Emx-httKO mice were persistently hyperactive, possibly due to defects in corticostriatal development. Importantly, deletion of htt from cortical pyramidal neurons did not yield age-related progressive cortical or striatal pathology.
Keywords: Huntingtin knockout, Development, Cortex, Striatum, Neuronal survival
1. Introduction
An expanded CAG repeat in the ubiquitously expressed Huntingtin gene (termed HD in humans) is causal for the neurodegeneration in the striatum and cortex in Huntington’s disease (HD). Among its functions, the protein derived from the normal HD allele (i.e. huntingtin, or Htt in humans) interacts with microtubules, the dynein/dynactin complex and kinesin to regulate the microtubule-dependent transport of proteins and organelles in neurons (Caviston et al., 2007; Colin et al., 2008; Gauthier et al., 2004; McGuire et al., 2006; Saudou and Humbert, 2016). Huntingtin also appears to play a role in dividing cells through its presence in spindle microtubules of the centrosome. Accordingly, huntingtin knockdown has been found to reduce cell division in vitro (Godin et al., 2010). Consistent with an important role of huntingtin in cell division in at least some brain regions, we have found that Hdh−/− embryonic stem cells injected into blastocysts readily survive in brainstem, but are greatly underrepresented in cortical and striatal areas, the major sites of neuron loss in HD (Reiner et al., 2001). Similarly, studies in zebrafish have shown that huntingtin plays a greater role in forebrain than midbrain and hindbrain development (Henshall et al., 2009). We have further found that at embryonic day 12.5, Hdh−/− neuroblasts are as abundant in cortex, striatum, and thalamus as they are in brainstem in mice with blastocyst injection of Hdh−/− embryonic stem cells (Reiner et al., 2003). This suggests that the underrepresentation of Hdh−/−neurons in cortex and striatum in our blastocyst injection chimeras may reflect, at least in part, death of Hdh−/− cells rather than or as well as deficient cell division. Moreover, Conforti et al. (2013) have found that Hdh−/− stem cells tend to adopt a glial rather than a neuronal fate. In addition to possible roles in neuron division, survival and fate choice during development, huntingtin may also play an important role in cortical neuron migration to final adult laminar position during development (Tong et al., 2011).
To further address the roles of huntingtin in neurogenesis, neuronal migration and neuronal survival during development of cerebral cortex, we evaluated the impact of early embryonic deletion of Hdh from dorsal telencephalic neuronal and glial progenitors using a cre-loxP strategy to inactivate the mouse Hdh gene in emx1-expressing cell lineages (Emx-httKO mice). Since emx1 is expressed in the pallial proliferative zone beginning at about E8 (Simeone et al., 1992; Yoshida et al., 1997), Emx-httKO mice sustain Hdh deletion from cortical pyramidal neuron and glial precursors and their progeny beginning as early as E8 and demonstrably by E10.5 (Gorski et al., 2002). Although the forebrain huntingtin deletion in Emx-httKO mice is primarily limited to cerebral cortex, we also examined striatum because of the impact of the cortex on striatum via the massive corticostriatal projection system (Reiner et al., 2010), and because a prior study by others showed early exuberance in corticostriatal connections in Emx-httKO mice (McKinstry et al., 2014). Moreover, because huntingtin is believed to be important for forebrain neuron survival (Reiner et al., 2001, 2003), and because therapeutics for Huntington’s disease designed to deplete huntingtin expression are under development (Crook and Housman, 2013), we also examined the impact of cortical deletion of huntingtin on cortical and striatal neuron abundance up to nearly 2 years of age. We found that embryonic deletion of huntingtin from developing pallium impaired cortical neurogenesis and/or neuronal survival during development, but had no notable effect on migration of cortical neurons to their correct laminar destinations. The resulting deficiency in cortical and striatal neurons beginning early in life was accompanied by a hyper-activity phenotype, but subsequent progressive age-related loss of cortical or striatal neurons was not observed. Thus, an early embryonic deletion of Hdh from cortical pyramidal neurons does not adversely affect the long-term survival of cortical or striatal neurons in adulthood.
2. Materials and methods
2.1. Animals
A total of 22 control mice and 16 Emx-httKO mice were used in the present study. Of these, five control (2 males, 3 females) and 5 Emx-httKO mice (2 males, 3 females) were used in in Western blot studies. The remaining mice were used in behavioral and histological studies. Fifteen control mice (11 males, 4 females) and 9 Emx-httKO mice (6 males, 3 females) were studied behaviorally and by immunolabeling, and 2 control males and 2 Emx-httKO males were studied behaviorally and by in situ hybridization histochemistry. Mice ranged in age from 2 months to 23.6 months, with a mean age of 11.6 months for control mice and a mean age of 12.2 months for Emx-httKO mice. No noteworthy male-female differences were seen in the results (other than greater weight in males), and so male-female data are combined. Control mice were littermates of the Emx-httKO mice, and included Hdh+/+ mice, Hdhflox/+ mice, and HdH+/− mice. As previously reported, mice with one floxed Hdh allele and one wild-type (WT) allele as well as hemizygous Hdh mice are indistinguishable from Hdh+/+ WT mice (Dragatsis et al., 2000; Duyao et al., 1995; Nasir et al., 1995; Zeitlin et al., 1995), as we also observed here. We refer to all of these different mice expressing WT huntingtin as control mice. All experiments were undertaken in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals, Society for Neuroscience Guidelines, and University of Tennessee Health Science Center Guidelines, and had institutional approval.
2.2. Western blot analysis
Total protein extract was obtained from cortex and striatum of three 5-month old and two 12-month old Hdhflox/− mice and three 5-month old and two 12-month old Emx-httKO mice by tissue homogenization in RIPA buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate) containing proteinase inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Roche). Insoluble debris was discarded after centrifugation, and protein concentration was determined by the Bradford assay (BIO-RAD, Hercules, CA). Approximately 30 μg of protein was separated by SDS-PAGE (8% or 12%) and transferred to nitrocellulose membranes. The membranes were blocked in 5% non-fat milk in PBS for 1 h at room temperature and incubated overnight at 4 °C with mouse monoclonal antibody against huntingtin (Millipore MAB 2166, 1:3000), rabbit polyclonal against BDNF (Alomone labs ANT-010, 1:1000), rabbit monoclonal against Erk1/2 (Cell Signaling mAb 4695, 1:1000), rabbit monoclonal against phospho-Erk1/2 (Cell Signaling mAb 4370, 1:2000), rabbit polyclonal against phospho-Akt (Cell Signaling Ab 9271, 1:1000), or mouse monoclonal against beta-tubulin (Chemicon, MAB 3408, 1:5000). The specificity of these antibodies for their target antigens has been demonstrated by the manufacturer by Western blot and in published studies by others (Harrington et al., 2012; Milman and Woulfe, 2013; Czech et al., 2014; Duarte-Neves et al., 2015; Gu et al., 2017; Patel et al., 2017). The membranes were then washed in PBS containing 0.2% Tween, and incubated with anti-mouse or anti-rabbit secondary antibodies at room temperature for 1 h. The membranes were subsequently washed in PBS containing 0.2% Tween, and protein bands were visualized by chemiluminescence (Pierce, Rockford, IL) and autoradiography. For quantification, membranes were scanned and band intensities were analyzed using ImageJ software (http://rsb.info.nih.gov/ij/). Final relative values were determined using the ratio of net band to net loading control.
2.3. Immunohistochemistry
Fifteen control mice (11 males, 4 females) and 9 Emx-httKO mice (6 males, 3 females) that had been studied behaviorally were subsequently used in immunolabeling studies. Under avertin anesthesia (0.2 ml/g body weight), these mice were perfused transcardially with 6% dextran in 0.1 M sodium phosphate buffer at pH 7.4 (PB), followed by 4% paraformaldehyde, 0.1 M lysine-0.1 M sodium periodate in 0.1 M PB. The brains were removed, stored overnight at 4 °C in 20% sucrose/10% glycerol, and sectioned frozen in the transverse plane at 35 μm on a sliding microtome. A one in six series of brain sections from each mouse was mounted as sectioned, and subsequently stained for cresyl violet. Immunohistochemical single-labeling using peroxidase-antiperoxidase (PAP) procedures described previously (Meade et al., 2002; Reiner et al., 2012a) was employed to visualize a variety of neurochemical features in mutant and control brains. To study the laminar organization of cerebral cortex, we used immunolabeling with a mouse monoclonal antibody (Sigma-Aldrich, C89848) to detect the lightly labeled calbindinergic neurons defining layers 2–3 (Van Brederode et al., 1991; Kondo et al., 1997; Fauser et al., 2013), a rabbit polyclonal antibody (Sigma/Aldrich, V2514) to detect VGLUT2+ fibers in layer 4 (Deng et al., 2013), the SMI-32 mouse monoclonal antibody (Covance, SMI-32R) and a rat monoclonal antibody against Ctip2 (Abcam, AB18465) to define neurons in layer 5 (Özdinler et al., 2011; Fauser et al., 2013), and a rabbit polyclonal antibody against FoxP2 (Abcam, AB16046) to define neurons in layer 6 (Özdinler et al., 2011). The specificity of these antibodies for their target antigens has been demonstrated by the manufacturer by Western blot and in published studies (Stillman et al., 2009; Hirano et al., 2011; Hashimoto et al., 2012; Huang et al., 2012; Lei et al., 2013).
To detect neuropathology and/or neuron deficiency, immunolabeling was performed using a mouse monoclonal anti-GFAP or using a mouse monoclonal anti-NeuN, respectively. The specificity and efficacy of these antibodies has been shown previously (Mullen et al., 1992; Wolf et al., 1996; Bloch et al., 2011; Hu et al., 2011). Immunolabeling for Substance P (SP) and for the D1 dopamine receptor was used to study the SP+/D1+ direct pathway striatal neurons and their projections to the globus pallidus internus (GPi) and substantia nigra (SN), while immunolabeling for enkephalin (ENK) was used to study the ENK+ indirect pathway neuron striatal projection system to the globus pallidus externus (GPe). Immunolabeling for DARPP32 was used to evaluate striatal projection neurons and their projections as a group. The anti-SP was a rabbit polyclonal antibody (ImmunoStar, AB1566) whose specificity has been documented previously (Russo et al., 2013; Zahm et al., 2013). The anti-D1 antibody was a rat monoclonal directed against the 97-amino acid C-terminal fragment of human D1 (Sigma-Aldrich, D187), whose specificity has been demonstrated previously (Levey et al., 1993; Hersch et al., 1995). The anti-ENK used was a rabbit polyclonal antibody against leucine-enkephalin (ImmunoStar, 20066) whose specificity has also been shown previously (Reiner, 1987; Reiner et al., 2007; Tripathi et al., 2010). Immunolabeling for DARPP32 was carried out using an anti-DARPP32 (generously provided by P. Greengard and H. Hemmings), whose specificity has been previously documented (Ouimet et al., 1984).
To characterize the regional specificity of cortical huntingtin deletion, we used immunolabeling for huntingtin using the D7F7 antibody from Cell Signaling (#5656). This antibody is selective for huntingtin and detects labeling for huntingtin in dendrites, terminals, and perikarya (Clemens et al., 2015), but the latter is often obscured by the density of neuropil labeling. We used D7F7 to assess the effect of the cortical huntingtin deletion immunohistochemically because anti-huntingtin antibodies that label perikarya tend to have substantial background labeling, which in this case would hinder interpretations. Finally, huntingtin is reportedly important for BDNF expression levels, which is highly expressed in cortical pyramidal neurons (Zuccato and Cattaneo, 2009; Reiner et al., 2012b). Accordingly, we used immunolabeling to characterize BDNF in cortical pyramidal neurons, using the N20 antiserum from Santa Cruz (Cat # sc546), whose specificity is demonstrated in Flores-Otero and Davis (2011).
2.4. In situ hybridization Histochemistry (ISHH)
Two control males and two Emx-httKO males that had been studied behaviorally were subsequently used for in situ hybridization histochemistry at 18.2 months of age. Fresh-frozen coronal sections were processed for preproenkephalin (PPE, the enkephalin precursor), preprotachykinin (PPT, the substance P precursor), the D1 dopamine receptor, the D2 dopamine receptor, and brain-derived neurotrophic factor (BDNF) mRNA detection by ISSH, using previously described methods (Sun et al., 2002; Wang et al., 2006; Dragatsis et al., 2009; Reiner et al., 2012a, 2012b). ISHH was performed on 20 μm thick fresh frozen cryostat sections through the cortex and striatum anterior to the anterior commissure. The sections were collected onto pre-cleaned Superfrost®/Plus microscope slides, dried on a slide warmer, and stored at −80 °C until used for ISHH. To process sections for ISHH, the slides were removed from −80 °C, quickly thawed and dried using a hair dryer. After fixation with 2% paraformaldehyde in saline sodium citrate (2× SSC) for 5 min, the sections were acetylated with 0.25% acetic anhydride/0.1 M triethanolamine hydrochloride (pH 8.0) for 10 min, dehydrated through a graded ethanol series, and air-dried. Digoxigenin-UTP labeled cRNA probes (i.e. riboprobes) were transcribed from plasmids with PPE, PPT, D1, D2 or BDNF cDNA inserts, generated by us using RT-PCR. Table 1 shows details on these probes and their target mRNA sequences. The BDNF riboprobe was directed against the protein-coding region of BDNF and part of the adjacent 3-prime untranslated sequence. Note that all BDNF transcripts share this sequence, found within exon IX of the BDNF gene, and thus our probe detected all BDNF transcripts (Aid et al., 2007). The sections were incubated with digoxigenin-labeled probe in hybridization buffer containing 50% formamide, 4× SSC, 1× Denhardt’s solution, 200 μg/ml denatured salmon sperm DNA, 250 μg/ml yeast tRNA, 10% dextran sulfate, and 20 mM dithiothreitol at 63 °C overnight. After hybridization, the slices were washed at 58 °C consecutively in 4× SSC, 50% formamide with 4× SSC, 50% formamide with 2× SSC, and then 2× SSC, followed by treatment with RNase A (20 μg/ml) for 30 min at 37 °C. Finally, sections were washed at 55 °C in 1× SSC, 0.5× SSC, 0.25× SSC, dehydrated through a graded ethanol series, and air-dried. Digoxigenin labeling was detected using anti-digoxigenin Fab fragments conjugated to alkaline phosphatase (AP), as visualized with nitroblue tetrazolium histochemistry (Roche, Indianapolis, IN). Sections were coverslipped with a 1% gelatin-based aqueous solution. The abundance of striatal neurons labeled for PPT, PPE D1 or D2 was analyzed using ImageJ.
Table 1.
ISHH probe design.
| Target mRNA | Riboprobe length | PCR primers | Nucleotide target | GenBank mRNA accession # |
|---|---|---|---|---|
| PPE | 817 bp | Sense: 5′-TTCCTGAGGCTTTGCACC-3′ | 312–1128 | 001002927 |
| Antisense: 5′-TCACTGCTGGAAAAGGGC-3′ | ||||
| PPT | 900 bp | Sense: 5′-TCGAACATGAAAATCCTCGTGGCC-3′ | 95–994 | D17584 |
| Antisense: 5′-CACATCATACAATGACTGAAGACC-3′ | ||||
| D1 | 694 bp | Sense: 5-CTCATAAGCTTTTACATCCCCG-3 | 1116–1809 | NM_010076 |
| Antisense: 5-GAGACATCGGTGTCATAGTCCA-3 | ||||
| D2 | 1086 bp | Sense: 5′-GAAGATCCTGCACTGCTGAGT-3 | 1412–2497 | NM_010077.1 |
| Antisense: 5′-ATGTTACAGAGTTGGAGCCCAG-3′ | ||||
| BDNF | 920 bp | Sense: 5′-GGCGCCCATGAAAGAAGTAAAC-3′ | 715–1634 | MN_007540 |
| Antisense: 5′-CGGCAACAAACCACAACATTAT-3′ |
2.5. Motor behavior
Seventeen control mice (13 males, 4 females) and 11 Emx-httKO mice (8 males, 3 females) were studied behaviorally using accelerating rotarod and open field analysis, in some cases at different points during the lifespan of individual mice. Rotarod analysis was carried out on Emx-httKO mice and control mice, using a San Diego instruments (San Diego, CA) rodent rotarod. For the rotarod task, RPM increased from 0 to 30 over a four-minute period, and 30 RPM was then maintained for another 2 min. The mice typically received a total of six rotarod sessions, separated by 3–5 min. Time to fall was the measure of rotarod performance. We also conducted automated 30-minute assessment of open field behavior at the same time points as for rotarod, using a Noldus EthoVision video tracking system to record and digitize the mouse movements (Noldus Information Technology, The Netherlands), and the SEE software of Drai and Golani (2001) to analyze the mouse motor behavior (Reiner et al., 2012a). The SEE software uses algorithms to dichotomous mouse movements into lingering episodes and progression segments, and allows rapid characterization of endpoints related to locomotion, and it is robust in characterizing behavioral differences among mouse strains (Drai et al., 2000; Drai and Golani, 2001; Kafkafi et al., 2001, 2003; Lipkind et al., 2004; Reiner et al., 2012a). Each animal was brought from its housing room, introduced into the arena and returned after the end of the 30 min session. The arena was a 200 cm diameter circular area with a non-porous gray floor and a 50 cm high gray wall. The gray floors and wall provided a high-contrast background, enabling video tracking of the mice. The arena was illuminated with two ceiling-mounted 40 W neon bulbs.
2.6. Stereology
Stereological neuron counts were carried out blindly for nine Emx-httKO mice (6 males, 3 females) and fifteen control mice (11 males, 4 females) that had been studied behaviorally. A one-in-twelve series of coronal sections immunolabeled for NeuN from the rostral end of striatum to the level of the anterior commissure was used for striatal and cortical neuron counts in each mouse. These same landmarks were used to define the limits of cerebral cortex and striatum for volumetric analysis. Unbiased stereological counts of striatal and cortical neurons were obtained using a Neurolucida Stereo Investigator system (Micro-Brightfield, Colchester, VT (Bu et al., 2016; Guley et al., 2016)). The dissector counting method was used, in which neurons were counted in counting frames assigned by the software throughout the areas pre-defined as striatum and cortex. Counts for the two sides of the brain were averaged for each case. In a subset of cases, stereological counts of cortical and striatal neurons were performed using the cresyl violet-stained sections. These studies showed that the two methods produced indistinguishable results.
2.7. Image analysis of immunolabeling in striatal target areas
To assess any possible pathology to the neurons of origin of the striatal projection systems, blinded computer-assisted image analysis was carried out on immunolabeled striatal terminals in each of the two main striatal projection targets in rodents, the GPe (n = 8 Emx-httKO, 14 control), GPi (n = 4 Emx-httKO, 5 control) and SN (n = 4 Emx-httKO, 5 control). For these studies, the extent and immunolabeling intensity of the ENK fiber plexus in GPe, and the extent and immunolabeling intensity of the SP fiber plexi in GPi and substantia nigra were analyzed. Images of individual fields were captured using a 4× objective, and analyzed using ImageJ (v. 1.61), as in prior studies (Meade et al., 2000; Sun et al., 2002; Reiner et al., 2007, 2012a). Fiber abundance for a given structure in a given case was expressed as the mean area occupied by labeled fibers for all measurements for that case. This provided measures of the abundance of labeled fibers per striatal target and of the intensity of peptide within them (reflecting peptide abundance). We have used this approach in prior studies on human HD brain or the brain in HD animal models (Figueredo-Cardenas et al., 1994; Deng et al., 2004; Reiner et al., 2012a).
2.8. Statistics
Data from the behavioral, immunohistochemical and stereological studies were analyzed statistically by unpaired two-tailed t-tests comparing control and Emx-httKO mice, with the exception that the relative performance of control and Emx-httKO mice on rotarod across trials was analyzed using one-way ANOVA. Regression analysis was used to evaluate any changes in any of the parameters over the age-range examined in the control and Emx-httKO mice. Regression analysis and t-tests were performed using Excel, and ANOVA was performed using SPSS. Analysis of covariance was used to determine if the slope of the regression line for age-related effects in the control mice differed from that for Emx-httKO mice for any of the morphological or behavioral parameters, using online tools (http://www.danielsoper.com/).
3. Results
3.1. Generation of Emx-httKO mice
A mutant mouse line (Mouse Genome Informatics designation, Htttm2Szi) was previously created in which lox sequences were inserted that flank the 1.3kB region upstream of the Hdh transcription initiation site and intron1 (Fig. 1A, B) (Dragatsis et al., 2000). We used this mouse line in combination with a previously described Emx1IREScre/+ mouse line (Mouse Genome Informatics designation, Emx1tm1(cre)Krj) to direct Hdh deletion from cortical pyramidal neurons (Gorski et al., 2002). To this end, we bred Hdh+/− mice (Mouse Genome Informatics designation, Htttm1Szi) with Emx1IREScre/+ mice, and the resulting Emx1IREScre/+; Hdh+/− offspring were then crossed with Hdhflox/flox mice (Mouse Genome Informatics designation, Htttm2Szi) for the generation of Em-x1IREScre/+; Hdhflox/− mice (referred to here as Emx-httKO mice for simplicity). As expression of emx1 begins in the developing cortex at E8 (Simeone et al., 1992; Yoshida et al., 1997) and is robust by E10.5 (Gorski et al., 2012), cre driven by an emx1 promoter deletes Hdh from cortical pyramidal neurons early in cortical development. We confirmed that cre expression is specific for pallium in Emx1IREScre/+ mice by crossing them to R26R reporter mice (Gtrosa26tm1Sor), which express β-galactosidase upon cre-mediated recombination. Staining of forebrain sections from Emx1IREScre/+; R26R/+ offspring by X-gal histochemistry showed that cre-driven LacZ expression in these mice is restricted to the cerebral cortex (Figs. 1C).
Fig. 1.

A mutant mouse line (Htttm2Szi) was created in which loxP sequences were inserted that flanked the 1.3 kB region upstream of the Hdh transcription initiation site and intron 1 (Dragatsis et al., 2000). Images A and B show a schematic of the wild-type Hdh allele and the floxed Hdh allele before recombination (A), and after recombination (B), the latter generating an Hdh allele lacking the promoter and exon 1. To evaluate the specificity of emx1-cre driven recombination in mouse forebrain that would be achieved by crossing Emx1IREScre/+ mice with Htttm2Szi mice, we crossed Emx1IREScre/+ mice with R26R reporter mice (Gtrosa26tm1Sor) possessing a floxed lacZ- gene that expresses β-galactosidase upon recombination by cre. Image C shows the results for a section through the telencephalon from an Emx1IREScre/+; R26R offspring that had been X-gal stained. The results confirm that cre expression in Emx1IREScre/+ mice is specific for cerebral cortex. LacZ labeling in striatum is in corticofugal fibers.
3.2. Specificity of huntingtin deletion
In the Emx-httKO mice, Western blot analysis for both 5 month old and 12 month old mice showed a significant 85.3% reduction of huntingtin protein in cerebral cortex of 5 control and 5 Emx-httKO mice (p = 0.00000005), confirming the efficacy of deletion from cortical pyramidal neurons (Fig. 2A, C). Residual cortical huntingtin is likely to be localized to interneurons, due to their origins from a subpallial lineage not expressing emx1, and/or to cells of the vasculature. Immunolabeling for huntingtin with the D7F7 antibody from Cell Signaling confirmed the greatly diminished expression of huntingtin in neuronal perikarya and the neuropil of cerebral cortex of Emx-httKO mice (Fig. 3A, B), with the only residual labeling being in presumptive cortical interneurons and in the terminals of the thalamic projection to layer 4. As also observed by McKinstry et al. (2014), Western blotting revealed a substantial and significant reduction (82.3%, p = 0.00000025) in huntingtin in striatum of the Emx-httKO mice as well (Fig. 2B, C). Immunolabeling with D7F7 confirmed this finding and showed a prominent reduction of huntingtin in the striatal neuropil of Emx-httKO mice, with perikaryal labeling remaining prevalent (Fig. 3C, D). Most conspicuous among the huntingtin-immunolabeled striatal neurons were perikarya the size and shape of cholinergic and parval-buminergic interneurons, which have been previously reported to express high levels of huntingtin (Fusco et al., 1999). The bulk of the prominent neuropil labeling in striatum thus appears to represent labeling of cortical terminals for huntingtin, as also suggested by McKinstry et al. (2014). Consistent with the expression of huntingtin by striatal projection neurons (Fusco et al., 1999) and its anterograde axonal transport (Block-Galarza et al., 1997; McKinstry et al., 2014), the terminals in the three major striatal target areas – GPe, GPi, and sub-stantia nigra - were rich in huntingtin in control mice (Fig. 4). The abundance of huntingtin in striatal terminals in GPe, GPi and the nigra in Emx-httKO mice appeared to be as ample as in age-matched control mice (Fig. 4), suggesting that huntingtin production by striatal neurons themselves was not diminished by the genetic deletion of Hdh from cortical pyramidal neurons. These findings further indicate that the diminished huntingtin in striatum of the Emx-httKO mice is likely to reflect loss of huntingtin from corticostriatal terminals rather than from striatal neurons. The deletion of Hdh from cortical neurons of the emx1 pyramidal neuron lineage was associated with a significant 37.9% reduction in BDNF levels in cerebral cortex (n = 5 control, 5 Emx-httKO, p = 0.000031) (Fig. 5A, D), caused by reduced expression of BDNF message by cortical pyramidal neurons (Fig. 5B, C), and as a result a reduction in BDNF protein in cortical pyramidal neurons (Fig. 6A, B). Levels of BDNF in layer 5 cortical neurons, where corticostriatal neurons reside (Reiner et al., 2010), appeared to be reduced by about 40–50% in both immunolabeled sections and ISHH sections. Western analyses were performed to evaluate the impact of the cortical deletion of huntingtin from pyramidal neurons on Akt and ERK signaling pathways, both of which are driven by BDNF. We found that both phospho-Akt and phospho-ERK were significantly reduced in cerebral cortex of Emx-httKO mice compared to control mice (n = 5 control, 5 Emx-httKO) (Fig. 5A, D), indicating reduced signaling by both BDNF-driven pathways. Phospho-Akt (pAkt) was reduced by 44.6% in Emx-httKO cortex (p = 0.000040), and phospho-ERK (pERK) was reduced by 70.8% in Emx-httKO cortex (p = 0.00000013). ERK itself was unchanged (Fig. 5A, D).
Fig. 2.
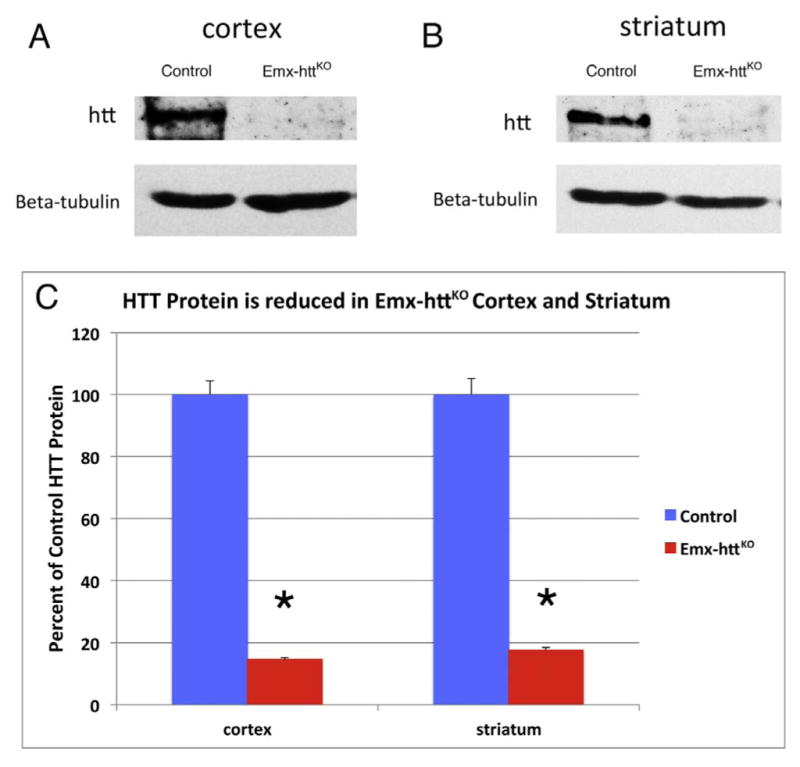
Western blot analyses of total protein lysates from cortex and striatum of control and Emx-httKO mice. Protein lysates were separated in 8% SDS-PAGE and transferred to nitrocellulose membrane. The upper panel in image A shows detection of huntingtin with a mouse monoclonal anti-huntingtin antibody 2166, and the lower panel shows anti-β-tubulin staining as a loading control, from 5-month-old control and Emx-httKO mice. Note that huntingtin protein is substantially reduced in cortex of Emx-httKO mice, confirming knockout efficacy. Huntingtin protein was also substantially reduced in striatum, as shown in image B comparing 5-month-old control and Emx-httKO mice. As explained in the text, this is likely to largely represent depletion of huntingtin from corticostriatal terminals, which appears to be the predominant source of huntingtin in striatum. The graph in image C shows densitometric analysis of Western blot results for huntingtin in cerebral cortex and striatum in 5 control and 5 Emx-httKO mice at 5- or 12-months of age, normalized to tubulin, confirming that the reduction of huntingtin in Emx-httKO cortex and striatum is highly significant (asterisks).
Fig. 3.

Images A–D show immunolabeling in control and Emx-httKO cortex at low power and in control and Emx-httKO striatum (Str) at slightly higher power using the D7F7 antibody against huntingtin, which labels huntingtin in axons, terminals, neuropil and the cytoplasm of perikarya. Note that huntingtin is reduced in the cortex in Emx-httKO mice except in the terminals of thalamic input in layer 4 and in presumptive interneurons. In striatum, huntingtin is reduced in the neuropil in Emx-httKO mice, but not in striatal perikarya themselves. Note that with the loss of corticostriatal huntingtin from the striatal neuropil, the huntingtin-immunolabeled striatal perikarya are more salient. The mice shown for immunolabeling were 10.8 months old. Scale bar in B applies to A as well, and the scale bar in D applies to C as well.
Fig. 4.

Immunolabeling with D7F7 showing huntingtin in terminals in the striatal target areas GPe (globus pallidus externus), GPi (globus pallidus internus) and SN (substantia nigra) in control and Emx-httKO mice. Note that anti-huntingtin labeling in striatofugal terminals in GPe, GPi and SN in Emx-httKO mice is indistinguishable from that in control mice, suggesting that striatal projection neuron huntingtin production and axonal transport was unaffected by the cortical huntingtin deletion.
Fig. 5.

Images showing cortical BDNF reduction in Emx-httKO mice by Western Blot (A) and in situ hybridization histochemistry (ISHH) (B, C). Image A shows Western blot analyses of total protein lysates from cortex of 5-month-old control and Emx-httKO mice. Protein lysates were separated in 12% SDS-PAGE and transferred to nitrocellulose membrane, which were then sequentially probed with anti-BDNF, anti-phospho-Akt, (pAkt) anti phospho-ERK1/2 (pERK), anti-ERK1/2 and anti-β-tubulin. Note that BDNF, phospho-Akt and phospho-ERK are substantially reduced in cortex of Emx-httKO mice. Images B and C show BDNF ISHH labeling of cerebral cortex from an 18-month old control and Emx-httKO mice. Consistent with the Western blot data, BDNF expression is reduced in cortical pyramidal neurons of layers 2/3 and 5 in the Emx-httKO mice. The graph in image D shows densitometric analysis of Western blot results for cerebral cortex in 5 control and 5 Emx-httKO mice (at 5 and 12 months of age), confirming that the reduction in BDNF, pAkt, and pERK (asterisks) in Emx-httKO cortex is highly significant.
Fig. 6.

Images showing cortical BDNF reduction in Emx-httKO mice by immunohistochemistry (IHC) (A, B). Images A and B show BDNF immunolabeling of cerebral cortex from 10.8-month old control and Emx-httKO mice, respectively, showing the reduced abundance of BDNF in cortical pyramidal neurons of layer 5 in the Emx-httKO mice. Scale bar in A applies to B as well.
3.3. Forebrain morphology and cortical organization
General forebrain morphology and cortical organization appeared normal in adult mice with early embryonic deletion of huntingtin from cortical pyramidal neurons. For example, NeuN immunostained sections cut transverse to the long axis of the brain show that at 10.5 months of age, the overall structure of the forebrain of Emx-httKO mice was indistinguishable from that in control mice at the same age (Fig. 7). Moreover, cortical lamination was normal in terms of the relative location of layer-specific cell populations such as the lightly labeled calbindinergic projection neurons in layers 2–3, VGLUT2+ fibers in layer 4, SMI-32+ and Ctip2+ neurons in layer 5, and FoxP2+ neurons in layer 6 (Fig. 8), as well as in terms of the thickness of these layers. To determine if the cortical huntingtin deletion had an adverse effect on cortical and/or striatal neuron abundance (either due to deficient neurogenesis or diminished survival), we performed blinded stereological assessment of cortical volume and neuron abundance (n = 15 control, 9 Emx-httKO). We found that cerebral cortex volume in Emx-httKO mice was slightly but significantly smaller than in control mice (8.7% reduction) overall across the 3.2 to 23.6 month lifespan examined (p = 0.0011). The volume difference did not progress with age, and the regression lines for volume as a function of age remained largely parallel for control and Emx-httKO mice throughout the ages examined (Fig. 9A). Similarly, cortical neuron abundance in mice with early embryonic deletion of huntingtin from cortical pyramidal neurons was significantly less than in control mice (22.3% reduced) overall across the 3.2 to 23.6 month lifespan examined (p = 0.0006) (Fig. 9C). This reduction was evident in both counts of NeuN-immunolabeled and cresyl violet-stained tissue. As in the case of cortical volume, the cortical neuron abundance difference did not progress significantly with age, and the regression lines for volume as a function of age remained largely parallel for control and Emx-httKO mice throughout the ages examined. Consistent with the absence of progressive cortical degeneration in Emx-httKO mice, no astrocytic reaction was evident in the Emx-httKO mice at any age (Fig. 8K, L).
Fig. 7.

Images A and B show low power views of NeuN immunostained transverse sections (medial to left) revealing that the overall structure of the forebrain in Emx-httKO mice (B) at 10.5 months of age is indistinguishable from control mice (A) at the same age. Images C and D present higher power views of motor cortex (medial to left) in NeuN immunostained transverse sections, showing that the structure of the cerebral cortex (C, D) in Emx-httKO mice at 10.5 months of age (D) is indistinguishable from control mice (C) at the same age.
Fig. 8.

Images of immunolabeled transverse sections showing that cortical lamination was normal in Emx-httKO mice in terms of the relative location of layer-specific cell populations such as calbindinergic neurons in layers 2–3 (A, B), VGLUT2+ fibers in layer 4 (C, D), SMI-32+ (E, F) and CTIP2+ (G, H) neurons in layer 5, and FoxP2+ neurons (I, J) in layer 6, which confirmed the absence of obvious laminar abnormalities. GFAP immunolabeling (K, L) did not reveal any differences between Emx-httKO and control mice.
Fig. 9.

Cortical volume was persistently less in Emx-httKO mice (n = 9) than in control mice (n = 15) throughout the life span of the mice, by about 9% overall (A). The trend was, however, not progressive, as the decline in cortical volume in Emx-httKO mice by 24 months of age was no different than in control mice, and in neither case was volume significantly correlated with age. Similarly, striatal volume was also persistently less in Emx-httKO than in control mice throughout the life span of the mice, by about 14% overall (B). The trend again was, however, not progressive, as the change in striatal volume in Emx-httKO mice by 24 months of age was no different than in control mice, and in neither case was volume significantly correlated with age. Cortical and striatal neuron abundance (C, D) in Emx-httKO mice were also less over the lifespan of the mice (cortex 22.3% less, striatum 20.5% less), but the difference did not become significantly enhanced with age, nor was neuron abundance in either control or Emx-httKO mice significantly correlated with age. Analysis of covariance confirmed that the slope of the regression lines did not differ significantly between control and Emx-httKO mice for any of these four parameters.
3.4. Striatal morphology and neurochemistry
The deletion of huntingtin from cortical pyramidal neurons during embryogenesis had an effect on striatal volume and neuron abundance. Striatal volume was significantly reduced by 14.0% (p = 0.0048) and neuronal abundance was significantly reduced by 20.5% (p = 0.0004). As in the case of cortex, age-related decline in striatal volume and neuron abundance was not statistically evident, and age-related patterns were not different than in the control mice in any case (Fig. 9B, D). Despite the reduced neuron abundance, striatal neurochemistry was normal in Emx-httKO mice, based on ISHH labeling for PPE, PPT, D1 and D2 perikarya in striatum, immunolabeling for D1 and DARPP32 in striatum, and immunolabeling for DARPP32, ENK, D1, and SP terminals in striatal target areas. The spatial abundance per square millimeter and labeling intensity of PPE perikarya per section in 18.2-month old Emx-httKO mice (n = 2) was indistinguishable from that in age-matched control mice (n = 2) (Fig. 10A, E), and the abundance of ENK+ terminals in GPe in Emx-httKO mice (54.9% occupancy of GPe) was not significantly different than that in control mice (53.0% occupancy of GPe) for a sample of 14 control and 8 cortical KO mice ranging in age from 8.4 to 23.6 months (p = 0.5061) (Fig. 10I, L). No significant age-related changes were seen in ENK+ striato-GPe terminal abundance in control or Emx-httKO mice. Similarly, the abundance and density of PPT perikarya per section in 18.2-month old Emx-httKO mice (n = 2) was comparable to that in age-matched control mice (n = 2) (Fig. 10C, G), and the abundances of SP+ terminals in the GPi (38.4% in control GPi versus 41.5% in Emx-httKO GPi) and the substantia nigra (40.0% in control SN versus 38.7% in Emx-httKO SN) of Emx-httKO mice were not significantly different from that in control mice, for a sample of 4 Emx-httKO mice and 5 control mice (GPi, p = 0.3708; SN, p = 0.7336). No significant age-related changes were seen in SP+ striato-GPi or striato-SN terminal abundance in control or Emx-httKO mice. Similar quantitative results were obtained for D2 (Fig. 10B, F) and D1 ISHH (Fig. 10D, H) as for PPE and PPT ISHH, respectively. Note that because of the volumetric reduction, striatal neuron density per square millimeter was only 8.3% less than control in the Emx-httKO mice. This may explain why the spatial density of PPT, PPE, D1 and D2 striatal neurons appeared indistinguishable from normal in Emx-httKO mice, despite the overall reduction in striatal neuron abundance.
Fig. 10.

Striatal neurochemistry is normal in Emx-httKO mice based on in situ hybridization histochemistry (ISHH) for preproenkephalin (PPE), preprotachykinin (PPT), D1 receptors and D2 receptors in striatum, and immunolabeling of enkephalinergic striatal terminals in GPe and of substance P-containing terminals in GPi and SN. As reflected by the ISHH images shown, Emx-httKO mice did not show obvious neurochemical abnormalities for striatal PPE (A, E) or D2 (B, F) in indirect pathway neurons or for striatal PPT (C, G) or D1 (D, H) in direct pathway neurons compared to control mice. All mice used for these images were 18.2 months old at the time of sacrifice. Similarly, immunolabeling for enkephalinergic (ENK) striatal terminals in GPe (I, L) and for substance P-containing striatal terminals in GPi (J, M) and SN (K, N) did not differ between control and Emx-httKO mice. Scale bar in image H is applicable to images A–H, and scale bars in images L, M and N are applicable to images I, J and K, respectively.
3.5. Behavioral data
The Emx-httKO mice were indistinguishable from control mice in weight throughout their lifespan, and both groups showed a significant but normal trend toward age-related weight gain (Fig. 11A). By contrast, Emx-httKO mice showed a mild and non-progressive mean rotarod deficit throughout their lifespan (Fig. 11B), for 23 WT and 14 Emx-httKO measured time points. Additionally, the Emx-httKO mice were persistently and significantly poorer on rotarod than control mice over the repeated trials on the day of testing, as assessed by one-way ANOVA (p = 0.00033). Emx-httKO mice were also abnormal in their open field behavior. In particular, they were hyperactive compared to control mice, traversing a 19.2% greater total distance than age-matched control mice (p = 0.0299) (Fig. 12A). Underlying this increase in distance were a significant 17.9% increase in speed (p = 0.0226), a significant 74.3% increase in the unit of locomotion referred to as progression segment length (p = 0.0087), a significant 34.4% increase in endurance (the ratio of distance traveled in the second 15 min compared to the first 15 min of the open field session) (p = 0.00002), and a significant 35.2% decrease in the number of stops per unit distance (p = 0.0018) (Fig. 12B–E). The increase in the length of progression segments was associated with a significant 22.1% decrease in the frequency of their occurrence (p = 0.0262) (Fig. 12F), which was not enough to offset the other factors driving the activity increase.
Fig. 11.

The Emx-httKO mice do not show weight loss compared to control mice over their lifespan (A), but do show a mild rotarod defect over their lifespan (B). Seventeen control mice (13 males, 4 females) and 11 Emx-httKO mice (8 males, 3 females) were studied for weight, in some cases at different points during the lifespan of individual mice, yielding 19 WT and 12 Emx-httKO measured time points. In the case of rotarod, seventeen control mice (13 males, 4 females) and 11 Emx-httKO mice (8 males, 3 females) were studied, in some cases at different points during the lifespan of individual mice, yielding 23 WT and 14 Emx-httKO measured time points. Overall means for control and Emx-httKO mice, irrespective of age, were compared by unpaired two-tailed t-test, while age-related trends were assessed by regression analysis and ANCOVA.
Fig. 12.

The Emx-httKO mice are hyperactive in open field, especially under 1 year of age, as revealed by their increased distance traveled (A), increased progression segment length (B), increased speed (C), increased endurance (D), and decreased number of stops (E). The increase in progression segment length was so great that the Emx-httKO mice actually performed significantly fewer progression segments (F) than did control mice. Seventeen control mice (13 males, 4 females) and 11 Emx-httKO mice (8 males, 3 females) were studied for open field analysis, in some cases at different points during the lifespan of individual mice, yielding 23 WT and 14 Emx-httKO measured time points. Control and Emx-httKO mice means were compared by unpaired two-tailed t-test, while age-related trends were assessed by regression analysis and ANCOVA.
4. Discussion
Our results in Emx-httKO mice show that early embryonic deletion of huntingtin from the developing pallium yields reduced numbers of cortical neurons and reduced cortical volume in adults but no evident abnormalities in cortical lamination. Striatal neurons were also reduced in their abundance, as was striatal volume, but striatal neurons were normal in their neurochemistry. Expression of huntingtin by cortical pyramidal neurons thus appears critical for development of both normal cortical neuron and striatal neuron abundance. For both cortex and striatum, it seems likely that the neuron reduction contributes importantly to the volume reduction we observed in Emx-httKO mice. Neither cortical nor striatal neurons, however, showed accelerated age-related loss in Emx-httKO mice compared to control mice out to nearly 2 years of age. The Emx-httKO mice were hyperactive in open field and slightly defective on rotarod at all ages, but normal in body weight. These findings are discussed in more detail below.
4.1. Effect on cortical and striatal development
Among its functions, huntingtin interacts with microtubules, the dynein/dynactin complex, and kinesin to regulate the microtubule-dependent transport of proteins and organelles in neurons (Caviston et al., 2007; Colin et al., 2008; Gauthier et al., 2004; McGuire et al., 2006; Saudou and Humbert, 2016). Huntingtin appears to also play a role in cell division by means of its presence in the spindle microtubules of the centrosome of dividing cells, and huntingtin knockdown has been found to reduce cell division in vitro (Godin et al., 2010). Genetic manipulations in mice have confirmed that huntingtin is critical for embryogenesis, with homozygous deletion of the mouse homologue of the human HD gene (i.e. Hdh) resulting in apoptosis in the embryonic ectoderm shortly after the onset of gastrulation, yielding embryos that are developmentally retarded and disorganized prior to death between embryonic days 8.5 and 10.5 (Duyao et al., 1995; Nasir et al., 1995; Zeitlin et al., 1995; Dragatsis et al., 1998; Dragatsis and Zeitlin, 2001). An important role of huntingtin in vesicular trafficking of nutrients across extraembryonic membranes appears to be a critical factor in the deleterious effect of huntingtin deletion in embryos (Dragatsis et al., 1998). While huntingtin levels that are 50% of normal (as in hemizygous Hdh deletion) do not obviously hinder development (Ambrose et al., 1994; Duyao et al., 1995; Nasir et al., 1995; Zeitlin et al., 1995; Persichetti et al., 1996), reduction of huntingtin to levels between 25% and 50% of normal during early development results in defective neurogenesis and profound malformations of cerebral cortex and striatum (White et al., 1997).
In the present study, we evaluated the impact of selective early embryonic deletion of huntingtin from cortical pyramidal neurons, using the cre-loxP system to inactivate the mouse huntingtin gene (Hdh) in emx1-expressing cell lineages beginning early in cortical development. Based on the prior findings implicating huntingtin in cell division and neuronal migration (Godin et al., 2010; Tong et al., 2011), we expected reduced cortical neuron numbers and laminar disorganization. We found about a 20% reduction in cortical neuron abundance and about a 10% reduction in cortical volume, but like McKinstry et al. (2014) we saw no evident alteration in laminar organization, in our case using layer-specific markers. It seems likely that the cortical neuron reduction was predominantly in pyramidal neurons, since cortical interneurons, which arise from the medial ganglionic eminence of the subpallium and migrate to the pallium (Parnavelas, 2000), would not have undergone huntingtin deletion, as consistent with our huntingtin immunolabeling results. McKinstry et al. (2014) did not note reduced neuron numbers in cortex at 3–5 weeks of age in Emx-httKO mice, but they assessed neuron abundance by areal density rather than by the 3-dimensional stereological methods we used. Although it is possible that the loss occurs after 5 weeks, our stereological counts indicate a shortfall in cortical neurons already at 3 months of age, which is not evident from neuronal areal packing density assessments alone. Our results are thus consistent with the prediction that cortical pyramidal neuron huntingtin deletion should affect cortical neuron abundance, due to the presumptively impaired neurogenesis caused by the huntingtin deletion from developing pallial neurons. In a prior study involving injection of Hdh−/− embryonic stem cells into mouse blastocysts, we found that Hdh−/− cortical neuroblasts may undergo premature death (Reiner et al., 2001, 2003), suggesting that such death could also contribute to the reduced abundance of cortical neurons in Emx-httKO mice. With regard to the apparent normalcy of cortical lamination, it may be that the previously reported defect in the migration of early born cells (Tong et al., 2011) is transient and self-correcting, or that any cells that migrate incorrectly either die or adopt the phenotype of the layer to which they incorrectly migrate. As cortical neurons appear specified shortly after they are born (Greig et al., 2013; Woodworth et al., 2016) and neuronal re-specification does not seem to occur in other disorders involving cortical laminar disorganization (D’Arcangelo et al., 1995; Ikeda and Terashima, 1997; Rice et al., 1998), it seems more likely that any migration abnormality of early born cells is either transient or resolved through death of ectopic neurons. More recently, Barnat et al. (2017) reported that with huntingtin deletion ~10% of cortical cells born after E15.5 fail to correctly migrate to layers 2–3 of cortex and are mis-localized to the deeper layers in adult mouse cerebral cortex. If such a migration defect occurred during cortical development in our Emx-httKO mice, it was not substantial enough to yield any evident thinning or mis-alignment of layers 2/3.
In the present study, we also observed a 20% reduction in striatal neuron abundance in Emx-httKO mice. The reduced expression of BDNF by cortical neurons, likely to stem at least in part from the loss of the positive effect of huntingtin on BDNF synthesis (Zuccato et al., 2001), may have been a major factor in the reduced striatal neuron abundance. BDNF produced by cortical neurons is transported axonally to the striatum and released (Altar et al., 1997; Zuccato et al., 2001; Gauthier et al., 2004; Zuccato and Cattaneo, 2007), promoting survival of neurons in striatum (Lessmann et al., 2003; Poo, 2001; Zuccato and Cattaneo, 2007). Consistent with a trophic effect of corticostriatal BDNF, cortex-specific embryonic BDNF knock-out or embryonic deletion of the BDNF receptor (TrkB) from striatal neurons results in striatal neuron deficiency (Gorski et al., 2003; Baquet et al., 2004; Strand et al., 2007; Baydyuk et al., 2011; Baydyuk and Xu, 2014). Thus, the reduced corticostriatal BDNF in Emx-httKO mice we observed may have caused diminished striatal neuron survival (Davies, 1996). As BDNF also exerts trophic effects on cortical neurons, its reduction in cortical pyramidal neurons in the mice with cortical deletion of Htt may also have contributed to the reduced neuron numbers in cerebral cortex. Note that since the glutamine repeat expansion in huntingtin causing HD can adversely affect its function (Barnat et al., 2017), our results with embryonic deletion of huntingtin from cortical neurons suggest the mutation in huntingtin could have an adverse developmental impact on neuron abundance and the connectivity of cortex and striatum, which may help explain the typically smaller brain size in HD gene carriers even at young ages much prior to any disease symptoms (Nopoulos et al., 2011; Lee et al., 2012), and could ultimately contribute to HD pathogenesis as well.
4.2. Effect on long-term cortical and striatal neuron survival
Huntingtin appears to exert a neuroprotective effect (Rigamonti et al., 2000; Kalchman et al., 1997; Hackam et al., 2000; Gervais et al., 2002), in part by promoting neuronal survival via a stimulatory effect on production of BDNF (Zuccato et al., 2001). Huntingtin may have this effect on BDNF production by means of an interaction with the transcriptional regulator Sp1, since the BDNF gene is known to possess an Sp1 response element in its proximal promoter region, and Sp1-dependent transcription is diminished by mutant huntingtin (Luthi-Carter et al., 2002). Corticostriatal BDNF exerts an important receptor-mediated, pro-survival effect in neurodegenerative diseases such as Huntington’s disease (Altar et al., 1997; Ivkovic and Ehrlich, 1999; Schuman, 1999; Reiner et al., 2012b). For this reason, it might be expected that deletion of huntingtin from cortical pyramidal neurons would have a long-term adverse effect on age-related cortical and striatal neuron survival. In the present study, however, we found that cortical and striatal neurons showed age-dependent survival in Emx-httKO mice that was no different than in control mice, despite the reduced cortical BDNF production and BDNF signaling. Perhaps after the early loss of a sensitive neuronal population, additional cell losses did not occur in Emx-httKO mice due to a unique sensitivity of these early-lost neurons or the activation of compensatory mechanisms.
Our present results are also surprising in light of prior studies by Dragatsis et al. (2000) using conditional knockout of Hdh in mouse forebrain during late embryonic or perinatal development (Table 2). These studies used conditional deletion of Hdh in two separate lines of mice with cre expression driven by the promoter for the alpha subunit of calcium-dependent calmodulin kinase-2 (Camk2a), either starting after E15 in one line or after postnatal day 5 in the other (Dragatsis and Zeitlin, 2000). Since the forebrain in rodents is still developing during the first postnatal week (which roughly corresponds to the third trimester in humans), the Hdh deletion in both lines of mice occurs during the late stages of forebrain development. Hdh deletion beginning around E15 resulted in an earlier onset of behavioral abnormality (clasping) and forebrain degeneration, while the later deletion of Hdh yielded a milder phenotype. Reactive astrocytosis and axonal degeneration were observed at 3–4 months of age in forebrain in E15 deletion mice, and some of these showed degeneration in striatum, caudolateral cerebrum and amygdala at 8 months (Dragatsis et al., 2000). As the neuronal death in forebrain occurs months after the elimination of forebrain Hdh expression at E15, it might be expected that cortical pyramidal neuron elimination of Hdh at E10.5 as in our study would also adversely affect long-term survival of at least cortical neurons. That we did not find this to be the case suggests that E15 forebrain Hdh deletion is not equivalent to the E10.5 pallium-specific Hdh deletion in our Emx-httKO mice. It is worth noting that the Emx1-cre driven deletion occurs in both neurons and glia (Gorski et al., 2002), whereas Camk2a transgene expression is usually restricted to neurons; perhaps an imbalance in the effects of Hdh deletion on neurons and glia leads to a different phenotype. It would be useful to know the particular fate of cortical pyramidal neurons with E15 forebrain Hdh deletion. It may be that they, like cortical pyramidal neurons in our Emx-httKO mice, do not show progressive degeneration. Alternatively, a compensation for huntingtin deletion (e.g. elevated expression of an alternative trophic factor) may occur with E10.5 deletion that does not with E15 deletion.
Table 2.
comparing major findings of the current study and other studies involving conditional deletion of huntingtin in mice.
| Current study | Dragatsis et al., 2000 | Dragatsis et al., 2000 | Wang et al., 2016 | Wang et al., 2016 | Pla et al., 2013 | Dietrich et al., 2017 | |
|---|---|---|---|---|---|---|---|
| Construct driving KO | emx1-driven cre | CAMK2a-driven cre | CAMK2a-driven cre | Nestin-driven CreER | CAAGG-driven CreER | CAMK2a-driven CreER | CAAGG-driven CreER |
| Selective for Hdh | Cortical pyramidal neurons | Forebrain neurons | Forebrain neurons | Neurons | Global | Forebrain neurons | Global |
| Deletion onset | E10.5 | PND5 | E15 | 2, 4, or 8 months | 2, 4, or 8 months | 2 month | 3, 6, or 9 months |
| Behavioral phenotype | Motor hyperactivity | Clasping by PND60, hypoactivity by 10–12 months of age | Clasping by PND21, hypoactivity by 10–12 months of age | No motor abnormalities up to 6 months post | No motor abnormalities up to 10 months post | No motor deficit up to 6 months post | Progressive rotarod impairment by 1 month post, clasping and tremors by 6 months post |
| Overall neuropathology | Smaller cortex and striatum by 3 months of age | Smaller brain | Smaller brain, temporal lobe degeneration by 4–6 months of age | None up to 3 months post | None up to 3 months post | Defective hippocampal neurogenesis | Progressive brain atrophy and thalamic calcification |
| Cortex neuropathology | Early and nonprogressive reduction in neuron abundance | Gliosis | Neuronal apoptosis at 10 months of age, gliosis | Not examined in detail | Not examined in detail | Not examined in detail | No overt neuronal loss at 14 months post |
| Striatum neuropathology | Early and nonprogressive reduction in neuron abundance | Gliosis | Neuronal apoptosis at 10 months of age, gliosis | Not examined in detail | Not examined in detail | Not examined in detail | Gliosis but no overt neuronal loss at 12 months post |
In this regard, comparison of our results with studies of the effects of adult deletion of huntingtin using inducible Cre/Lox systems are also of interest (Table 2). Wang et al. (2016) recently reported that huntingtin deletion from neurons at 2, 4 or 8 months of age, using Hdhflox/flox mice expressing a nestin promotor-driven CreER treated with tamoxifen, yielded no change in body weight and no motor abnormalities up to 6–7 months after depletion and no evident brain pathology or volume reduction up to 3 months after depletion. Careful morphometric studies such as performed here were, however, not conducted, and so the possibility of neuron loss in cortex or striatum cannot be excluded. By contrast, Pla et al. (2013) found that depletion of huntingtin from hippocampal neurons at 2 months of age, using a similar approach but with a CAMK2a promotor-driven CreER, caused a deficit in the maturation and survival of adult-generated hippocampal neurons, due to a decline in BDNF-mediated trophic support of the new neurons. Whether overall reductions in cortical and hippocampal neuron abundance occurred, or only in newborn hippocampal neurons, was not addressed. In a more extensive study (Dietrich et al., 2017), adult Hdhflox/− mice carrying the tamoxifen-inducible CAAGG-CreER™ allele were injected with tamoxifen to induce global cre-mediated recombination and huntingtin elimination at 3, 6 or 9 months of age. A progressive rotarod impairment was evident by one month after tamoxifen treatment, and severe gait abnormalities, hind limb clasping on tail suspension, and resting tremors were seen by 16 months of age in all mice with deletion. Neuropathological changes included progressive brain weight loss and bilateral thalamic lesions. Neither overt cortical nor overt striatal neuron loss was observed up to over a year after huntingtin deletion. These studies collectively show that adult huntingtin deletion can have adverse effects on brain morphology and function, but they do not resolve if adult deletion of huntingtin from cortical pyramidal neurons adversely affects their survival over a mouse lifetime, or if it is as well tolerated as after early embryonic deletion as in Emx-httKO mice.
4.3. Behavioral abnormalities in Emx-httKO mice
We observed rotarod impairment, open field hyperactivity, but normal weight maintenance in Emx-httKO mice. Of interest, mice with early embryonic deletion of the torsin gene (Dyt1) from the cortical pyramidal neuron lineage similarly show motor impairments and hyperactivity (Yokoi et al., 2008). Mutation of the Dyt1 gene is associated with early onset generalized dystonia, and the authors attributed their phenotype to some unknown cortical abnormality. It seems likely that abnormalities in cortical and corticostriatal development with early embryonic huntingtin deletion from cortical pyramidal neurons are the basis of the motor deficit and hyperactivity we observed. Along these lines, McKinstry et al. (2014) reported that in Emx-httKO mice, created with the same approach as we used, cortical and corticostriatal excitatory synapses form and mature at an accelerated pace through postnatal day 21 (P21). This exuberant synaptic connectivity within cortex was lost by 5 weeks, but retained yet at 5 weeks in the case of corticostriatal synapses. It may be that this exuberant corticostriatal connectivity, if persistent, could account for the hyperactivity we saw in our mice. Given the current understanding of basal ganglia functional organization, preferentially increased input to the striatal go-neurons of the direct pathway would be one way in which exuberant corticostriatal connectivity in cortical Htt-KO mice could yield hyper-activity (Deng et al., 2014). It is also possible that the massive loss of huntingtin from corticostriatal terminals in the Emx-httKO mice accounts for the phenotype. Huntingtin has been reported to associate with synaptic vesicles and facilitate neurotransmitter release in pre-synaptic excitatory synaptic terminals (DiFiglia et al., 1995; Rozas et al., 2011). If the huntingtin depletion preferentially diminished neurotransmitter release from cortical terminals ending on indirect pathway neurons, for example those of the pyramidal tract type corticostriatal neurons (Deng et al., 2015), hyperactivity would be the predicted outcome (Albin et al., 1989).
4.4. Implications for HD therapy
One possible avenue for HD treatment involves gene therapy to prevent production of the mutant protein, typically using either anti-sense DNA oligonucleotides (ASOs) against mutant huntingtin mRNA, or RNA that interferes with huntingtin expression (RNAi) (Crook and Housman, 2013; Ramaswamy and Kordower, 2012). Several studies have shown that direct delivery of RNAi molecules to the forebrain via an AAV vehicle can reduce mutant Htt production and slow phenotypic progression in mouse HD models (DiFiglia et al., 2007; Wang et al., 2005). Nuclease resistant ASOs have been developed that target human Htt message, and shown to be effective following intraventricular infusion in reducing mutant human Htt and improving motor function in mice (Kordasiewicz et al., 2012; Southwell et al., 2014; Stanek et al., 2013). Given the above evidence of an important role of wild-type (WT) huntingtin in neuronal survival, and the evidence that mutant huntingtin is more deleterious on a background of reduced WT huntingtin expression (Auerbach et al., 2001), safety concerns remain for gene therapy approaches that seek to reduce mutant huntingtin at the expense of also reducing WT huntingtin. Several studies have, however, shown that knocking down both mutant and WT huntingtin by 60–75% can achieve the benefit of mutant knockdown without an evident harm of WT allele co-knockdown (Boudreau et al., 2009), and others have shown that knockdown of WT huntingtin alone in striatum is well tolerated in mice (Grondin et al., 2012; McBride et al., 2008). Our present results show that complete deletion of huntingtin from cortical pyramidal neurons and glia beginning early in cortical development is without evident harmful consequences for the long-term postnatal survival of cortical and striatal neurons. Although some early compensation for the loss of huntingtin may be critical to this effect, our findings nonetheless do not support the view that the complete elimination of WT huntingtin from pyramidal neurons is invariably harmful for cortex and striatum. Whether elimination of WT huntingtin from pyramidal neurons is harmful when done in adults, or whether striatal neuron huntingtin depletion at any point in the lifespan is harmful for striatum, needs fuller examination. Moreover, even if neither cortical nor striatal huntingtin depletion are individually harmful, combined cortical and striatal huntingtin depletion might compromise striatum by the double hit of huntingtin insufficiency and corticostriatal BDNF reduction (Reiner et al., 2012b).
Acknowledgments
We are thankful to CO-ED for a CURE organized by Lauren, Daylan, Kristian and Jim Crimm, for generous contribution to our work. We thank Marion Joni, Ting Wong, and Reagan Marienga for their excellent technical assistance with this work. This research was supported by NS-057722 (AR), NS-028721 (AR), NS-098137 (AR), and The Methodist Hospitals Endowed Professorship in Neuroscience (AR), EY-14998 (KRJ).
References
- Aid T, Kazanyseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85:525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- Altar CA, Ning CA, Bliven T, Juhasz M, Conner M, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Ambrose CM, Duyao MP, Barnes G, Bates GP, Lin CS, Srinidhi J, Baxendale S, Hummerich H, Lehrach H, Atherr M, Wasmuth J, Buckler A, Church D, Housman D, Berks M, Micklem G, Durbin R, Dodge A, Read A, Gusella J, MacDonald ME. Evidence against simple inactivation due to an expanded CAG repeat. Somat Cell Mol Genet. 1994;20:27–38. doi: 10.1007/BF02257483. [DOI] [PubMed] [Google Scholar]
- Auerbach W, Hurlbert MS, Hilditch-Maguire P, Wadghiri YZ, Wheeler VC, Cohen SI, Joyner AL, MacDonald ME, Turnbull DH. The HD mutation causes progressive lethal neurological disease in mice expressing reduced levels of huntingtin. Hum Mol Genet. 2001;10:2515–2523. doi: 10.1093/hmg/10.22.2515. [DOI] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnat M, Le Friec J, Benstaali C, Humbert S. Huntingtin-mediated multipolar-bipolar transition of newborn cortical neurons is critical for their postnatal neuronal morphology. Neuron. 2017;93:99–114. doi: 10.1016/j.neuron.2016.11.035. [DOI] [PubMed] [Google Scholar]
- Baydyuk M, Xu B. BDNF signaling and survival of striatal neurons. Front Cell Neurosci. 2014;8:254. doi: 10.3389/fncel.2014.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baydyuk M, Russell T, Liao GY, Zang K, An JJ, Reichardt LF, Xu B. TrkB receptor controls striatal formation by regulating the number of newborn striatal neurons. Proc Natl Acad Sci U S A. 2011;108:1669–1674. doi: 10.1073/pnas.1004744108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch J, Kaeser M, Sadeghi Y, Rouiller EM, Redmond DE, Jr, Brunet JF. Doublecortin-positive cells in the adult primate cerebral cortex and possible role in brain plasticity and development. J Comp Neurol. 2011;519:775–789. doi: 10.1002/cne.22547. [DOI] [PubMed] [Google Scholar]
- Block-Galarza J, Chase KO, Sapp E, Vaughn KT, Vallee RB, DiFiglia M, Aronin N. Fast transport and retrograde movement of huntingtin and HAP1 in axons. Neuroreport. 1997;8:2247–2251. doi: 10.1097/00001756-199707070-00031. [DOI] [PubMed] [Google Scholar]
- Boudreau RL, McBride JL, Martins I, Shen S, Xing Y, Carter BJ, Davidson BL. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol Ther. 2009;17:1053–1063. doi: 10.1038/mt.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu W, Ren H, Deng Y, Del Mar N, Guley N, Moore B, Honig MG, Reiner A. Mild traumatic brain injury produces neuron loss that can be rescued by modulating microglial activation using a CB2 receptor inverse agonist. Front Neurosci. 2016;10:449. doi: 10.3389/fnins.2016.00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviston JP, Ross JL, Antony SM, Tokito M, Holzbaur EL. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc Natl Acad Sci U S A. 2007;104:10045–10050. doi: 10.1073/pnas.0610628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens LE, Weber JJ, Wlodkowski TT, Yu-Taeger L, Michaud M, Calaminus C, Eckert SH, Gaca J, Weiss A, Magg JC, Jansson EK, Eckert GP, Pichler BJ, Bordet T, Pruss RM, Riess O, Nguyen HP. Olesoxime suppresses calpain activation and mutant huntingtin fragmentation in the BACHD rat. Brain. 2015;138:3632–3653. doi: 10.1093/brain/awv290. [DOI] [PubMed] [Google Scholar]
- Colin E, Zala D, Liot G, Rangone H, Borrell-Pagès M, Li XJ, Saudou F, Humbert S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008;27:2124–2134. doi: 10.1038/emboj.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti P, Camnasio S, Mutti C, Valenza M, Thompson M, Fossale E, Zeitlin S, MacDonald ME, Zuccato C, Cattaneo E. Lack of huntingtin promotes neural stem cells differentiation into glial cells while neurons expressing huntingtin with expanded polyglutamine tracts undergo cell death. Neurobiol Dis. 2013;50:160–170. doi: 10.1016/j.nbd.2012.10.015. [DOI] [PubMed] [Google Scholar]
- Crook ZR, Housman DE. Surveying the landscape of Huntington’s disease mechanisms, measurements, and medicines. J Huntington Dis. 2013;2:405–436. doi: 10.3233/JHD-130072. [DOI] [PubMed] [Google Scholar]
- Czech DP, Lee J, Correia J, Loke H, Möller EK, Harley VR. Transient neuroprotection by SRY upregulation in dopamine cells following injury in males. Endocrinology. 2014;155:2602–2612. doi: 10.1210/en.2013-2158. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- Davies AM. The neurotrophic hypothesis: where does it stand? Philos Trans R Soc Lond Ser B Biol Sci. 1996;351:389–394. doi: 10.1098/rstb.1996.0033. [DOI] [PubMed] [Google Scholar]
- Deng YP, Penney JB, Young AB, Albin RL, Anderson KD, Reiner A. Differential loss of striatal projection neurons in Huntington’s disease: a quantitative immunohistochemical study. J Chem Neuroanat. 2004;27:143–164. doi: 10.1016/j.jchemneu.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Deng YP, Wong T, Bricker-Anthony C, Deng B, Reiner A. Loss of corticostriatal and thalamostriatal synaptic terminals precedes striatal projection neuron pathology in heterozygous Q140 Huntington’s disease mice. Neurobiol Dis. 2013;60:89–107. doi: 10.1016/j.nbd.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng YP, Wong T, Wan JY, Reiner A. Differential early loss of thalamostriatal and corticostriatal input to striatal projection neuron types in the Q140 Huntington’s disease knock-in mouse model. Front Syst Neurosci. 2014;8:198. doi: 10.3389/fnsys.2014.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Lanciego J, Goff LK, Coulon P, Salin P, Kachidian P, Lei W, Del Mar N, Reiner A. Differential organization of cortical inputs to striatal projection neurons of the matrix compartment in rats. Front Syst Neurosci. 2015;9:51. doi: 10.3389/fnsys.2015.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich P, Johnson IM, Alli S, Dragatsis I. Elimination of huntingtin in the adult mouse leads to progressive behavioral deficits, bilateral thalamic calcification, and altered brain iron homeostasis. PLoS Genet. 2017;13:e1006846. doi: 10.1371/journal.pgen.1006846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sena-Esteves M, Chase K, Sapp E, Pfister E, Sass M, Yoder J, Reeves P, Pandey RK, Rajeev KG, Manoharan M, Sah DW, Zamore PD, Aronin N. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci U S A. 2007;104:17204–17209. doi: 10.1073/pnas.0708285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragatsis I, Zeitlin S. CaMKIIalpha-Cre transgene expression and recombination patterns in the mouse brain. Genesis. 2000;26:133–135. doi: 10.1002/(sici)1526-968x(200002)26:2<133::aid-gene10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Zeitlin S. A method for the generation of conditional gene repair mutations in mice. Nucleic Acids Res. 2001;29:E10. doi: 10.1093/nar/29.3.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragatsis I, Efstratiadis A, Zeitlin S. Mouse mutant embryos lacking huntingtin are rescued from lethality by wild-type extraembryonic tissues. Development. 1998;125:1529–1539. doi: 10.1242/dev.125.8.1529. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Levine M, Zeitlin S. Inactivation of the mouse Huntington’s disease gene in the brain and testis results in progressive neurodegeneration and sterility. Nat Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Goldowitz D, Del Mar N, Deng YP, Meade CA, Liu L, Sun Z, Dietrich P, Yue J, Reiner A. CAG repeat lengths ≥335 attenuate the phenotype in the R6/2 Huntington’s disease transgenic mouse. Neurobiol Dis. 2009;33:315–330. doi: 10.1016/j.nbd.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drai D, Golani I. SEE: a tool for the visualization and analysis of rodent exploratory behavior. Neurosci Biobehav Rev. 2001;25:409–426. doi: 10.1016/s0149-7634(01)00022-7. [DOI] [PubMed] [Google Scholar]
- Drai D, Benjamini Y, Golani I. Statistical discrimination of natural modes of motion in rat exploratory behavior. J Neurosci Methods. 2000;96:119–131. doi: 10.1016/s0165-0270(99)00194-6. [DOI] [PubMed] [Google Scholar]
- Duarte-Neves J, Gonçalves N, Cunha-Santos J, Simões AT, den Dunnen WF, Hirai H, Kügler S, Cavadas C, Pereira de Almeida L. Neuropeptide Y mitigates neuropathology and motor deficits in mouse models of Machado–Joseph disease. Hum Mol Genet. 2015;24:5451–5463. doi: 10.1093/hmg/ddv271. [DOI] [PubMed] [Google Scholar]
- Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, Ge P, Vonsattel JP, Gusella JF, Joyner AL, MacDonald ME. Inactivation of the mouse HD gene homolog Hdh. Science. 1995;269:407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- Fauser S, Häussler U, Donkels C, Huber S, Nakagawa J, Prinz M, Schulze-Bonhage A, Zentner J, Haas CA. Disorganization of neocortical lamination in focal cortical dysplasia is brain-region dependent: evidence from layer-specific marker expression. Acta Neuropathol Commun. 2013;1:47. doi: 10.1186/2051-5960-1-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueredo-Cardenas G, Anderson KD, Chen Q, Veeman CL, Reiner A. Relative survival of striatal projection neurons and interneurons after intrastriatal injection of quinolinic acid in rats. Exp Neurol. 1994;129:37–56. doi: 10.1006/exnr.1994.1145. [DOI] [PubMed] [Google Scholar]
- Flores-Otero J, Davis RL. Synaptic proteins are tonotopically graded in postnatal and adult type I and type II spiral ganglion neurons. J Comp Neurol. 2011;519:1455–1475. doi: 10.1002/cne.22576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco FR, Chen Q, Lamoreaux WJ, Figueredo-Cardenas G, Jiao Y, Coffman JA, Surmeier DJ, Honig MG, Carlock LR, Reiner A. Cellular localization of huntingtin in striatal and cortical neurons in rats: lack of correlation with neuronal vulnerability in Huntington’s disease. J Neurosci. 1999;19:1189–1202. doi: 10.1523/JNEUROSCI.19-04-01189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier LR, Charrin B, Borrell-Pages M, Dompierre J, Rangone H, Cordelieres F, De Mey J, MacDonald M, Lessmann HS. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Singaraja R, Xanthoudakis S, Gutekunst CA, Leavitt BR, Metzler M, Hackam AS, Tam J, Vaillancourt JP, Houtzager V, Rasper DM, Roy S, Hayden MR, Nicholson DW. Recruitment and activation of caspase-8 by the Huntingtin-interacting protein Hip-1 and a novel partner Hippi. Nat Cell Biol. 2002;4:95–105. doi: 10.1038/ncb735. [DOI] [PubMed] [Google Scholar]
- Godin JD, Colombo K, Molina-Calavita M, Keryer G, Zala D, Charrin BC, Dietrich P, Volvert ML, Guillemot F, Dragatsis I, Bellaiche Y, Saudou F, Nguyen L, Humbert S. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron. 2010;67:392–406. doi: 10.1016/j.neuron.2010.06.027. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qui M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Zeiler SR, Tamowski S, Jones KR. Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J Neurosci. 2003;23:6856–6865. doi: 10.1523/JNEUROSCI.23-17-06856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD. Molecular logic of neocortical projection neuron specification, development and diversity. Nat Rev Neurosci. 2013;14:755–769. doi: 10.1038/nrn3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grondin R, Kaytor MD, Ai Y, Nelson PT, Thakker DR, Heisel J, Weatherspoon MR, Blum JL, Burright EN, Zhang Z, Kaemmerer WF. Six-month partial suppression of Huntingtin is well tolerated in the adult rhesus striatum. Brain. 2012;135:1197–1209. doi: 10.1093/brain/awr333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Zhang H, Ji B, Jiang H, Zhao T, Jiang R, Zhang Z, Tan S, Ahmed A, Gu Y. Vesicle miR-195 derived from endothelial cells inhibits expression of serotonin transporter in vessel smooth muscle cells. Sci Rep. 2017;7:43546. doi: 10.1038/srep43546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guley NG, Rogers JT, Del Mar NA, Deng Y, Islam RM, D’Surney L, Ferrell J, Deng B, Hines-Beard JB, Bu W, Ren H, Elberger AJ, Marchetta JG, Rex TS, Honig MG, Reiner A. A novel closed-head model of mild traumatic brain injury using focal primary overpressure blast to the cranium in mice. J Neurotrauma. 2016;33:403–422. doi: 10.1089/neu.2015.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackam AS, Yassa AS, Singaraja R, Metzler M, Gutekunst CA, Gan L, Warby S, Wellington CL, Vaillancourt J, Chen N, Gervais FG, Raymond L, Nicholson DW, Hayden MR. Huntingtin interacting protein 1 induces apoptosis via novel caspase-dependent death effector domain. J Biol Chem. 2000;275:41299–41308. doi: 10.1074/jbc.M008408200. [DOI] [PubMed] [Google Scholar]
- Harrington AM, Brierly SM, Isaacs N, Hughes PA, Castro J, Blackshaw LA. Sprouting of colonic afferent central terminals and increased spinal mitogen-activated protein kinase expression in a mouse model of chronic visceral hypersensitivity. J Comp Neurol. 2012;520:2241–2255. doi: 10.1002/cne.23042. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Ito R, Kitamura N, Namba K, Hisano Y. Epha4 controls the midline crossing and contralateral axonal projections of inferior olive neurons. J Comp Neurol. 2012;520:1702–1720. doi: 10.1002/cne.23008. [DOI] [PubMed] [Google Scholar]
- Henshall TL, Tucker B, Lumsden AL, Nornes S, Lardelli MT, Richards RI. Selective neuronal requirement for huntingtin in the developing zebrafish. Hum Mol Genet. 2009;18:4830–4842. doi: 10.1093/hmg/ddp455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch SM, Ciliax BJ, Gutekunst CA, Rees HD, Heilman CJ, Yung KKL, Bolam JP, Ince E, Yi H, Levey AI. Electron microscopic analysis of D1 and D2 dopamine receptor proteins in the dorsal striatum and their synaptic relationships with motor corticostriatal afferents. J Neurosci. 1995;15:5222–5237. doi: 10.1523/JNEUROSCI.15-07-05222.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano AA, Brandstätter JH, Morgans CW, Brecha NC. SNAP25 expression in mammalian retinal horizontal cells. J Comp Neurol. 2011;519:972–988. doi: 10.1002/cne.22562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Yu Y, Shimoda Y, Watanabe K, Liu Y. Loss of neural recognition molecule NB-3 delays the normal projection and terminal branching of developing corticospinal tract axons in the mouse. J Comp Neurol. 2012;520:1227–1245. doi: 10.1002/cne.22772. [DOI] [PubMed] [Google Scholar]
- Hu H, Li J, Gagen CS, Gray NW, Zhang Z, Qi Y, Zhang P. Conditional knockout of protein O-mannosyltransferase-2 reveals tissue-specific roles of O-mannosyl glycosylation in brain development. J Comp Neurol. 2011;519:1320–1337. doi: 10.1002/cne.22572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Terashima T. Corticospinal tract neurons are radially malpositioned in the sensory-motor cortex of the shaking rat Kawasaki. J Comp Neurol. 1997;383:370–380. [PubMed] [Google Scholar]
- Ivkovic S, Ehrlich ME. Expression of the striatal DARPP-32/ARPP-21 phenotype in GABAergic neurons requires neurotrophins in vivo and in vitro. J Neurosci. 1999;19:5409–5419. doi: 10.1523/JNEUROSCI.19-13-05409.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafkafi N, Mayo C, Drai D, Golani I, Elmer G. Natural segmentation of the locomotor behavior of drug-induced rats in a photobeam cage. J Neurosci Methods. 2001;109:111–121. doi: 10.1016/s0165-0270(01)00392-2. [DOI] [PubMed] [Google Scholar]
- Kafkafi N, Lipkind D, Benjamini Y, Mayo CL, Elmer GI, Golani I. SEE locomotor behavior test discriminates C57BL/6J and DBA/2J mouse inbred strains across laboratories and protocol conditions. Behav Neurosci. 2003;117:464–477. doi: 10.1037/0735-7044.117.3.464. [DOI] [PubMed] [Google Scholar]
- Kalchman MA, Koide HB, McCutcheon K, Graham RK, Nichol K, Nishiyama K, Kazemi-Esfarjani P, Lynn FC, Wellington C, Metzler M, Goldberg YP, Kanazawa I, Gietz RD, Hayden MR. HIP1: a human homologue of S. cerevisiae sla2p interacts with membrane-associated huntingtin in the brain. Nat Genet. 1997;16:44–53. doi: 10.1038/ng0597-44. [DOI] [PubMed] [Google Scholar]
- Kondo M, Sumino R, Okado H. Combinations of AMPA receptor subunit expression in individual cortical neurons correlate with expression of specific calcium-binding proteins. J Neurosci. 1997;17:1570–1581. doi: 10.1523/JNEUROSCI.17-05-01570.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS, Hung G, Bennett CF, Cleveland D. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74:1031–1044. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Mathews K, Schlaggar B, Perlmutter J, Paulsen JS, Epping E, Burmeister L, Nopoulos P. Measures of growth in children at risk for Huntington disease. Neurology. 2012;79:668–674. doi: 10.1212/WNL.0b013e3182648b65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei W, Deng Y, Liu B, Mu S, Guley NM, Wong T, Reiner A. Confocal laser scanning microscopy and ultrastructural study of VGLUT2 thalamic input to striatal projection neurons in rats. J Comp Neurol. 2013;521:1354–1377. doi: 10.1002/cne.23235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann V, Gottmann K, Malcangio M. Neurotrophin secretion: current facts and future prospects. Prog Neurobiol. 2003;69:341–374. doi: 10.1016/s0301-0082(03)00019-4. [DOI] [PubMed] [Google Scholar]
- Levey AI, Hersch SM, Rye DB, Sunahara RK, Niznik HB, Kitt CA, Price DL, Magnio R, Brann MR, Ciliax BJ. Localization of D1 and D2 dopamine receptors in brain with subtype-specific antibodies. Proc Natl Acad Sci U S A. 1993;90:8861–8865. doi: 10.1073/pnas.90.19.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkind D, Sakov A, Kafkafi N, Elmer GI, Benjamini Y, Golani I. New replicable anxiety-related measures of wall vs center behavior of mice in the open field. J Appl Physiol. 2004;97:347–359. doi: 10.1152/japplphysiol.00148.2004. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan Kooperberg EY, Krainc CD, Young AB, Tapscott SJ, Olson JM. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet. 2002;11:1911–1926. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- McBride JL, Boudreau RL, Harper SQ, Staber PD, Monteys AM, Martins I, Gilmore BL, Burstein H, Peluso RW, Polisky B, Carter BJ, Davidson BL. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci U S A. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire JR, Rong J, Li SH, Li XJ. Interaction of Huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J Biol Chem. 2006;281:3552–3559. doi: 10.1074/jbc.M509806200. [DOI] [PubMed] [Google Scholar]
- McKinstry SU, Karadeniz YB, Worthington AK, Hayrapetyan VY, Ozlu MI, Serafin-Molina K, Risher WC, Ustunkaya T, Dragatsis I, Zeitlin S, Yin HH, Eroglu C. Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J Neurosci. 2014;34:9455–9472. doi: 10.1523/JNEUROSCI.4699-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade CA, Figueredo-Cardenas G, Fusco FR, Nowak TS, Pulsinelli W, Reiner A. Transient global ischemia in rats yields striatal projection neuron and inter-neuron loss resembling that in Huntington’s disease. Exp Neurol. 2000;166:307–323. doi: 10.1006/exnr.2000.7530. [DOI] [PubMed] [Google Scholar]
- Meade CA, Deng YP, Fusco F, Del Mar N, Hersch S, Goldowitz D, Reiner A. Localization of neuronal intranuclear inclusions (NIIs) in the striatum and cortex of the Bates R6/2 transgenic mouse. J Comp Neurol. 2002;449:241–269. doi: 10.1002/cne.10295. [DOI] [PubMed] [Google Scholar]
- Milman P, Woulfe J. Novel variant of neuronal intranuclear rodlet immunoreactive for 40 kDa huntingtin associated protein and ubiquitin in the mouse brain. J Comp Neurol. 2013;521:3832–3846. doi: 10.1002/cne.23381. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Nasir J, Floresco SB, O’Kusky JR, Borowski A, Richman JM, Zeisler J, Phillips AG, Hayden MR. Targeted disruption of the HD gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- Nopoulos PC, Aylward EH, Ross CA, Mills JA, Langbehn DR, Johnson HJ, Magnotta VA, Pierson RK, Beglinger LJ, Nance MA, Barker RA, Paulsen JS PREDICT-HD Investigators and Coordinators of the Huntington Study Group. Smaller intracranial volume in prodromal Huntington’s disease: evidence for abnormal neurodevelopment. Brain. 2011;134:137–142. doi: 10.1093/brain/awq280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet CC, Miller PE, Hemmings HC, Jr, Walaas SI, Greengard P. DARPP-32, a dopamine- and adenosine 3′:5′-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. III Immunocytochemical localization. J Neurosci. 1984;4:111–124. doi: 10.1523/JNEUROSCI.04-01-00111.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özdinler PH, Benn S, Yamamoto TH, Güzel M, Brown RH, Jr, Macklis JD. Corticospinal motor neurons and related subcerebral projection neurons undergo early and specific neurodegeneration in hSOD1G93A transgenic ALS mice. J Neurosci. 2011;31:4166–4177. doi: 10.1523/JNEUROSCI.4184-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnavelas JG. The origin and migration of cortical interneurons: new vistas. Trends Neurosci. 2000;23:126–311. doi: 10.1016/s0166-2236(00)01553-8. [DOI] [PubMed] [Google Scholar]
- Patel SJ, Trivedi GL, Darie CC, Clarkson BD. The possible roles of B-cell novel protein-1 (BCNP1) in cellular signalling pathways and in cancer. J Cell Mol Med. 2017;21:456–466. doi: 10.1111/jcmm.12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persichetti F, Carlee L, Faber PW, McNeil SM, Ambrose CM, Srinidhi J, Anderson MA, Barnes GT, Gusella JF, MacDonald ME. Differential expression of normal and mutant Huntington’s disease gene alleles. Neurobiol Dis. 1996;3:183–190. doi: 10.1006/nbdi.1996.0018. [DOI] [PubMed] [Google Scholar]
- Pla P, Orvoen S, Benstaali C, Dodier S, Gardier AM, David DJ, Humbert S, Saudou F. Huntingtin acts noncell-autonomously on hippocampal neurogenesis and controls anxiety-related behaviors in adult mouse. PLoS One. 2013;8:e73902. doi: 10.1371/journal.pone.0073902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Kordower JH. Gene therapy for Huntington’s disease. Neurobiol Dis. 2012;48:243–254. doi: 10.1016/j.nbd.2011.12.030. [DOI] [PubMed] [Google Scholar]
- Reiner A. The distribution of proenkephalin-derived peptides in the central nervous system of turtle. J Comp Neurol. 1987;259:65–91. doi: 10.1002/cne.902590106. [DOI] [PubMed] [Google Scholar]
- Reiner A, Del Mar N, Meade CA, Yang H, Dragatsis I, Zeitlin S, Goldowitz D. Neurons lacking huntingtin differentially colonize brain and survive in chimeric mice. J Neurosci. 2001;21:7608–7618. doi: 10.1523/JNEUROSCI.21-19-07608.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Dragatsis I, Zeitlin SO, Goldowitz D. Wild-type huntingtin plays a role in brain development and neuronal survival. Mol Neurobiol. 2003;28:259–275. doi: 10.1385/MN:28:3:259. [DOI] [PubMed] [Google Scholar]
- Reiner A, Del Mar N, Deng YP, Meade CA, Sun Z, Goldowitz D. R6/2 neurons with intranuclear inclusions survive for prolonged periods in the brains of chimeric mice. J Comp Neurol. 2007;505:603–737. doi: 10.1002/cne.21515. [DOI] [PubMed] [Google Scholar]
- Reiner A, Hart NM, Lei WL, Deng YP. Corticostriatal projection neurons –dichotomous types and dichotomous functions. Front Neuroanat. 2010;4:142. doi: 10.3389/fnana.2010.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Lafferty DC, Wang HB, Del Mar N, Deng YP. The group 2 metabotropic glutamate receptor agonist LY379268 rescues neuronal, neurochemical and motor abnormalities in R6/2 Huntington’s disease mice. Neurobiol Dis. 2012a;47:75–91. doi: 10.1016/j.nbd.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Wang HB, Del Mar N, Sakata K, Yoo W, Deng YP. BDNF may play a differential role in the protective effect of the mGluR2/3 agonist LY379268 on striatal projection neurons in R6/2 Huntington’s disease mice. Brain Res. 2012b;1473:161–172. doi: 10.1016/j.brainres.2012.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DS, Sheldon M, D’Arcangelo G, Nakajima K, Goldowitz D, Curran T. Disabled-1 acts downstream of Reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development. 1998;125:3719–3729. doi: 10.1242/dev.125.18.3719. [DOI] [PubMed] [Google Scholar]
- Rigamonti D, Bauer JH, De-Fraja C, Conti L, Sipione S, Sciorati C, Clementi E, Hackam A, Hayden MR, Li Y, Cooper JK, Ross CA, Govoni S, Vincenz C, Cattaneo E. Wild-type huntingtin protects from apoptosis upstream of cas-pase-3. J Neurosci. 2000;20:3705–3713. doi: 10.1523/JNEUROSCI.20-10-03705.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas JL, Gomez-Sanchez L, Tomas-Zapico C, Lucas JJ, Fernandez-Chacon R. Increased neurotransmitter release at the neuromuscular junction in a mouse model of polyglutamine disease. J Neurosci. 2011;31:1106–1113. doi: 10.1523/JNEUROSCI.2011-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo D, Clavenzani P, Sorteni C, Bo Minelli L, Botti M, Gazza F, Panu R, Ragionieri L, Chiocchetti R. Neurochemical features of boar lumbosacral dorsal root ganglion neurons and characterization of sensory neurons innervating the urinary bladder trigone. J Comp Neurol. 2013;521:342–366. doi: 10.1002/cne.23177. [DOI] [PubMed] [Google Scholar]
- Saudou F, Humbert S. The biology of huntingtin. Neuron. 2016;89:910–926. doi: 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Schuman EM. Neurotrophin regulation of synaptic transmission. Curr Opin Neurobiol. 1999;9:105–109. doi: 10.1016/s0959-4388(99)80013-0. [DOI] [PubMed] [Google Scholar]
- Simeone A, Acampora D, Gulisano M, Stornaiuolo A, Boncinelli E. Nested expression domains of four homeobox genes in developing rostral brain. Nature. 1992;358:687–690. doi: 10.1038/358687a0. [DOI] [PubMed] [Google Scholar]
- Southwell AL, Skotte NH, Kordasiewicz HB, Østergaard ME, Watt AT, Carroll JB, Doty CN, Villanueva EB, Petoukhov E, Vaid K, Xie Y, Freier SM, Swayze EE, Seth PP, Bennett CF, Hayden MR. In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol Ther. 2014;22:2093–20106. doi: 10.1038/mt.2014.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanek LM, Yang W, Angus S, Sardi PS, Hayden MR, Hung GH, Bennett CF, Cheng SH, Shihabuddin LS. Antisense oligonucleotide-mediated correction of transcriptional dysregulation is correlated with behavioral benefits in the YAC128 mouse model of Huntington’s disease. J Huntington Dis. 2013;2:217–228. doi: 10.3233/JHD-130057. [DOI] [PubMed] [Google Scholar]
- Stillman AA, Krsnik Z, Sun J, Rasin MR, State MW, Sestan N, Louvi A. Developmentally regulated and evolutionarily conserved expression of SLITRK1 in brain circuits implicated in Tourette syndrome. J Comp Neurol. 2009;513:21–37. doi: 10.1002/cne.21919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand AD, Baquet ZC, Aragaki AK, Holmans P, Yang L, Cleren C, Beal MF, Jones L, Kooperberg C, Olson JM, Jones KR. Expression profiling of Huntington’s disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J Neurosci. 2007;27:11758–11768. doi: 10.1523/JNEUROSCI.2461-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Del Mar N, Meade C, Goldowitz D, Reiner A. Differential changes in striatal projection neurons in R6/2 mice transgenic for Huntington’s disease. Neurobiol Dis. 2002;11:369–385. doi: 10.1006/nbdi.2002.0554. [DOI] [PubMed] [Google Scholar]
- Tong Y, Ha T, Liu L, Nishimoto A, Reiner A, Goldowitz D. Spatial and temporal requirements for huntingtin (Htt) in neuronal migration and survival during brain development. J Neurosci. 2011;31:14794–14799. doi: 10.1523/JNEUROSCI.2774-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi A, Prensa L, Cebrián C, Mengual E. Axonal branching patterns of nucleus accumbens neurons in the rat. J Comp Neurol. 2010;518:4649–4673. doi: 10.1002/cne.22484. [DOI] [PubMed] [Google Scholar]
- Van Brederode JF, Helliesen MK, Hendrickson AE. Distribution of the calcium-binding proteins parvalbumin and calbindin-D28k in the sensorimotor cortex of the rat. Neuroscience. 1991;44:157–171. doi: 10.1016/0306-4522(91)90258-p. [DOI] [PubMed] [Google Scholar]
- Wang YL, Liu W, Wada E, Murata M, Wada K, Kanazawa I. Clinico-pathological rescue of a model mouse of Huntington’s disease by siRNA. Neurosci Res. 2005;53:241–249. doi: 10.1016/j.neures.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Wang HB, Laverghetta AV, Foehring RF, Deng YP, Sun Z, Yamamoto K, Lei WL, Jiao Y, Reiner A. Single-cell RT-PCR, in situ hybridization histochemical, and immunohistochemical studies of substance P and enkephalin co-occurrence in striatal projection neurons in rats. J Chem Neuroanat. 2006;31:178–199. doi: 10.1016/j.jchemneu.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Wang G, Liu X, Gaertig MA, Li S, LI XJ. Ablation of huntingtin in adult neurons is nondeleterious but its depletion in young mice causes acute pancreatitis. Proc Natl Acad Sci U S A. 2016;113:3359–3364. doi: 10.1073/pnas.1524575113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JK, Auerbach W, Duyao MP, Vonsattel JP, Gusella JF, Joyner AL, MacDonald ME. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat Genet. 1997;17:404–410. doi: 10.1038/ng1297-404. [DOI] [PubMed] [Google Scholar]
- Wolf HK, Buslei R, Schmidt-Kastner R, Schmidt-Kastner PK, Pietsch T, Wiestler OD, Bluhmke I. NeuN: a useful neuronal marker for diagnostic histopathology. J Histochem Cytochem. 1996;44:1167–1171. doi: 10.1177/44.10.8813082. [DOI] [PubMed] [Google Scholar]
- Woodworth MB, Greig LC, Liu KX, Ippolito GC, Tucker HO, Macklis JD. Ctip1 regulates the balance between specification of distinct projection neuron subtypes in deep cortical layers. Cell Rep. 2016;15:999–1012. doi: 10.1016/j.celrep.2016.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi F, Dang MT, Mitsui S, Li J, Li Y. Motor deficits and hyperactivity in cerebral cortex-specific Dyt1 conditional knockout mice. J Biochem. 2008;143:39–47. doi: 10.1093/jb/mvm191. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Suda Y, Matsuo I, Miyamoto N, Takeda N, Kuratani S, Aizawa S. Emx1 and Emx2 functions in development of dorsal telencephalon. Development. 1997;124:101–111. doi: 10.1242/dev.124.1.101. [DOI] [PubMed] [Google Scholar]
- Zahm DS, Parsley KP, Schwartz ZM, Cheng AY. On lateral septum-like characteristics of outputs from the accumbal hedonic “hotspot” of pecina and berridge with commentary on the transitional nature of basal forebrain “boundaries”. J Comp Neurol. 2013;521:50–68. doi: 10.1002/cne.23157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the HD gene homologue. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog Neurobiol. 2007;81:294–330. doi: 10.1016/j.pneurobio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009;5:311–322. doi: 10.1038/nrneurol.2009.54. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in HD. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]