

Abstract
Objective
The goal of this study was to comprehensively characterize CD4+ CD28+ T cells overexpressing CD11a and KIR genes, and examine the relationship between this T cell subset, genetic risk, and disease activity in lupus.
Methods
The size of the CD4+CD28+KIR+CD11ahi T cell subset was determined by flow cytometry, and total genetic risk for lupus was calculated in 105 female patients using 43 confirmed genetic susceptibility loci. Primary CD4+CD28+KIR+CD11ahi T cells were isolated from lupus patients or were induced from healthy individuals using 5-azacytidine. Genome-wide DNA methylation was analyzed using an array-based approach, and the transcriptome was assessed by RNA sequencing. Transcripts in the CDR3 region were used to assess the TCR repertoire. Chromatin accessibility was determined using ATAC-seq.
Results
A total of 31,019 differentially methylated sites were identified in induced KIR+CD11ahi T cells with >99% being hypomethylated. RNA sequencing revealed a clear pro-inflammatory transcriptional profile. TCR repertoire analysis suggests less clonotype diversity in KIR+CD11ahi compared to autologous KIR-CD11alow T cells. Similarly, primary KIR+CD11ahi T cells isolated from lupus patients were hypomethylated and characterized by a pro-inflammatory chromatin structure. We show that the genetic risk for lupus was significantly higher in African-American compared to European-American lupus patients. The demethylated CD4+CD28+KIR+CD11ahi T cell subset size was a better predictor of disease activity in young (age ≤ 40) European-American patients independent of genetic risk.
Conclusion
CD4+CD28+KIR+CD11ahi T cells are demethylated and characterized by pro-inflammatory epigenetic and transcriptional profiles in lupus. Eliminating these cells or blocking their pro-inflammatory characteristics might present a novel therapeutic approach for lupus.
Keywords: Lupus, T cells, DNA methylation, Genetic risk, Chromatin accessibility
1. Introduction
Systemic lupus erythematosus is a chronic multisystem autoimmune disease characterized by periods of disease flares and remission. Genetic susceptibility might explain at least in part familial aggregation of the disease, and plays a role in determining age of disease onset, severity, and disease heterogeneity [1–3]. Epigenetic aberrancies also play a role in the pathogenesis of lupus [4]. Lupus T cells are characterized by reduced expression and activity of the maintenance DNA methyltransferase DNMT1, resulting in T cell hypomethylation. Several factors might be mechanistically involved in reduced DNMT1 expression and the methylation defect in lupus T cells [5]. These include defective ERK pathway signaling, and increased expression and activity of PP2A, mTOR, and GADD45a [6,7].
CD4+ T cells from lupus patients are characterized by robust demethylation in interferon-regulated genes [8]. In addition, CD4+ T cells in lupus are characterized by overexpression of several methylation sensitive genes, including CD11a, CD70, Perforin, CD40L, and the KIR gene cluster [9]. Normal CD4+ T cells treated with DNA demethylating agents overexpress these same genes similar to lupus T cells, and become autoreactive in vitro [10]. Further, demethylated T cells or T cells with induced defect in DNMT1 expression can cause autoimmunity in animal models [11–13].
Using multi-color flow cytometry, a novel CD4+ CD28+ T cell subset characterized by cell surface CD11ahi and KIR expression was recently identified in patients with active lupus [14]. This T cell subset also expresses other methylation sensitive genes known to be overexpressed on lupus T cells, including CD70 and CD40L. Indeed, treating T cells from normal healthy individuals with DNA demethylating agents results in expansion of this T cell subset [14]. The goal of this study was to characterize this novel T cell subset to reveal the complete repertoire of genes that identify this subset and therefore understand its functional role upon disease pathogenesis. In addition, we aimed to determine if expansion of this T cell subset interacts with genetic risk to predict disease activity in lupus patients.
2. Methods
2.1. Lupus patients
Female participants previously diagnosed with lupus were included in this study. All patients fulfilled the American College of Rheumatology classification criteria for SLE [15]. A Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) score was calculated at the clinical visit concurrently with enrollment in the study and blood sampling draw. Patients who had received cyclophosphamide within the past 6 months or who were on methotrexate were excluded from this study, as cyclophosphamide and methotrexate alter the expression of cell surface molecules and DNA methylation patterns, respectively [16,17]. Patients were recruited from the University of Michigan Health System and Henry Ford Health System. The institutional review boards at the participating institutions approved this study. All participants signed an informed consent prior to enrollment.
2.2. CD4+CD28+KIR+CD11ahi T cell subset size measurement
Whole blood samples were separated by Ficoll-Paque (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) gradient centrifugation to isolate granulocytes for genotyping and peripheral blood mononuclear cells (PBMCs) for the analysis of the CD4+ CD28+ KIR+CD11ahi T cell subset size. The isolated PBMCs were stained with fluorochrome-conjugated antibodies, fixed with Fixation Buffer (BioLegend, San Diego, CA, USA) and subsequently analyzed by flow cytometry using an iCyt Synergy SY3200 Cell Sorter (Sony Biotechnology Inc., San Jose, Ca, USA) and WinList 8.0 software (Verity Software House, Topsham, ME, USA). The T cell subset size was defined as the percentage of CD3+ CD4+ CD28+ cells expressing both KIR+ and CD11ahi markers. The gating strategy for measuring this T cell subset size is shown in Supplementary Fig. 1.
Flow cytometry staining was performed using the following fluorochrome-conjugated antibodies: APC anti-human CD11a (clone: HI111), APC/Cy7 anti-human CD4 (clone: RPA-T4), Pacific Blue anti-human CD3 (clone: UCHT1) and PE/Cy5 anti-human CD28 (clone: CD28.2) (BioLegend, San Diego, Ca, USA); PE anti-human CD158a,h (clone: EB6B), PE anti-human CD158b1/b2,j (clone: GL183) and PE anti-human CD158i (clone: FES172) (Beckman Coulter, Marseille, France); PE anti-human CD158b (clone: CH-L) and PE anti-human NKB1 (clone: DX9) Becton Dickinson, Franklin Lakes, New Jersey, USA); PE anti-human CD158d (clone: 181703) (R&D Systems Inc, Minneapolis, MN, USA).
2.3. Genotyping and calculation of total genetic risk score for lupus
Granulocytes were isolated from whole blood as described above, and DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA). Genotyping was performed on the Infinium ImmunoArray-24 v2.0 BeadChip (Illumina, San Diego, CA, USA) to assess genetic variation at 39 confirmed lupus risk loci, including 34 risk loci covered by the array and 5 loci in high linkage disequilibrium (LD; r2 > 0.9) with surrogate variants included on the array. An additional 4 lupus risk loci were assessed by TaqMan genotyping assays (Life Technologies Corporation, Carlsbad, CA, USA) using the following probes: rs3768792 (IKZF2): reverse 5′-ACCAAAGTCCTACC-CAGAAAACTCT[G/A]AACTTTCCTGTCTTCCATCATAATT, rs2289583 (SCAMP5) forward 5′-TACAGAGGCATTCATGGGGAGGGAG[A/C] ACTGTTTTTTTTTTACAGATGGGTC, rs887369 (CXorf21) forward 5′-CCTTCTGCTCCAGGTACTCATTCAG[A/C]ACTGCATTAGAAATAGGATTCT GCA, rs1734787 (MECP2) forward 5′-TTCTCATGGGTACTTTAAGCAG-TAC[A/C]TCTGTTGGCAAAACACAAATGTTGT. Total genetic risk score for lupus was then calculated in each patient as previously described [18] using the formula: , where n is the number of lupus risk alleles (0, 1 or 2) and OR is the allelic odds ratio for each lupus risk locus. The genetic variants used to calculate the total genetic risk score are shown in Supplementary Table 1.
2.4. In vitro generation of CD4+CD28+KIR+CD11ahi T cells
We recruited 8 healthy women for these studies. PBMCs were isolated using density gradient centrifugation, and then stimulated overnight with PHA. Cells were then cultured with and without 1 μM 5-azacytidine for 72 h. Magnetic bead separation was used to enrich for CD4+ T cells, then multi-color flow cytometry was used to isolate CD4+CD28+KIR+CD11ahi and CD4+CD28+KIR-CD11alow cells.
2.5. Genome-wide DNA methylation and RNA sequencing
DNA and RNA were extracted and subsequently used to assess the DNA methylome and RNA transcriptome, using the Illumina Infinium HumanMethylation450 BeadChip or Infinium MethylationEPIC BeadChip array, and RNA-seq, respectively. RNA sequencing libraries were created using the TruSeq Stranded mRNA library kit (Illumina, San Diego, CA, USA) from purified total RNA and 100bp, single-end mRNA reads were sequenced on HiSeq 2500 sequencers.
Genome-wide DNA methylation analysis was performed using GenomeStudio Methylation module software (Illumina, San Diego, CA, USA). Raw image files were read into the software and determined to meet QC if at least 99% of probes had a detection P-value <0.05. Significantly differentially methylated loci were selected using the following criteria: |ΔBeta| > 10%, detection P-value < 0.05, |’DiffScore’| > 22, and no SNP within 10 bp of the probe target site. Raw RNA-seq transcript reads were pre-processed using Trimmomatic (v0.32) and mapped to human genome GRCh38.p2 using STAR (v2.4.2a) [19,20]. Differential expression analysis was accomplished using the exact test in edgeR (v3.6.8) [21]. We selected transcripts with an FDR <0.05 and |fold-change| > 1.5 as being significantly differentially expressed.
2.6. Assay for transposase-accessible chromatin using sequencing (ATAC-seq)
ATAC-seq was performed to assess genome-wide chromatin accessibility as previously described [22,23]. CD4+ CD28+ KIR+ C-D11ahi and autologous CD4+ CD28+ KIR-CD11alow T cell subsets were isolated from 7 female lupus patients. 50,000 cells from each subset were used for the transposition reaction, and DNA libraries were prepared. Following quality assessment as previously described [22], sequencing was performed on a HiSeq2500 Illumina sequencer (2 samples per lane), generating 50bp pair-end reads. Reads were pre-processed to remove adapter reads, and then aligned to the reference genome. Chromatin accessibility peaks were identified and then differential peaks between CD4+CD28+KIR+CD11ahi and CD4+ CD28+ KIR-CD11alow T cell subsets were identified. Chromatin accessibility peaks with differential accessibility of 1.5-fold or more were annotated and subjected to further analysis. Pre-processing steps and analysis of ATAC-seq data were performed as we previously reported [23].
2.7. Bioinformatics analysis
Functional enrichment analyses were performed using FGNet and GeneTerm Linker Bioconductor packages for the R statistical programming language [24,25], and Database for Annotation, Visualization and Integrated Discovery (DAVID) [26,27]. FGNet was used to create functional gene networks using the results of an initial functional enrichment analysis by GeneTerm Linker, which generated gene-term sets based on ontologies for biological processes, molecular functions, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The functional gene networks were considered significant with an adjusted P value <0.05 and a silhouette width >0, representing a greater functional coherence of genes and gene-term sets. Literature mining was conducted using IRIDESCENT [28–30]. Briefly, the software uses names and synonyms derived from public databases (e.g., OMIM, Entrez, ChemID, Gene Ontology) combined with acronym resolution to identify co-mentioned terms and their frequencies within over 27 million MEDLINE abstracts to develop and score a network of related entities. The distribution of delta-beta values of genes was first plotted and then a cutoff based upon the inflection point was chosen. The inflection point is where the rate of change of the slope reaches zero, suggesting a natural division point between the sites that differ most between conditions. The inflection point was approximately at a delta-beta value of 0.18 and there were 235 genes to the left (i.e., with a delta-beta >0.18). IRIDESCENT then identified the literature relationships between these 235 genes and their relatedness to lupus as a disease.
2.8. TCR repertoire analysis
Sequences containing the CDR3 region of the T cell receptor β chain were extracted from unaligned RNA sequencing reads using MiTCR [31]. Sequences from both the CD4+CD28+KIR+CD11ahi and the CD4+CD28+KIR-CD11alow T cell subsets were extracted, and only sequences with a minimum sequencing quality score of 25 were analyzed. CDR3 sequences were then assigned clonotypes by identifying V and J segments. The TCR repertoire was evaluated based on total number of clonotypes expressed for each sample, and relative expression levels of individual clones were compared between both T cell subsets.
2.9. Statistics analysis
Statistical analysis of the correlation between CD4+CD28+KIR+CD11ahi T cell subset size and SLEDAI score was performed using SAS (v9.4) software (SAS Institute Inc., Cary, NC, USA). European-American and African-American patients were analyzed separately using linear regression and evaluated using a two-tailed t-test. The SLEDAI score was selected as the dependent variable and subset size with and without normalization for genetic risk was the variable of interest. CD4+CD28+KIR+CD11ahi T cell subset size was calculated as a percentage value. The normalized value was calculated as (subset size/genetic risk score)× 100.
3. Results
3.1. In vitro generated CD4+CD28 KIR+CD11ahi T cells are demethylated and characterized by a pro-inflammatory transcriptional profile
We previously demonstrated that CD4+CD28+KIR+CD11ahi T cells express proteins of other methylation sensitive genes known to be hypomethylated in lupus T cells, such as CD70 and CD40L, and that treating CD4+ T cells with the DNA methylation inhibitor 5-azacytidine results in the generation of CD4+ CD28+ KIR+CD11ahi T cells [14]. To assess the extent of DNA demethylation in this T cell subset, we evaluated genome-wide DNA methylation changes in experimentally derived CD4+CD28+KIR CD11ahi T cells compared to autologous CD4+CD28+KIR-CD11alow T cells (n = 8)We identified 31,018 differentially methylated sites across the genome between the two T cell subsets, with 30,736 (99.1%) hypomethylated in CD4+CD28+KIR+CD11ahi T cells. This finding confirms our hypothesis that KIR+CD11ahi expression in CD4+ T cells identifies a demethylated T cell subset, and that this demethylated T cell subset can be distinguished and isolated using flow cytometry. We used literature mining analysis to assess relatedness of the demethylated genes in CD4+CD28+KIR+CD11ahi T cells to lupus. Of 235 genes with a delta-beta of >0.18 used in the analysis, 227 were mentioned at least once in the literature, and 22 of those were mentioned with lupus (P = 0.002, Chi-Square test with Yates correction) (Fig. 1). The relative importance of IL-10 and MMP9 stand out in this analysis, both for the magnitude of their hypomethylation in the KIR+C-D11ahi T cell subset and their high interconnectedness within this network. Both MMP9 and IL10 genes have been previously shown to be hypomethylated in CD4+ T cells from lupus patients [32,33]. Indeed, treating normal CD4+ T cells with 5-azacytidine resulted in IL10 mRNA overexpression [33].
Fig. 1.
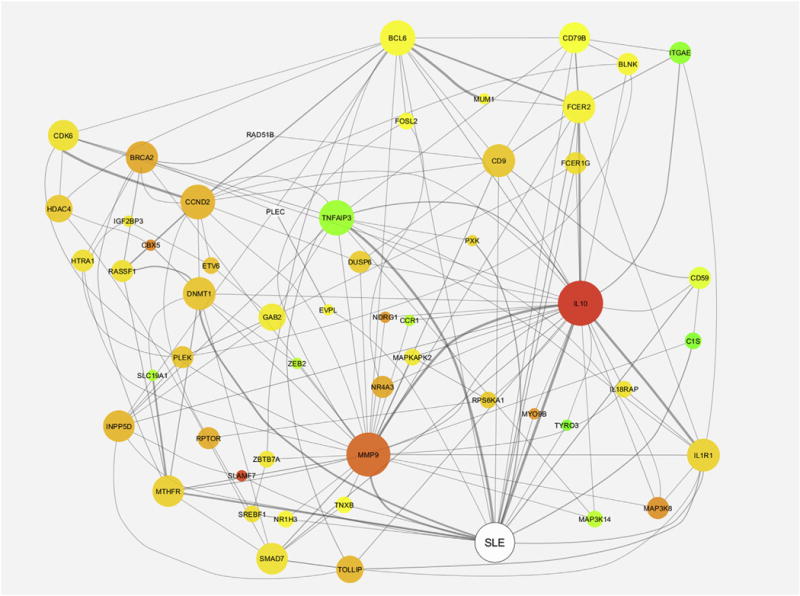
Literature relatedness of lupus and the 227 genes with high delta-beta values. The node color corresponds to each gene’s relative delta-beta value (higher = red, lower = green). The node size corresponds to how interconnected each gene is within this subnetwork (the larger, the more interconnected), and the edge width corresponds to the strength of the relationship between the entities (the thicker the line, the more literature co-mentions found).
To determine if DNA demethylation in CD4+CD28+KIR+C-D11ahi T cells is associated with increased transcriptional activity we performed RNA-seq in a subset of the same cells used for DNA methylation analysis (n = 6). RNA-seq identified 1897 differentially expressed genes in the demethylated T cell subset compared to the autologous KIR-CD11alow T cells isolated from the same tissue culture wells. Consistent with the demethylated nature of this T cell subset, 1620 (85.4%) differentially expressed genes were overexpressed in KIR+CD11ahi T cells. Importantly, these RNA-seq data showed significant overexpression of multiple KIR genes including KIR2DL1, KIR2DL3, KIR2DL4, KIR2DP1, KIR2DS4, KIR3DL1, KIR3DL2, KIR3DL3, KIR3DX1 (74.5, 82.6, 37.4, 67.9, 64.9, 42.3, 7.0, 28.7, and 17.2-fold, respectively), which serves as a validation for the RNA-seq data. Other methylation sensitive genes known to be demethylated and overexpressed in lupus T cells were also overexpressed in this experimentally derived KIR+CD11ahi T cell subset, including CD70 (3.3-fold, P = 2.29E-07), and perforin (3.6-fold, P = 2.50E-08). A number of pro-inflammatory cytokine genes, adhesion molecules, Fc-gamma receptor genes, Toll-like receptor genes, HLA molecules, and metalloproteinases were also significantly overexpressed. Of particular interest, and similar to what is known in lupus T cells, the experimentally derived demethylated KIR+CD11ahi T cell subset demonstrates reduced IL2 mRNA expression (3.3-fold, P 0.0079).
We then identified genes that were hypomethylated and also overexpressed in the KIR+CD11ahi T cell subset. These dysregulated genes are likely mechanistically linked to the DNA methylation defect in this T cell subset, and would be expected to help us understand the pathogenicity of this T cell subset as it relates to the DNA methylation defect in lupus. There were 718 hypomethylated and overexpressed genes in the KIR+CD11ahi compared to autologous KIR-CD11alow T cell subset (Fig. 2A). Bioinformatics analysis of these 718 genes revealed significant enrichment in pro-inflammatory gene ontologies, pathways, and gene meta-groups. The most significant gene ontologies enriched in this subset of genes point to a positive regulation of the immune response, and the most significant pathway was “graft versus host disease” (Table 1). Gene meta-group analysis of genes that are both hypomethylated and overexpressed in the in vitro generated KIR+C-D11ahi compared to autologous KIR-CD11alow T cells are shown in (Fig. 2B).
Fig. 2.
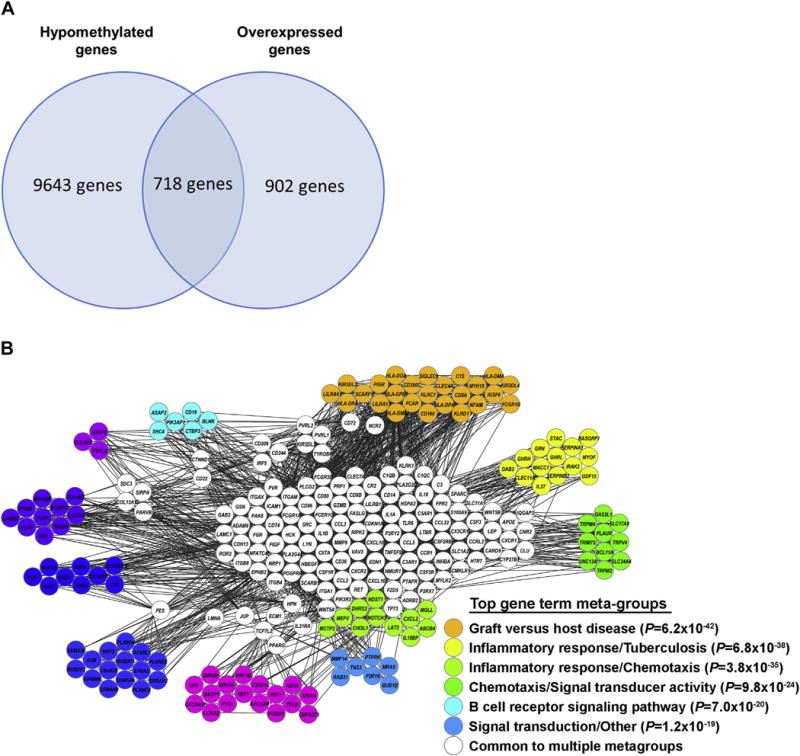
(A) Number of shared and unique hypomethylated and overexpressed genes in CD4+CD28+KIR+CD11ahi T cells. (B) Gene meta-group analysis of genes that are both hypomethylated and overexpressed in the in vitro generated KIR+CD11ahi compared to autologous KIR-CD11alow T cells.
Table 1.
Functional annotation analysis of genes hypomethylated and overexpressed in in vitro induced CD4+CD28+KIR+CD11ahi cells.
| Term | Fold Enrichment | P value (FDR adjusted) |
|---|---|---|
| Gene Ontology Analysis | ||
| GO:0002376~immune system process | 2.91 | 4.07E-24 |
| GO:0006955~immune response | 3.27 | 7.53E-22 |
| GO:0006952~defense response | 3.17 | 1.17E-17 |
| GO:0009611~response to wounding | 3.36 | 1.59E-17 |
| GO:0009605~response to external stimulus | 2.60 | 3.67E-16 |
| GO:0006954~inflammatory response | 3.95 | 4.26E-15 |
| GO:0006950~response to stress | 1.92 | 1.28E-11 |
| GO:0048583~regulation of response to stimulus | 3.02 | 2.83E-11 |
| GO:0050776~regulation of immune response | 4.00 | 3.16E-10 |
| GO:0050778~positive regulation of immune response | 4.92 | 5.19E-10 |
| Pathway Analysis | ||
| hsa05332:Graft-versus-host disease | 8.59 | 2.24E-09 |
| hsa04640:Hematopoietic cell lineage | 4.35 | 1.30E-05 |
| hsa04940:Type I diabetes mellitus | 6.10 | 3.02E-05 |
| hsa04612:Antigen processing and presentation | 4.04 | 1.03E-04 |
| hsa05330:Allograft rejection | 6.02 | 2.14E-04 |
| hsa04650:Natural killer cell mediated cytotoxicity | 2.96 | 7.85E-04 |
| hsa05320:Autoimmune thyroid disease | 4.25 | 0.0039 |
| hsa04514:Cell adhesion molecules (CAMs) | 2.69 | 0.0056 |
| hsa05322:Systemic lupus erythematosus | 2.99 | 0.0065 |
| hsa04672:Intestinal immune network for IgA production | 4.02 | 0.0095 |
3.2. CD4+CD28+KIR+CD11ahi T cell clonality
TCR repertoire analysis, using RNA-seq data, suggests that the KIR+CD11ahi T cell subset is polyclonal, but with less diverse clonotypes compared to autologous KIR-CD11alow T cells (mean number of TCR clones 673±72 versus 837±39; P = 0.001) (Fig. 3).
Fig. 3.
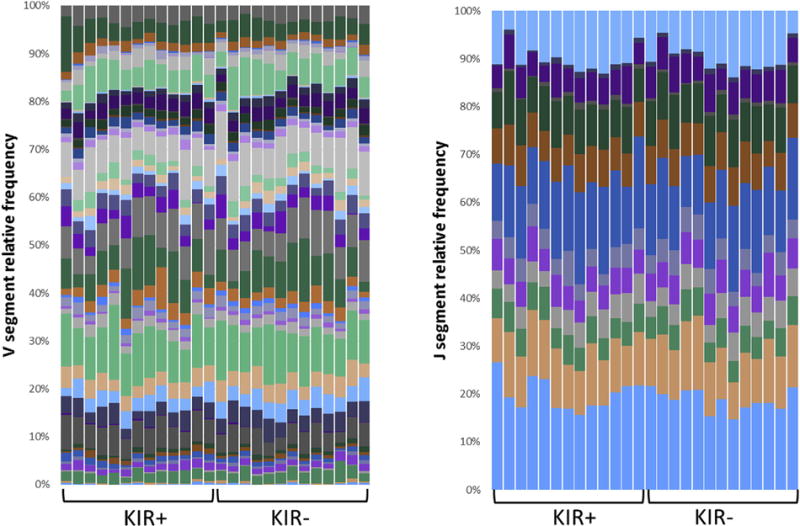
TCR V segment and J segment clonotype diversity in in vitro generated KIR+CD11ahi compared to autologous KIR-CD11alow T cells. RNA-seq data were used to infer and identify T cell clonotypes from CDR3 sequences. Columns represent individual samples, and the colors in the bars within each column indicate different V and J segments.
3.3. CD4+CD28+KIR+CD11ahi T cells isolated from lupus patients are characterized by a pro-inflammatory chromatin accessibility pattern
Primary CD4+CD28+KIR+CD11ahi and autologous CD4+CD28+KIR-CD11alow T cells were isolated from 8 female lupus patients (age range: 28–61 years; mean age: 40 years). Genome-wide DNA methylation analysis showed that CD4+CD28+KIR+C-D11ahi T cells isolated from lupus patients were demethylated compared to autologous CD4+CD28+KIR-CD11alow T cells, consistent with our findings in experimentally demethylated T cells. Indeed, out of 37,255 differentially methylated sites between the two T cell subsets, 34,540 sites (93%) were hypomethylated in CD4+CD28+KIR+CD11ahi T cells. To confirm that demethylation in CD4+CD28+KIR+CD11ahi T cells translates to increased chromatin accessibility, we assessed genome-wide chromatin accessibility in CD4+CD28+KIR+CD11ahi T cells isolated from lupus patients compared to autologous CD4+CD28+KIR-CD11alow T cells, using ATAC-seq. We identified 1685 genetic loci with differential chromatin accessibility between the two T cell subsets. 1233 (73%) and 452 (27%) loci demonstrated increased and decreased chromatin accessibility, respectively, in the CD4+CD28+KIR+CD11ahi compared to the CD4+CD28+KIR-CD11alow T cell subset. These data are consistent with overall increased chromatin accessibility in the demethylated CD4+CD28+KIR+CD11ahi T cell subset. Indeed, all differentially methylated CG sites in CD4+CD28+KIR+CD11ahi T cells, which are included on the methylation array and are within a peak of increased chromatin accessibility, were hypomethylated in CD4+CD28+KIR+CD11ahi compared to CD4+CD28+KIR-CD11alow T cells (105 CG sites). Gene ontology analysis of loci associated with increased chromatin accessibility in CD4+CD28+ KIR+CD11ahi cells revealed an overall pattern of a pro-inflammatory phenotype of this T cell subset, further supporting a pathogenic role for this demethylated T cell subset in lupus (Table 2). Of particular interest, chromatin accessibility analysis revealed positive regulation of integrin-mediated cell-cell adhesion in this T cell subset, as well as positive regulation of cytokine production including IFN-γ, IL-17, and IL-21, but no evidence at the chromatin level to support that these cells can produce Th2 cytokines.
Table 2.
Gene ontology analysis of genes associated with increased chromatin accessibility in CD4+CD28+KIR+CD11ahi compared to the CD4+CD28+KIR-CD11alow T cells isolated from lupus patients (derived from ATAC-seq). Gene ontologies with fold enrichment of at least 3 and false discovery rate of <10% are shown.
| Gene Ontology Term | Genes | Fold Enrichment | P Value | FDR |
|---|---|---|---|---|
| GO:0033634~positive regulation of cell-cell adhesion mediated by integrin | CXCL13, PODXL, CCL5, PIEZO1 | 14.24 | 0.0016 | 2.95 |
| GO:0032740~positive regulation of interleukin-17 production | PRKCQ, NOD2, IL23R, IL15, LY9, IL21, IL2 | 9.59 | 3.75E-05 | 0.07 |
| GO:0042523~positive regulation of tyrosine phosphorylation of Stat5 protein | IL3, CSF2, IL23R, ERBB4, FYN, GHR, IL2 | 7.33 | 2.23E-04 | 0.41 |
| GO:2000484~positive regulation of interleukin-8 secretion | HYAL2, CD58, F2RL1, CD2, F2R | 6.85 | 0.0047 | 8.32 |
| GO:0048008~platelet-derived growth factor receptor signaling pathway | PTPRJ, NRP1, BCR, CSRNP1, ARID5B, TIPARP, PDGFC, RAPGEF1, IQGAP1 | 5.52 | 1.42E-04 | 0.26 |
| GO:0015026~coreceptor activity | RGMA, ACVR2A, CD86, NRP1, CD80, CXCR4, CXCR6, TGFBR3, ITGB1 | 5.34 | 1.85E-04 | 0.29 |
| GO:0045672~positive regulation of osteoclast differentiation | FOS, IL23R, KLF10, IFNG, CCL5, TMEM64 | 5.09 | 0.0053 | 9.32 |
| GO:0005164~tumor necrosis factor receptor binding | TRAF2, TNFSF10, TNFSF15, TNFSF14, FASLG, TNFSF9, EDA, TRAF3 | 4.91 | 9.03E-04 | 1.43 |
| GO:0030032~lamellipodium assembly | ARHGEF4, CDH13, GOLPH3, NCK1, RAC1, CAPZB, PARVB, FGD4 | 4.59 | 0.0014 | 2.51 |
| GO:0032266~phosphatidylinositol-3-phosphate binding | DAB2IP, PARD3, PLEKHA5, ZFYVE26, NCF4, ZFYVE28, DENND1A, PHLDA3 | 4.45 | 0.0017 | 2.64 |
| GO:0042110~T cell activation | CD86, CD80, NEDD4, FYN, NCK1, CD2, CD276, TNFSF14, PPP3CA, WAS, DPP4 | 4.26 | 1.89E-04 | 0.35 |
| GO:0005251~delayed rectifier potassium channel activity | KCNC2, KCNQ3, KCNA2, KCNA4, KCNA3, KCNA6, KCNV2, KCNG1 | 3.95 | 0.0034 | 5.32 |
| GO:0032467~positive regulation of cytokinesis | KIF14, CUL3, KIF3B, CXCR5, SVIL, KIF20B, PRKCE, CDC25B | 3.75 | 0.0047 | 8.33 |
| GO:0007223~Wnt signaling pathway, calcium modulating pathway | GNAO1, GNG2, AGO4, PPP3CA, TNRC6B, PLCB1, TCF7L2, CTNNB1 | 3.65 | 0.0054 | 9.61 |
| GO:0010811~positive regulation of cell-substrate adhesion | LIMS1, HSD17B12, RAC1, CCDC80, NID1, EDIL3, PRKCE, ITGB1 | 3.65 | 0.0054 | 9.61 |
| GO:0045785~positive regulation of cell adhesion | PTPRJ, PTGER4, TNFRSF18, ITGA2, CCL5, NRG1, ADAM8, ADGRG1, TNFSF18 | 3.41 | 0.0043 | 7.60 |
| GO:0014068~positive regulation of phosphatidylinositol 3-kinase signaling | ERBB4, NEDD4, FYN, F2RL1, PDGFC, SEMA4D, CCL5, PRR5L, NRG1, TGFB2, F2R | 3.01 | 0.0032 | 5.72 |
3.4. CD4+CD28+KIR+CD11ahi T cell subset size, genetic risk, and disease activity in lupus patients
DNA methylation defects in T cells play an important role in the pathogenesis of lupus, and T cell DNA demethylation levels correlate with disease activity in lupus patients. In addition, multiple genetic risk loci for lupus have been identified. Therefore, we hypothesized that expansion of the demethylated CD4+CD28+KIR+CD11ahi T cell subset interacts with total genetic risk for lupus to determine disease activity in lupus patients. We determined the size of the demethylated CD4+CD28+KIR+CD11ahi T cell subset by flow cytometry in 105 lupus patients including 68 European-American patients and 37 African-American patients (Supplementary Table 2).
Genotyping across 43 confirmed lupus susceptibility loci was performed to calculate total genetic risk. Genetic risk score was significantly higher in African-American compared to European-American lupus patients in our cohort (7.81±0.12 versus 7.31±0.12 (mean ± SEM), P = 0.0079) (Fig. 4A). As expected, African-American patients had a higher mean SLEDAI score compared to European-American patients (4.19±0.86 versus 1.94±0.29 (mean ± SEM), P = 0.003).
Fig. 4.
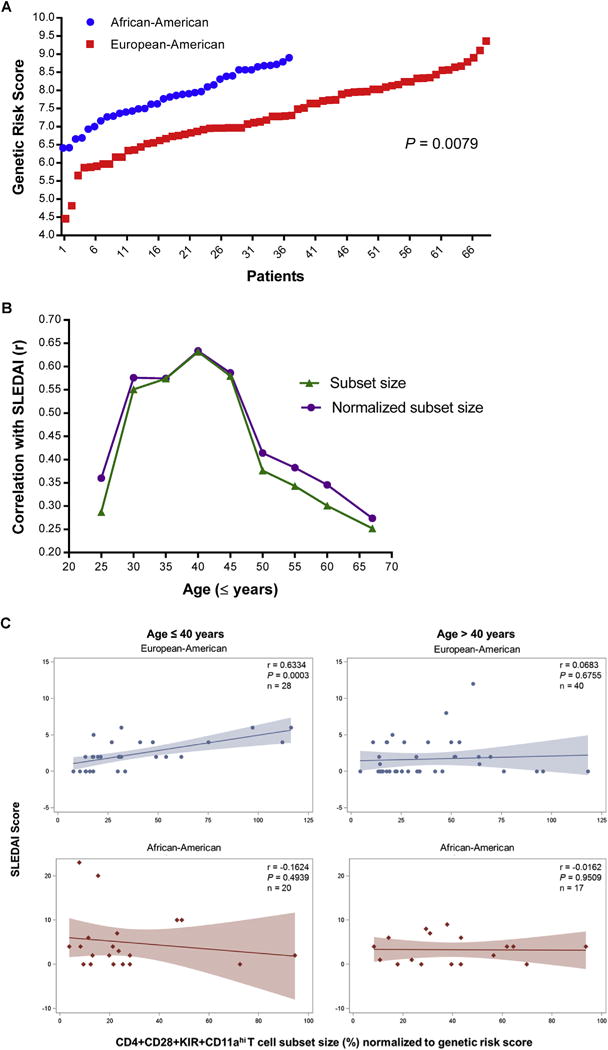
(A) Genetic risk score for lupus was significantly higher in African-American compared to European-American lupus patients. (B) Correlation of SLEDAI score with CD4+CD28+KIR+CD11ahi T cell subset size and CD4+CD28+KIR+CD11ahi T cell subset size normalized by genetic risk score in European-American lupus patients stratified by age (n = 68). (C) Correlation of SLEDAI score with CD4+CD28+KIR+CD11ahi T cell subset size normalized by genetic risk score in European-American and African-American lupus patients (age ≤40 or >40 years) using linear regression. r: correlation coefficient; P: two-tailed t-test P-value. n = number of patients included in regression analysis.
The CD4+CD28+KIR+CD11ahi T cell subset size correlated with disease activity in European-American (r = 0.25, P = 0.038), but not in African-American lupus patients (r = − 0.18, P = 0.29). Linear regression models suggest that this correlation is largely independent of genetic risk, as the subset size was only a slightly better predictor of disease activity when normalized for total genetic risk in each individual (European-American: r = 0.27, P = 0.024; African-American: r = −0.15, P = 0.37). To determine if age influenced the relationship between SLEDAI score and subset size, we assessed age-dependent correlation between the subset size and SLEDAI scores, and show that the CD4+CD28+KIR+CD11ahi T cell subset size, whether normalized to genetic risk score or not, is a better predictor for disease activity in European-American lupus patients ≤40 years of age (Fig. 4B). Indeed, we find no correlation between disease activity and the subset size in patients >40 years of age (Fig. 4C, Supplementary Fig. 2). No correlation between the subset size and disease activity was detected in any age group in African-American patients (Fig. 4C and Supplementary Fig. 2).
4. Discussion
The role of CD4+ T cell DNA hypomethylation in the pathogenesis of lupus has been thoroughly investigated [4]. We described a demethylated T cell subset characterized by KIR and CD11a overexpression in lupus patients. The KIR locus is comprised of 15 KIR genes, which are most commonly expressed on natural killer cells. However, because regulation of the KIR locus is methylation sensitive, KIR genes have previously been shown to be expressed in the context of lupus [14,34]. Furthermore, KIR expression on CD4+ T cells has been shown to play an active role in T cell cytotoxicity, demonstrated by increased macrophage killing and interferon gamma production by KIR+ T cells, which are both hallmarks of lupus pathogenesis [34]. In addition, expression of KIR on CD8+ T cells, but not CD4+ T cells has been shown to be correlated with age in healthy individuals. This increased expression of KIR genes on T cells, coupled with the decreased global DNA methylation, could be relevant to the increasing risk of developing lupus with age.
We show that the subset size of CD4+CD28+KIR+CD11ahi T cells is associated with disease activity in young (age ≤40) European-American, but not African-American women with lupus. We found no correlation between disease activity and the subset size in patients >40 years of age in either ethnic group. This could be due to the known effect of estrogens on inducing oxidative stress [35], which has been linked to DNA demethylation in CD4+ T cells [36]. Indeed, CD4+ T cells exposed to oxidative stress can induce a lupus-like disease in animal models [37]. It is not clear why we did not observe an association between the size of the CD4+CD28+KIR+CD11ahi T cell subset and disease activity in African-American patients. Of interest, African-American lupus patients had a significantly higher genetic risk for lupus compared to European-American patients (Fig. 4A), which is consistent with a higher prevalence and generally more severe disease in African-Americans. This is also consistent with data derived from population based genetic analysis in African and European populations [38]. We should note that the variants used in genetic risk calculation were detected or confirmed in a large European-derived cohort of 7219 lupus patients and 15,991 healthy controls [39]. As of the time of conducting this study, no GWAS data had been published in African-American lupus patients. Ethnicity-specific epigenetic variation could also play a role in the differences between African-American and European-American lupus patients. It has previously been documented that there are significant differences in DNA methylation between African-American and European-American lupus patients in naïve CD4+ T cells [40]. As the CD4+CD28+KIR+CD11ahi T cell subset is characterized by hypomethylation of pro-inflammatory genes, and DNA methylation has been shown to play a role in regulating the KIR locus, this variation could lead to differential KIR expression patterns between ethnicities.
We comprehensively characterized the CD4+CD28+KIR+C-D11ahi T cell subset. When this cell subset is induced in vitro via 5-azacytidine treatment, we observed global hypomethylation compared to autologous CD4+CD28+KIR-CD11alow T cells from the same individuals. Moreover, demethylation in CD4+CD28+KIR+CD11ahi T cells coupled with increased chromatin accessibility leads to increase expression of pro-inflammatory genes that could play a mechanistic role in lupus flares.
5. Conclusion
Our findings indicate that the novel CD4+CD28+KIR+CD11ahi T cell subset is demethylated and characterized by pro-inflammatory chromatin accessibility and transcriptional profiles in lupus patients. We identified a number of pathways and specific genes overexpressed in this T cell subset. These data suggest that eliminating CD4+CD28+KIR+CD11ahi T cells or blocking some of the specific pro-inflammatory characteristics we identified might provide novel therapeutic options for lupus.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health grants number U19AI110502 and R01AI097134.
Appendix A. Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.jaut.2017.09.011.
Footnotes
Conflict of interest
None of the authors has any financial conflict of interest to disclose.
Competing interest
None declared.
References
- 1.Chen L, Morris DL, Vyse TJ. Genetic advances in systemic lupus erythematosus: an update. Curr Opin Rheumatol. 2017;29:423–433. doi: 10.1097/BOR.0000000000000411. [DOI] [PubMed] [Google Scholar]
- 2.Webb R, Kelly JA, Somers EC, Hughes T, Kaufman KM, Sanchez E, et al. Early disease onset is predicted by a higher genetic risk for lupus and is associated with a more severe phenotype in lupus patients. Ann Rheum Dis. 2011;70:151–156. doi: 10.1136/ard.2010.141697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanchez E, Nadig A, Richardson BC, Freedman BI, Kaufman KM, Kelly JA, et al. Phenotypic associations of genetic susceptibility loci in systemic lupus erythematosus. Ann Rheum Dis. 2011;70:1752–1757. doi: 10.1136/ard.2011.154104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teruel M, Sawalha AH. Epigenetic variability in systemic lupus erythema-tosus: what we learned from genome-wide DNA methylation studies. Curr Rheumatol Rep. 2017;19:32. doi: 10.1007/s11926-017-0657-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deng C, Kaplan MJ, Yang J, Ray D, Zhang Z, McCune WJ, et al. Decreased Ras-mitogen-activated protein kinase signaling may cause DNA hypomethylation in T lymphocytes from lupus patients. Arthritis Rheum. 2001;44:397–407. doi: 10.1002/1529-0131(200102)44:2<397::AID-ANR59>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 6.Long H, Yin H, Wang L, Gershwin ME, Lu Q. The critical role of epigenetics in systemic lupus erythematosus and autoimmunity. J Autoimmun. 2016;74:118–138. doi: 10.1016/j.jaut.2016.06.020. [DOI] [PubMed] [Google Scholar]
- 7.Oaks Z, Perl A. Metabolic control of the epigenome in systemic Lupus erythematosus. Autoimmunity. 2014;47:256–264. doi: 10.3109/08916934.2013.834495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coit P, Jeffries M, Altorok N, Dozmorov MG, Koelsch KA, Wren JD, et al. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J Autoimmun. 2013;43:78–84. doi: 10.1016/j.jaut.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel DR, Richardson BC. Dissecting complex epigenetic alterations in human lupus. Arthritis Res Ther. 2013;15:201. doi: 10.1186/ar4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richardson BC, Strahler JR, Pivirotto TS, Quddus J, Bayliss GE, Gross LA, et al. Phenotypic and functional similarities between 5-azacytidine-treated T cells and a T cell subset in patients with active systemic lupus erythematosus. Arthritis Rheum. 1992;35:647–662. doi: 10.1002/art.1780350608. [DOI] [PubMed] [Google Scholar]
- 11.Quddus J, Johnson KJ, Gavalchin J, Amento EP, Chrisp CE, Yung RL, et al. Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors, 5-azacytidine or procainamide, is sufficient to cause a lupus-like disease in syngeneic mice. J Clin Investig. 1993;92:38–53. doi: 10.1172/JCI116576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yung RL, Quddus J, Chrisp CE, Johnson KJ, Richardson BC. Mechanism of drug-induced lupus. I. Cloned Th2 cells modified with DNA methylation inhibitors in vitro cause autoimmunity in vivo. J Immunol. 1995;154:3025–3035. [PubMed] [Google Scholar]
- 13.Sawalha AH, Jeffries M, Webb R, Lu Q, Gorelik G, Ray D, et al. Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun. 2008;9:368–378. doi: 10.1038/gene.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strickland FM, Patel D, Khanna D, Somers E, Robida AM, Pihalja M, et al. Characterisation of an epigenetically altered CD4(+) CD28(+) Kir(+) T cell subset in autoimmune rheumatic diseases by multiparameter flow cytometry. Lupus Sci Med. 2016;3:e000147. doi: 10.1136/lupus-2016-000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 16.McCune WJ, Golbus J, Zeldes W, Bohlke P, Dunne R, Fox DA. Clinical and immunologic effects of monthly administration of intravenous cyclophosphamide in severe systemic lupus erythematosus. N Engl J Med. 1988;318:1423–1431. doi: 10.1056/NEJM198806023182203. [DOI] [PubMed] [Google Scholar]
- 17.Kim YI, Logan JW, Mason JB, Roubenoff R. DNA hypomethylation in inflammatory arthritis: reversal with methotrexate. J Lab Clin Med. 1996;128:165–172. doi: 10.1016/s0022-2143(96)90008-6. [DOI] [PubMed] [Google Scholar]
- 18.Sawalha AH, Wang L, Nadig A, Somers EC, McCune WJ, Lupus C Michigan, et al. Sex-specific differences in the relationship between genetic susceptibility, T cell DNA demethylation and lupus flare severity. J Autoimmun. 2012;38:J216–J222. doi: 10.1016/j.jaut.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsou PS, Wren JD, Amin MA, Schiopu E, Fox DA, Khanna D, et al. Histone deacetylase 5 is overexpressed in scleroderma endothelial cells and impairs angiogenesis via repression of proangiogenic factors. Arthritis Rheumatol. 2016;68:2975–2985. doi: 10.1002/art.39828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aibar S, Fontanillo C, Droste C, De Las Rivas J. Functional Gene Networks: R/Bioc package to generate and analyse gene networks derived from functional enrichment and clustering. Bioinformatics. 2015;31:1686–1688. doi: 10.1093/bioinformatics/btu864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fontanillo C, Nogales-Cadenas R, Pascual-Montano A, De las Rivas J. Functional analysis beyond enrichment: non-redundant reciprocal linkage of genes and biological terms. PLoS One. 2011;6:e24289. doi: 10.1371/journal.pone.0024289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 27.da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wren JD, Bekeredjian R, Stewart JA, Shohet RV, Garner HR. Knowledge discovery by automated identification and ranking of implicit relationships. Bioinformatics. 2004;20:389–398. doi: 10.1093/bioinformatics/btg421. [DOI] [PubMed] [Google Scholar]
- 29.Wren JD, Garner HR. Shared relationship analysis: ranking set cohesion and commonalities within a literature-derived relationship network. Bioinformatics. 2004;20:191–198. doi: 10.1093/bioinformatics/btg390. [DOI] [PubMed] [Google Scholar]
- 30.Wren JD. Extending the mutual information measure to rank inferred literature relationships. BMC Bioinf. 2004;5:145. doi: 10.1186/1471-2105-5-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolotin DA, Shugay M, Mamedov IZ, Putintseva EV, Turchaninova MA, Zvyagin IV, et al. MiTCR: software for T-cell receptor sequencing data analysis. Nat Methods. 2013;10:813–814. doi: 10.1038/nmeth.2555. [DOI] [PubMed] [Google Scholar]
- 32.Jeffries MA, Dozmorov M, Tang Y, Merrill JT, Wren JD, Sawalha AH. Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics Off J DNA Methylation Soc. 2011;6:593–601. doi: 10.4161/epi.6.5.15374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao M, Tang J, Gao F, Wu X, Liang Y, Yin H, et al. Hypomethylation of IL10 and IL13 promoters in CD4+ T cells of patients with systemic lupus erythematosus. J Biomed Biotechnol. 2010;2010:931018. doi: 10.1155/2010/931018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basu D, Liu Y, Wu A, Yarlagadda S, Gorelik GJ, Kaplan MJ, et al. Stimulatory and inhibitory killer Ig-like receptor molecules are expressed and functional on lupus T cells. J Immunol. 2009;183:3481–3487. doi: 10.4049/jimmunol.0900034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kypreos KE, Zafirovic S, Petropoulou PI, Bjelogrlic P, Resanovic I, Traish A, et al. Regulation of endothelial nitric oxide synthase and high-density lipoprotein quality by estradiol in cardiovascular pathology. J Cardiovasc Pharmacol Ther. 2014;19:256–268. doi: 10.1177/1074248413513499. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Gorelik G, Strickland FM, Richardson BC. Oxidative stress, T cell DNA methylation, and lupus. Arthritis & Rheumatol. 2014;66:1574–1582. doi: 10.1002/art.38427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strickland FM, Li Y, Johnson K, Sun Z, Richardson BC. CD4(+) T cells epigenetically modified by oxidative stress cause lupus-like autoimmunity in mice. J Autoimmun. 2015;62:75–80. doi: 10.1016/j.jaut.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris DL, Sheng Y, Zhang Y, Wang YF, Zhu Z, Tombleson P, et al. Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat Genet. 2016;48:940–946. doi: 10.1038/ng.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bentham J, Morris DL, Cunninghame Graham DS, Pinder CL, Tombleson P, Behrens TW, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet. 2015;47:1457–1464. doi: 10.1038/ng.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coit P, Ognenovski M, Gensterblum E, Maksimowicz-McKinnon K, Wren JD, Sawalha AH. Ethnicity-specific epigenetic variation in naive CD4+T cells and the susceptibility to autoimmunity. Epigenetics Chromatin. 2015;8:49. doi: 10.1186/s13072-015-0037-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.