

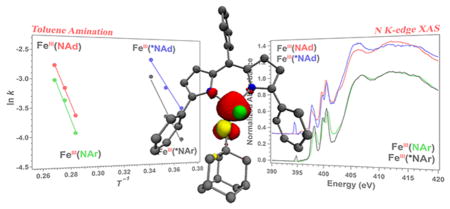
Abstract
Reduction of previously reported iminyl radical (ArL)FeCl(•N(C6H4-p-tBu)) (2) with potassium graphite furnished the corresponding high-spin (S = 5/2) imido (ArL)Fe(N(C6H4-p-tBu)) (3) (ArL = 5-mesityl-1,9-(2,4,6-Ph3C6H2)dipyrrin). Oxidation of the three-coordinate imido (ArL)Fe(NAd) (5) with chlorotriphenylmethane afforded (ArL)FeCl(•NAd) (6) with concomitant expulsion of Ph3C-(C6H5)CPh2. The respective aryl/alkyl imido/iminyl pairs (3, 2; 5, 6) have been characterized by EPR, zero-field 57Fe Mössbauer, magnetometry, single crystal X-ray diffraction, XAS, and EXAFS for 6. The high-spin (S = 5/2) imidos exhibit characteristically short Fe–N bonds (3: 1.708(4) Å; 5: 1.674(11) Å), whereas the corresponding iminyls exhibit elongated Fe–N bonds (2: 1.768(2) Å; 6: 1.761(6) Å). Comparison of the pre-edge absorption feature (1s → 3d) in the X-ray absorption spectra reveals that the four imido/iminyl complexes share a common iron oxidation level consistent with a ferric formulation (3: 7111.5 eV, 2: 7111.5 eV; 5: 7112.2 eV, 6: 7112.4 eV) as compared with a ferrous amine adduct (ArL)FeCl(NH2Ad) (7: 7110.3 eV). N K-edge X-ray absorption spectra reveal a common low-energy absorption present only for the iminyl species 2 (394.5 eV) and 6 (394.8 eV) that was assigned as a N 1s promotion into a N-localized, singly occupied iminyl orbital. Kinetic analysis of the reaction between the respective iron imido and iminyl complexes with toluene yielded the following activation parameters: Ea (kcal/mol) 3: 12.1, 2: 9.2; 5: 11.5, 6: 7.1. The attenuation of the Fe–N bond interaction on oxidation from an imido to an iminyl complex leads to a reduced enthalpic barrier [Δ(ΔH‡) ≈ 5 kcal/mol]; the alkyl iminyl 6 has a reduced enthalpic barrier (1.84 kcal/mol) as compared with the aryl iminyl 2 (3.84 kcal/mol), consistent with iminyl radical delocalization into the aryl substituent in 2 as compared with 6.
Graphical Abstract
1. INTRODUCTION
Late transition metal complexes featuring metal–ligand multiple bonds are investigated intensely to probe their efficacy for functional group transfer to organic substrates.1–15 Within this class of coordination complexes, iron complexes to which the nitrene functional group (R–N) is bound have now been characterized spanning a large range of oxidation states and electronic structures (FeII S = 0;16 FeIII S = 1/2, S = 3/2,17–23 S = 5/2;24 FeIII(•NR) S = 2;25,26 FeIV S = 0, S = 1;27–31 FeV S = 1/232,33). Of these examples, only the high-spin, ferric imido24 and iminyl complexes25,26 have been demonstrated to be competent for nitrene group transfer into aliphatic or olefinic substrates. The unique electronic configuration of the two amination agents warrants further investigation into how the electronic structure of the reactive intermediate impacts functional group transfer from complexes bearing metal–ligand multiple bonds.
Key to the success of the two examples for which C–H bond amination is observed is the utilization of a weak-field ancillary ligand, ArL, where ArL = 5-mesityl-1,9-(2,4,6-Ph3C6H2)-dipyrrin.26 We previously have shown that reaction of the ferrous chloride precursor (ArL)FeCl (1) with aryl azide N3(C6H4-p-tBu) results in formation of the ferric iminyl complex (ArL)FeCl(•N(C6H4-p-tBu)) (2) (Scheme 1), where evidence of the iminyl radical can be seen in disruption of the iminyl aryl substituent.25,26 For example, reaction of 1 with N3(C6H5) results in bimolecular, radical coupling of the phenyl iminyl ligands to afford the diferric product [(ArL)FeCl]2(μ-N(Ph)(C6H5)N), allowing for unambiguous spectroscopic assignment of the ferric state. A room temperature solution magnetic moment determination of 2 (5.3(1) μB) in conjunction with agreement of the spectroscopic Mössbauer parameters between 2 and the bimolecularly coupled product suggest the quintet 2 is comprised of a high-spin ferric center, antiferromagnetically coupled to an iminyl radical [Stotal = (Sferric = 5/2) − (Siminyl = 1/2) = 2]. Although this electronic structure had been suggested previously,34–37 2 constitutes the first structurally and spectroscopically authenticated example of a transition metal stabilized iminyl complex. The isolated iminyl 2 was competent for both intermolecular amination of toluene and azirdination of styrene in stoichiometric reactions.26 Employing the less sterically demanding 1,9-adamantyl substituted dipyrrin ligand (AdL) allowed for the nitrene-transfer reaction to be run catalytically using N3Ad as an oxidant (Scheme 2).26,38,39 Although no reactive intermediate could be isolated or observed during catalysis, the similarity between the kinetic isotope effects observed for both the stoichiometric and catalytic amination processes suggested a common ferric iminyl reactive intermediate was operative.
Scheme 1.

Synthesis of Imido (3, 5) and Iminyl (2, 6) Species
Scheme 2.

Inter- and Intramolecular Amination Reactions
We have previously demonstrated that oxidative group transfer with both alkyl (N3Ad) and aryl (N3Mes) azides to the monovalent iron complex (ArL)Fe (4) resulted in isolable ferric imido products (ArL)Fe(NAd) (5) (Scheme 1) and (ArL)Fe(NMes).24 The imido complexes were characterized and described as possessing metal-ligand multiple bond character with structural features similar to those reported for ferric imidos.17–23 Unlike those literature precedents, the imido complexes were spectroscopically determined to be high-spin (S = 5/2). Akin to their ferric iminyl congeners,25,26 these imido species were shown to be competent for both H-atom abstraction and nitrene transfer to C–H bond substrates.
Given the common core electronic structure (Sferric = 5/2) between the imido and iminyl molecular species previously characterized,24–26 we sought to address the following questions through a comparative examination of the two distinct molecular oxidation levels: (1) what are the redox stabilities of iminyl 2 and imido 5? Are their respective imido and iminyl congeners electrochemically or chemically accessible? (2) We proposed that the aryl-substituent in iminyl 2 is critical to stabilization of the iminyl functionality via delocalization of the radical throughout the aromatic ring.26 If chemical oxidation of the alkyl imido 5 is possible, would the oxidation of the complex be iron-borne resulting in an FeIV oxidation state akin to the hydroxylating ferryl compound I in cytochrome P450?40,41 Or would a similar ferric-stabilized iminyl radical [e.g., FeIII(•NR)] be observed without the ability to delocalize the radical, as proposed for the intra- and intermolecular amination catalysts? Furthermore, can the two redox resonance forms be spectroscopically distinguished [FeIV(NR) ↔ FeIII(•NR)]? (3) Lastly, if the redox isomers are accessible for both alkyl and aryl imido/iminyl moieties, how is the change in molecular oxidation level manifested in C–H bond amination efficacy? Herein we report our findings concerning the interconversion of the imido/iminyl redox forms, describe their comparative metal oxidation states using X-ray absorption spectroscopy, and provide a kinetic assessment of their reactivity in intermolecular amination.
2. RESULTS AND DISCUSSION
2.1. Redox Stability of the Imido/Iminyl Units
Cyclic voltammetry (CV) was performed on iminyl 2 and imido 5 to assess their redox stability (Figure 1). CV examination of iminyl 2 in dichloromethane shows a quasi-reversible reduction event at −1.10 V versus [Cp2Fe]+/0 (Figure 1). The anticipated imido complex may well dissociate the chloride ligand, akin to the three-coordinate (ArL)Fe(NMes).24 The quasi-reversible couple suggests a chemical process indeed occurs following reduction. The electrochemical behavior of imido 5 was also investigated, and its cyclic voltammogram reveals a quasi-reversible oxidation event at −374 mV versus [Cp2Fe]+/0 (Figure 1). Given that the reduction of 2 and oxidation of 5 constitute the same nominal redox couple, it is worth noting the large cathodic shift afforded by coordination of the chloride ligand and the aryl iminyl moiety (Δ = −0.676 V). The reduction potential for adamantyl imido 5 (E1/2 = −0.37 V) is also mildly anodically shifted as compared with the more electron-rich analogues reported ([(tris(carbeno)borate)Fe(NAd)]+ −0.64 V,27 [(bis-(phosphino)pyrazolylborate)Fe(NAd)]+ −0.72 V29), though the comparison is not straightforward given the coordination number change between species (three- vs four-coordinate).
Figure 1.

Cyclic voltammograms of 2 (blue) and 5 (red) obtained in CH2Cl2 at 25 °C, with 0.1 M (nBu4N)(PF6); 100 mV/s; referenced to [Cp2Fe]+/0 couple.
2.2. Synthesis and Spectroscopic Characterization of Imido Fe(N(C6H4-p-tBu)) and Iminyl FeCl(•N(C6H4-p-tBu)) Pair
While oxidative group transfer to monovalent 4 can afford imido complexes with sterically encumbered alkyl and aryl azides,24 no terminal imido complex could be obtained upon treatment of 2 with N3(C6H4-p-tBu) and only the tetraazide adduct was obtained (ArL)Fe(N4(C6H4-p-tBu)2) from net consumption of 2 equiv of azide.42 Given the mild reduction potential for 2 (−1.10 V vs [Cp2Fe]+/0), chemical reduction was thus pursued as a means for generation of the terminal imido analogue. Accordingly, reduction of 2 with KC8 in thawing benzene-d6 resulted in the formation of a new paramagnetic complex via 1H NMR. The molecular structure of three-coordinate iron imido (ArL)Fe(N(C6H4-p-tBu)) (3) was obtained by X-ray diffraction studies on single crystals grown from a concentrated toluene solution layered with pentane at −35 °C (Figure 2a). The molecular structure features a distorted trigonal planar iron(III) center supported by the dipyrrin ligand and the aryl imido. The Fe–N3 distance of 1.708(4) Å in 3 is longer than the iron imido bond in 5 (1.674(11) Å), and nearly identical to that found in the mesityl imido (ArL)Fe(NMes) (1.708(2) Å), though far shorter than the corresponding distance in 2 (1.768(2) Å). The pertinent bond distances to compare 2 and 3 are presented in Figure 2b. Notably, all of the carbon–carbon bonds within the imide aryl ring in 3 are equivalent within error, indicating the aromaticity of the imido aryl framework has been restored from the iminyl 2 upon reduction.
Figure 2.

(a) Solid state molecular structure of 3 with thermal ellipsoids at the 35% probability level. Positional disorder, solvent molecules, and H atoms are removed for clarity. Color scheme: Fe, orange; N, blue; C, gray. (b) Selected bond metrics (Å) for iminyl 2 in red and imido 3 in black. Mulliken spin density plots (α–β) and values calculated for 3 (c) and 2 (d).
Further spectroscopic characterization of 3 affords data in line with that observed for the alkyl imido 5.24 Akin to 5, complex 3 features a symmetric electric field gradient, displaying a zero-field 57Fe Mössbauer spectrum with a single transition (δ: 0.44 mm/s, |ΔEQ|: 0.00 mm/s) at 200 K that becomes broad and weakly absorbing at lower temperature (Figure 3a). In accord with the observed temperature dependent phenomena (vide supra), 3 does not exhibit an EPR signal above ca. 50 K. The frozen toluene EPR spectrum of 3 (2.8 K) displays a rhombic signal (geff = 8.62, 5.35, 3.10), well reproduced by an S = 5/2 simulation treating each intra-doublet transition as an effective S = 1/2 system (in accordance with the weak-field limit) and including the effects of both rhombicity (E/D) and D-strain (simulation parameters: g = 2.0, |E/D| = 0.05, line width = 15 G). The inclusion of D-strain is necessary to account for the randomness in spatial conformation of the frozen solution and provide an effective powder pattern for a given S and E/D.43 The data and corresponding simulation are shown in Figure S5. Accordingly, solid-state magnetometry of 3 is consistent with a high-spin state as evidenced by a μeff of 4.6 μB (χMT = 4.76 cm3 K/mol) at 295 K (Figure S4). The value of χMT over the temperature range surveyed is consistent with an S = 5/2 configuration (spin-only value anticipated is 4.38 cm3 K/mol). The spin state suggested by the magnetic susceptibility data is further corroborated by variable-temperature, variable-field magnetization data collected on heating from 1.8 to 10 K and at increasing fields of 1–7 T (Figure S4 inset). Magnetization saturation occurs at 3.5 μB at 1.8 K and 7 T. The lower than expected saturation (5 μB for an ideal S = 5/2 with g = 2) and the observation of non-superimposable isofield curves indicate the presence of zero-field splitting and rhombicity, which was quantified by fitting the data to the spin Hamiltonian Ĥ= DŜz2 + E(Ŝx2 − Ŝy2) + gisoμB S·H. The fit parameters considering an S = 5/2 system that best reproduce the data are g = 2.00, D = 13.4 cm−1, |E/D| = 0.29.44
Figure 3.

(a) Zero-field 57Fe Mössbauer spectra [δ, |ΔEQ|, Γ (mm/s)] of iminyl 2 (top left, red trace: 0.28, 2.22, 0.14), iminyl 6 (bottom left, blue trace: 0.26, 2.05, 0.24); imido 3 (top right, red trace: 0.44, 0.00, 0.74); and imido 5 (bottom right, blue trace: 0.37, 0.00, 0.50). (b) Overlaid Fe Kedge XANES with inset Fe 1s → Fe 3d pre-edge features for 3 (solid red), 2 (dashed red), 5 (solid blue), 6 (dashed blue), 7 (solid green). (c) EXAFS for 6 with data shown in blue and theoretical fit in black dashes.
The original spin-state assignment for iminyl 2 was based on room temperature, solution magnetic moment determination.26 Thus, we sought to probe the ground spin state electronic structure for this molecule to corroborate the quintet assignment. Solid-state magnetometry was collected on a sample of 2, cleanly isolated following removal of H-atom sources via treatment of reaction glassware with trimethylsilyl chloride, use of perdeuterated solvents, and immediate purification of azide prior to synthesis. The magnetic susceptibility reveals a well-isolated ground state with no thermal transitions apparent between 50 and 300 K. Both the room temperature effective magnetic moment of 5.1 μB (χMT = 3.6 cm3 K/mol) and reduced magnetization data can be well modeled as an S = 2 spin system (Figure S1). The spin state suggested by the magnetic susceptibility data is further corroborated by variable-temperature, variable-field magnetization data collected on heating from 1.8 to 10 K and at increasing fields of 1 to 7 T (Figure S1 inset). Magnetization saturation occurs at 3.5 μB at 1.8 K and 7 T. The lower than expected saturation (4 μB for an ideal S = 2 with g = 2) and the observation of non-superimposable isofield curves indicate the presence of zero-field splitting and rhombicity, which was quantified by fitting the data to the spin Hamiltonian Ĥ= DŜz2 + E(Ŝx2 − Ŝy2) + gisoμB S·H. The fit parameters considering an S = 2 system that best reproduce the data are g = 2.12, D = −8.8 cm−1, |E/D| = 0.33.44 As noted above, the magnetic susceptibility for 2 plateaus between 50 and 300 K without variation in the susceptibility. Thus, the strength of the antiferromagnetic coupling between the ferric site (SFe = 5/2) and iminyl moiety must exceed ca. 200 cm−1, in line with the value predicted via DFT methods45 and supporting a broken-symmetry electronic structure of 2 (Jcalc = −673 cm−1).26
For the iminyl/imido pair of 2 and 3, respectively, the data collectively support an assignment of 2 as a high-spin FeIII center antiferromagnetically coupled to an iminyl centered radical. Upon reduction of 2, the added electron populates an orbital that is principally imide-based to restore aromaticity to the imide aryl π-system, maintaining the ferric oxidation state. The unusual stability of the ferric iminyl state can potentially be attributed to the resonance stabilization afforded via π-delocalization within the iminyl aryl unit. To gain insight into the location of the redox, single-point DFT45 calculations on the iminyl 2 and imido 3 complexes were performed. The calculated spin density plots (α – β) for 2 and 3, shown in Figure 2c–d, illustrate the contributions to the total spin contributed by iron (2: 57%; 3: 70%) and the imido unit (2: 34%; 3: 26%). The total spin localization within the imido aryl substituent in 3 is equal to the spin contribution from the imido N (12% N, 14% Ar), but increases upon oxidation to the iminyl 2 (14% N, 20% Ar). Based on these findings, the aryl component of the imido/iminyl fragment can play a critical role in spin delocalization, though it was as of yet unclear if an alkyl iminyl complex could adopt a similar electronic configuration to 2 without a means for stabilizing the N-centered radical.
2.3. Synthesis and Spectroscopic Characterization of an Alkyl Iminyl (ArL)FeCl(•NAd)
To synthesize the alkyl iminyl analogue of 2, a solution of chlorotriphenylmethane was added to a solution of imido 5 in thawing benzene-d6 and allowed to stir for 1 h at room temperature. After 1 h elapsed and the reaction solution changed in color from pink to maroon, assessment of the reaction progress by 1H NMR revealed consumption of 5 with concomitant formation of a new paramagnetic species and Ph3C(C6H5)CPh2. The reaction volatiles were removed in vacuo, and the resultant precipitate was washed with pentane. Dissolution of the maroon solids in benzene followed by lyophilization yielded a maroon powder (80% yield). The zero-field 57Fe Mössbauer spectrum of the new material displayed a clean quadrupole doublet (δ: 0.26 mm/s, |ΔEQ|: 2.05 mm/s) with parameters very similar to those observed for 2 (δ: 0.28 mm/s, |ΔEQ|: 2.22 mm/s) (Figure 3a). The similar Mössbauer spectra suggest a common iron oxidation state, spin state, and geometric configuration between 2 and the oxidation product of 5, leading us to formulate the product as (ArL)FeCl(•NAd) (6). The magnetic susceptibility of 6 is shown in Figure 4, revealing a μeff of 4.4 μB at 295 K (χMT = 2.46 cm3 K/mol) consistent with an S = 2 ground state electronic configuration. The spin state suggested by the magnetic susceptibility data is further corroborated by variable-temperature, variable-field magnetization data collected on heating from 1.8 to 10 K and at increasing fields of 1–7 T (Figure 4, inset). Magnetization saturation occurs at 2.6 μB at 1.8 K and 7 T. The lower than expected saturation (4 μB for an ideal S = 2 with g = 2) and the observation of non-superimposable isofield curves indicate the presence of zero-field splitting and rhombicity, which was quantified by fitting the data to the spin Hamiltonian Ĥ= DŜz2 + E(Ŝx2 − Ŝy2) + gisoμB S·H. The fit parameters considering an S = 2 system that best reproduce the data are g = 1.95, D = 6.9 cm−1, and |E/D| = 0.33.44 Thus, the data support a view of 6 as an FeIII iminyl complex even in the absence of an iminyl aromatic π–framework throughout which the nitrogen-centered radical can delocalize.
Figure 4.

Variable-temperature susceptibility data of (ArL)FeCl(•NAd) (6) collected at 1.0 T, with χMT = 2.46 cm3K/mol at 295 K. (Inset) Reduced magnetization data collected at three fields (1, 4, 7 T) over the temperature range 1.8–10 K. Magnetization fit parameters obtained with PHI:44 S = 2, g = 1.95, D = 6.9 cm−1, |E/D|= 0.33.
2.4.1. Extended X-ray Absorption Fine Structure Analysis of Iminyl 6
The reactive nature of 6 precluded obtaining suitable single crystals for X-ray diffraction analysis. Thus, XAS and EXAFS analysis on a benzene solution of 6 was undertaken. The Fourier transform of the EXAFS region of the XAS spectrum for 6 to k = 10 Å−1 (resolution = 0.15 Å) is well-modeled via a first shell simulation considering two pyrrole nitrogens, one chloride, and one iminyl nitrogen scattering paths (Figure 3c). The values are shown in Table 1 and are nearly identical to the crystallographically determined bond lengths of 2 (Fe–Npyrrole 2.013(1) Å, 1.997(1) Å; Fe–Cl 2.20(1) Å; Fe–Niminyl 1.768(2) Å). The resolution of the data is sufficient to differentiate the Npyrrole and Niminyl scattering paths, although it is not sufficient to detect asymmetry in the individual Fe–Npyrrole scattering paths. The close structural similarity of 6 to 2 strongly suggests that oxidation of imido 5 occurs at the imido nitrogen, preserving the high-spin ferric electronic configuration at iron.
Table 1.
EXAFS Simulation of 6a
| Path | CN | R, Å | σ2, Å2 | E0 | F, % |
|---|---|---|---|---|---|
| Fe–Npyrrole | 2 | 1.987(8) | 0.0003(6) | −7.61 | 29.5 |
| Fe–Cl | 1 | 2.222(5) | 0.0011(4) | ||
| Fe–Niminyl | 1 | 1.761(6) | 0.0004(7) |
EXAFS were fit in OPT using paths calculated by FEFF7. Coordination numbers (CN) were held constant while distances (R) and Debye–Waller factors (σ2) were allowed to float. Parenthetical values correspond to fitting errors in R and σ2 in the final significant digit. Errors in coordination number are estimated at 25%. Fits were performed over the entire (0 to 6.0 Å) Fourier transform window. Goodness of fit is measured by F, defined as . Additional fitting details are included in the Supporting Information.
2.4.2. Fe and N K-edge X-ray Absorption near Edge Spectroscopy on Imidos (3, 5) and Iminyls (2, 6)
In light of the challenges associated with assignment of the metal oxidation state in the presence of high-lying, ligand-based molecular orbitals, samples of both imido/iminyl pairs (3 and 2; 5 and 6) were investigated by X-ray absorption near edge spectroscopy, and the results shown in Figure 3b. Note that, given the coordination number change as well as potential change in iron-imido/iminyl covalency, comparison of the rising edge energy will not unambiguously reflect the respective metal oxidation levels for the three-coordinate imido and four-coordinate iminyl complexes. Iron K-edge pre-edge absorption features are typically assigned to Fe 1s → 3d excitations, although in the present case appreciable ligand character can be expected in the acceptor molecular orbitals. Nevertheless, the maxima of the resolved pre-edge features demonstrate that upon reduction of iminyl 2 (7111.5 eV) to imido 3 (7111.5 eV), or oxidation of imido 5 (7112.2 eV) to iminyl 6 (7112.4 eV), very little change in Fe physical oxidation state is inferred. For comparison, the energy-weighted maximum of the resolved pre-edge bands of the ferrous product of 1 and adamantyl amine, (ArL)FeCl(NH2Ad) (7), is shifted to lower energy (7110.3 eV), consistent with a more reduced iron center in this complex as compared to the other species studied. Comparing the aryl and alkyl imido/iminyl pairs, the pre-edge absorption features for the alkyl species (5, 6) are shifted to higher energy relative to those of the aryl congeners (3, 2). These shifts are 0.7 eV for the imido complexes and 0.9 eV for the iminyl complexes. These energy differences are nearly as great as the difference between the ferrous complex examined (7) and the ferric imidos (~1.2 eV).
To provide further evidence for the iminyl radicals in 2 and 6, the nitrogen orbital containing the iminyl was probed directly using nitrogen K-edge X-ray absorption spectroscopy. As seen in Figure 5, pre-edge features at 400.5 and 399.8 eV are observed for all species (2, 3, 5, and 6) that can be assigned as transitions into high-lying antibonding molecular orbitals of N 2p parentage. Additional lower energy features present at 398.3 and 397.7 eV for the aryl (3 and 2) and alkyl (5 and 6) imido/iminyl pairs, respectively, represent transitions into the Fe 3d-based antibonding singly occupied molecular orbitals that are predicted to contain substantial mixing with N 2p orbitals (both dipyrrin and imido/iminyl N-donors). Most importantly, new, low energy pre-edge features found at 394.5 and 394.8 eV are observed exclusively in the iminyl species 6 and 2, respectively. In a N K-edge XAS calibration study of PNP-pincer supported Ni complexes, a similar low energy pre-edge feature was identified upon oxidation of the backbone amide to an aminyl donor.46 This new pre-edge feature present at 396.4 eV was assigned, through corroboration with TDDFT calculations, as a transition from N 1s into the nitrogen localized aminyl-based SOMO on the PNP ligand framework. Hence, we assign the pre-edge features observed here at 394.5 and 394.8 eV as transitions into the nitrogen-localized iminyl radicals present in 6 and 2, respectively.
Figure 5.

N K-edge XAS for imido (3, 5) and iminyl (2, 6) complexes; (inset) N 1s → iminyl N 2p pre-edge features for 2 (black) and 6 (blue).
2.4.3. Electronic Structure Calculations Corroborate Assignment of 6 as an Alkyl Iminyl Species
A geometry optimization45 of 6 was carried out to produce coordinates from which to carry out electronic structure calculations. The complete structure of 6 including full aryl substituents was optimized using the BP8647,48 functional with the segmented all-electron relativistically contracted (SARC) Ahlrich’s def2-TZVP-ZORA basis set.49,50 Optimization of 6 employed the broken symmetry (BS) approach, assuming partitioning between 5 Fe-based and 1 ligand-based spin BS (5,1). The structure produced at this level of theory closely reproduces the experimental bond lengths, yielding the following primary coordination sphere interatomic distances: Fe–Npyrrole 2.002 Å, 1.981 Å; Fe–Cl 2.200 Å; Fe–Niminyl 1.707 Å (Figure 6a). These coordinates, along with optimized structures for 2, 3, 5, and 7, were used in B3LYP calculations of Mössbauer δ51 values as well as time-dependent DFT (TDDFT) calculations of Fe Kedge XAS52 pre-edge bands (Table 2, Figure S21). Calculated spectroscopic parameters accord strongly with experiment–after calibration with the remaining compounds in the test set (Figures S22, S23), the value of δ for 6 is matched by theory, and the error in the 1s → 3d excitation is 0.1 eV.
Figure 6.

Calculated Mulliken spin density (a) and qualitative molecular orbital scheme (b) for 6. Spin density is plotted at an isovalue of 0.009 au. Molecular orbitals are plotted at isovalues of 0.04 au.
Table 2.
Experimental and Calculated Spectroscopic Parameters
Accord between calculated and experimental geometric and electronic structural parameters further supports assignment of 6 as a stable alkyl analogue of iminyl 2. Electronic structure calculations support preservation of a high-spin d5 FeIII that is strongly antiferromagnetically coupled to AdN• (calculated J = −2204 cm−1) (Figure 6b).57,58 Moreover, comparison of the orbital mixing coefficients in the calculated unrestricted corresponding orbitals (UCOs) for iminyl species 2 and 6 that is localized to the N-centered β SOMO of the antiferromagnetically coupled pair reveals a ratio of 1.91 (62.3% in 6 vs 32.6% in 2). This ratio corresponds well to the experimental 2.02 (0.141 in 6 vs 0.071 in 2) ratio of intensities of the iminyl pre-edge features found in the N Kedge XAS.
2.5. Comparative Kinetic Analysis for the Imido/Iminyl Complexes Undergoing C–H Bond Activation with Toluene
The isolation of iron imido/iminyl redox pairs provides a unique opportunity to investigate the effect of altering the valence of the transferred atom during a C–H functionalization reaction without affecting the transition metal oxidation state. Dissolution of iminyl complexes 2 or 6 in toluene results in immediate reaction, as demonstrated by the change in UV/vis absorption over time (Figure 7). The change in absorbance at λmax was fit as an exponential decay, and the resulting observed rate constant is shown in Table 3. In contrast, the UV/vis spectrum of the imido complexes 3 or 5 in toluene is identical to that observed in benzene and does not change over the course of 4 h at room temperature. In fact, a similar rate of reaction to those exhibited by the iminyl species is not achieved until the solution is heated to 80 °C, indicating a substantially higher barrier toward reactivity with toluene for the imidos 3 and 5. Assuming a simple kinetic profile wherein the change in absorbance at λmax is directly related to the consumption of starting material (imido or iminyl) and the reaction course is not significantly altered by temperature, the activation energy can be extracted from these data for each complex and is presented in Table 3.
Figure 7.

(Top left) UV/vis traces of the reaction of 2 with toluene at 22°C with spectra recorded every 10 min for 4 h. (Bottom left) Normalized absorbance at λmax (554 nm) during the course of the reaction at various temperatures. (Right) 1H NMR spectra progression of a toluene solution of equimolar imido 5 and iminyl 6 at 60 °C at the time elapsed indicated. Chemical shifts corresponding to 5 are highlighted in red, 6 in blue, and (ArL)FeCl (1) in green.
Table 3.
Rate Constants, Half-Lives, and Activation Energies for Reaction of Iron Complexes with Toluenea
| complex | T (°C) | k (s−1) | t1/2 (min) | Ea (kcal/mol) |
|---|---|---|---|---|
| 5 | 80 | 0.0264 | 26 | 11.5 |
| 90 | 0.0427 | 16 | ||
| 100 | 0.0633 | 11 | ||
| 3 | 80 | 0.0195 | 36 | 12.1 |
| 90 | 0.0343 | 20 | ||
| 100 | 0.0745 | 9 | ||
| 6 | 22 | 0.0299 | 23 | 7.1 |
| 32 | 0.0433 | 16 | ||
| 42 | 0.0722 | 10 | ||
| 2 | 22 | 0.0166 | 42 | 9.2 |
| 32 | 0.0256 | 27 | ||
| 42 | 0.0528 | 13 |
Exponential decay of Δ (λmax) versus time was fit independently at each temperature.
Upon oxidation of the nitrogen atom from the imide to the iminyl state, the barrier for reaction with toluene decreased by an average of 3.7 kcal/mol. The additional radical density present on the nitrogen atom can account for the enhancement of reactivity, more closely resembling the electronic structure at the transition state for hydrogen atom abstraction. This simple Hammond-type analysis is in line with the higher activation energy for the aryl imido and iminyl complexes 3 and 2, respectively, which feature electron delocalization through the aromatic fragment as opposed to the localized spin density at the transferring nitrogen atom of 5 and 6. Alternatively, the higher molecular oxidation state may also contribute to the increase in reactivity via a Bell–Evans–Polanyi-type correlation,59 wherein the rate enhancement upon oxidation is attributable to the increased driving force of the reaction as a result of a higher oxidation potential or enhanced N–H bond strength of the metal-amide product. Regardless, the activation parameters for reaction of these iron imide complexes with toluene demonstrate a higher enthalpic barrier upon reduction or upon substitution with an aromatic imide substituent, suggesting that both a higher metal-imide bond order and delocalization through the aromatic π-system stabilize the iron imide with respect to hydrogen atom abstraction.
Comparison of the independent rates of reaction for 5 and 6 suggests that a rate enhancement of 2 orders of magnitude occurs upon oxidation. Indeed, heating an equimolar ratio of 5 and 6 to 60 °C in toluene results in selective consumption of the iminyl complex 6 and formation of a new set of paramagnetic resonances via 1H NMR (Figure 7). The appearance of these chemicals shifts can be attributed to the formation of (ArL)FeCl (1) and occurs concomitantly with a peak at 3.72 ppm corresponding to N-benzyl-adamantylamine. The competition experiment validates the independent rate studies and demonstrates that the iminyl complex 6 is significantly more reactive toward C–H bonds than its imido congener 5. The increased reactivity of 6 is not limited to C–H amination: imido 5 is stable in neat styrene up to 100 °C, whereas iminyl 6 reacts smoothly with 200 equiv of styrene at room temperature to yield the desired aziridine in 36% yield. Further, hydrogen atom abstraction from cyclohexene occurs at room temperature with iminyl 6 to produce the H2NAd adduct (ArL)FeCl(H2NAd) (7) quantitatively, whereas the imido congener 5 requires heating to 80 °C and arrests at formation of the amide adduct (ArL)Fe(HNAd).24
2.6. Reaction Pathways and Thermodynamic Considerations
2.6.1. Frontier Molecular Orbital Considerations
The electronic structure of the imido and iminyl differ in the extent to which both N 2p orbitals engage in π-bonding with iron orbitals of appropriate symmetry. The imido complexes feature nearly linear Fe–N–C bond linkages, signifying maximal, though non-degenerate, π interactions: πx [σ*(dxz–Lσ) ± Npx] which includes an Fe–Ldipyrrin σ* component, and the orthogonal πy (Fe dyz ± Npy). The high-spin imido complexes give rise to a (πx)2(πy)2(πy*)1(πx*)1 configuration. The iminyl complexes, however, lack conjugation of the two π manifolds. Indeed, a simple description for the iminyl consistent with the iron oxidation levels observed by 57Fe Mössbauer and XAS consists of only the πx interaction, leaving a largely N-centered radical for 6. While the iminyl radical in 6 lacks the enhanced stabilization afforded by delocalization into the aromatic ring of 2, the radical is nonetheless stabilized by coupling antiferro-magnetically to the five iron α spins (see Figure 6). This electronic configuration manifests in the larger Fe–N bond lengths observed, as well as the enhanced reactivity of the iminyl with respect to the imido congener.
We have previously proposed a reaction sequence for intermolecular amination using imido 5. We can amend that orbital description to describe qualitatively the reactivity differences observed between the imido and iminyl congeners. The large KIEs observed in the reactions of both imido (3, 5) and iminyls (2, 6) with toluene suggest that the C–H bond breaking event contributes significantly to the rate limiting step of the reaction sequence illustrated for 6 in Figure 8a.
Figure 8.

Frontier molecular orbital description of (ArL)FeCl(•NAd) (6) reaction profile (a) for H atom abstraction from PhCH3 (b) and the subsequent radical rebound event (c).
A consideration of the frontier molecular orbitals (FMOs) that complete this process includes the Fe–N π interaction (πx), the N 2py orbital containing the iminyl radical, and the toluene C–H σ bond (Figure 8b). The N-radical (2py)1 dictates the primary orbital interaction with the incoming toluene C–H substrate given its steric accessibility and more favorable energetic overlap. Thus, the frontier orbital interaction with respect to the iron-iminyl is lower in energy than the analogous imido amination reaction which proceeds from the (πy*)1 orbital.
The H-atom abstraction step is highlighted in Figure 8b, where the homolyzed C–H orbital (yielding PhC•H2 and H 1s) combines with N 2py to yield three orbital combinations: (σN–H)2 (•CH2Ph)1(σ*N–H)0. Unlike the imido amination sequence where H-atom abstraction leads to metal reduction, the H-atom abstraction step for the iminyl is completely N-centered, leaving the iron oxidation level unchanged. The H-atom abstraction step conserves total spin-angular momentum as the resulting radical pair complex remains in a quintet spin configuration. The recombination step (Figure 8c) should proceed combining the benzylic radical with the remaining πx [σ*(dxz–Lσ) ± Npx] pair yielding three product orbitals: (σN–C)2(Fe 3dxz)2(σ*N–C)0, constituting a net reduction of the iron center, yet again conserving the total spin-angular momentum for the entire reaction sequence.
That the iminyl complexes will activate the C–H bonds of toluene at lower temperatures than their respective imido congeners is a direct result of the diminished Fe–N bond order. Thus, the oxidizing orbital (N 2py)1 is lower in energy than the corresponding imido reaction partner (πy*), leading to a better energetic overlap between H-atom donor and acceptor orbitals. Furthermore, the entire amination sequence for the iminyls conserves the total spin angular momentum while the imido complexes must change their electronic configurations (S = 5/2 → S = 3/2) concomitantly with iron reduction.
The presence of metal-stabilized iminyl radicals has now been observed9,36 or implicated in H-atom abstraction reactiv-ity12,13,21,37 and nitrene transfer catalysis7,8,10,11,60,61 in a number of systems.15 The supporting organic moiety on the bound nitrene can promote stabilization via heteroatom donation, akin to Fischer-type carbene ligands,10,11 or promote reactivity by concentration of radical density on the iminyl N-atom as is the case in 6.
2.6.2. Thermodynamic Considerations
In considering a stepwise mechanism for C–H amination, the initial hydrogen atom abstraction from substrate can be represented via a thermodynamic square scheme that partitions the driving force of the reaction into the constituent proton and electron transfer contributions, as shown in eq 1:
| (1) |
where the final constant represents the standard potential for proton reduction in aprotic solvent.62 Electrochemical analysis of 2 revealed a quasi-reversible reduction potential of −1.05 V versus [Cp2Fe]+/0. Given this reduction potential, the productive activation of toluene (C–H BDE: 90 kcal/mol)63 by 2 would require a pKa of 40 in the resulting (ArL)FeCl-(NH(C6H4-p-tBu)) complex. This estimation is in line with both the experimentally determined pKa of Smith’s iron(III)–amide (pKa = 37(3))27 and the calculated pKa of Borovik’s manganese(II)–hydroxide (pKa ≥ 36) complexes,64 as well as others. The higher calculated pKa value for 2 as compared to Smith’s cationic tris(carbeno)borate-supported iron(IV) imide can be rationalized via the difference in overall charge between these molecules and permits the activation of the stronger C–H bond of toluene.
The ability of 6 to functionalize toluene allows a rough estimation of the Fe═N bond strength via eq 2:
| (2) |
where E0 is the standard reduction potential of 6. Although the appropriate bond dissociation energies have not been determined for N-benzyl-adamantyl amine, experimental values for N-methyl-benzyl amine are readily available63 and provide an Fe–N bond strength of approximately 94 kcal/mol. This value is similar to the experimentally determined ΔH0(Fe═O) range of Nocera’s pacman iron(IV) oxo porphyrin system of 65–85 kcal/mol which can promote two-electron oxidation of substrate, but not C–H hydroxylation chemistry.65
A comparison of the activation energies for the imide/iminyl pairs demonstrated a lower barrier for reaction with toluene at the iminyl state. A Bell–Evans–Polanyi correlation between the rate of reaction and thermodynamic driving force would suggest that hydrogen atom transfer is therefore less favorable at the lower valent state. The higher Fe–N bond order, coupled with the absence of electrochemical reduction of 5 under experimental conditions (to −2.3 V versus [Cp2Fe]+/0), indicate that the overall functionalization process is likewise less favorable at the imide state. These data support that the enhancement of reactivity at the iminyl state is both kinetic and thermodynamic in nature, due to the higher radical density present at the N-atom and weaker iron–nitrogen interaction via the broken symmetry electronic structure, respectively.
3. CONCLUSIONS
We have previously demonstrated that the (dipyrrin)iron platform is capable of stabilizing both high-spin ferric imido species, as well as the highly unusual ferric iminyl complex which features a high-spin iron center antiferromagnetically coupled to the iminyl radical. Both of the high-spin complexes are capable of effecting nitrene transfer to C–H bonds and olefinic substrates. The foregoing results demonstrate that the imido/iminyl redox isomers are chemically interchangeable. Furthermore, the elusive alkyl iminyl intermediate (ArL)FeCl-(•NAd), implicated in both inter- and intramolecular C–H bond functionalization catalytic processes, was isolated and spectroscopically characterized. Comparison of the Mössbauer spectroscopy, magnetometry, and bond lengths of the alkyl iminyl complex with the aryl iminyl congener revealed striking similarities, suggesting a common electronic structure and indicating the nitrene aryl component is not necessary to stabilize the iminyl oxidation level via radical delocalization. Furthermore, oxidation of the imido complexes to their iminyl redox isomers largely localizes the oxidation event to the NR fragment. The iminyl complexes feature N K-edge pre-edge absorptions found to be characteristic for these N-based radical ligands, arising from N 1s promotion into a singly occupied N 2p iminyl orbital. Lastly, having access to both alkyl and aryl NR redox isomers, we were able to assess how the change in molecular oxidation level manifests in changes to C–H bond amination efficacy. Oxidation of the imido complexes to their iminyl redox isomers results in a rate enhancement of 2 orders in magnitude. The rate enhancement is largely enthalpic (entropic barriers are nearly equal). The ability of the imido or iminyl component to delocalize the spin-density throughout an aryl substituent also results in a greater barrier toward functional group transfer. We infer this latter to mean that concentration of radical density upon the transferring group can play a large role in C–H bond functionalization processes.
Supplementary Material
Acknowledgments
T.A.B. gratefully acknowledges support by grants from the NSF (CHE-0955885) and NIH (GM-115815), and from Harvard University. M.W. would like to thank the NSF for a Predoctoral Graduate Fellowship. K.M.L. gratefully acknowledges support from the NSF in the form of a CAREER award (CHE-1454455) and from the ACS Petroleum Research Fund (55181-DNI6). This work is based in part on research conducted at the Stanford Synchrotron Radiation Lightsource (SSRL), which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the Department of Energy’s Office of Biological and Environmental Research, and by NIH/HIGMS (including P41GM103393). The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of NIGMS or NIH.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b08714.
X-ray crystal structure images; magnetic data; cyclic voltammograms; X-ray absorption spectra; Mössbauer, UV–vis, and 1H NMR spectra (PDF)
Crystallographic data (CIF)
References
- 1.Nugent WA, Mayer JM. Metal-Ligand Multiple Bonds: The Chemistry of Transition Metal Complexes Containing Oxo, Nitrido, Imido, or Alkylidyne Ligands. Wiley-Interscience; New York: 1988. [Google Scholar]
- 2.Saouma CT, Peters JC. Coord Chem Rev. 2011;255:920. doi: 10.1016/j.ccr.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berry JF. Comments Inorg Chem. 2009;30:28. [Google Scholar]
- 4.Zhang L, Deng L. Chin Sci Bull. 2012;57:2352. [Google Scholar]
- 5.Bess EN, Deluca RJ, Tindall DJ, Oderinde MS, Roizen JL, Du Bois J, Sigman MS. J Am Chem Soc. 2014;136:5783. doi: 10.1021/ja5015508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu W, Groves JT. J Am Chem Soc. 2010;132:12847. doi: 10.1021/ja105548x. [DOI] [PubMed] [Google Scholar]
- 7.Bagh B, Broere DLJ, Sinha V, Kuijpers PF, van Leest NP, de Bruin B, Demeshko S, Siegler MA, van der Vlugt JI. J Am Chem Soc. 2017;139:5117. doi: 10.1021/jacs.7b00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujita D, Sugimoto H, Shiota Y, Morimoto Y, Yoshizawa K, Itoh S. Chem Commun. 2017;53:4849. doi: 10.1039/c7cc01840a. [DOI] [PubMed] [Google Scholar]
- 9.Goswami M, Lyaskovskyy V, Domingos SR, Buma WJ, Woutersen S, Troeppner O, Ivanović-Burmazović I, Lu H, Cui X, Zhang XP, Reijerse EJ, DeBeer S, van Schooneveld MM, Pfaff FF, Ray K, de Bruin B. J Am Chem Soc. 2015;137:5468. doi: 10.1021/jacs.5b01197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuijpers PF, Tiekink MJ, Breukelaar WB, Broere DLJ, Van Leest NP, van der Vlugt JI, Reek JNH, De Bruin B. Chem - Eur J. 2017;23:7945. doi: 10.1002/chem.201700358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuijpers PF, van der Vlugt JI, Schneider S, De Bruin B. Chem - Eur J. 2017;23:13819. doi: 10.1002/chem.201702537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spasyuk DM, Carpenter SH, Kefalidis CE, Piers WE, Neidig ML, Maron L. Chem Sci. 2016;7:5939. doi: 10.1039/c6sc01433j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou W, Patrick BO, Smith KM. Chem Commun. 2014;50:9958. doi: 10.1039/c4cc04545a. [DOI] [PubMed] [Google Scholar]
- 14.Prier CK, Zhang RK, Buller AR, Brinkmann-Chen S, Arnold FH. Nat Chem. 2017;9:629. doi: 10.1038/nchem.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suarez AIO, Lyaskovskyy V, Reek JNH, van der Vlugt JI, De Bruin B. Angew Chem, Int Ed. 2013;52:12510. doi: 10.1002/anie.201301487. [DOI] [PubMed] [Google Scholar]
- 16.Brown SD, Peters JC. J Am Chem Soc. 2005;127:1913. doi: 10.1021/ja0453073. [DOI] [PubMed] [Google Scholar]
- 17.Bart SC, Lobkovsky E, Bill E, Chirik PJ. J Am Chem Soc. 2006;128:5302. doi: 10.1021/ja057165y. [DOI] [PubMed] [Google Scholar]
- 18.Betley TA, Peters JC. J Am Chem Soc. 2003;125:10782. doi: 10.1021/ja036687f. [DOI] [PubMed] [Google Scholar]
- 19.Brown SD, Betley TA, Peters JC. J Am Chem Soc. 2003;125:322. doi: 10.1021/ja028448i. [DOI] [PubMed] [Google Scholar]
- 20.Cowley RE, DeYonker NJ, Eckert NA, Cundari TR, DeBeer S, Bill E, Ottenwaelder X, Flaschenriem C, Holland PL. Inorg Chem. 2010;49:6172. doi: 10.1021/ic100846b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cowley RE, Holland PL. Inorg Chem. 2012;51:8352. doi: 10.1021/ic300870y. [DOI] [PubMed] [Google Scholar]
- 22.Lu CC, Saouma CT, Day MW, Peters JC. J Am Chem Soc. 2007;129:4. doi: 10.1021/ja065524z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scepaniak JJ, Young JA, Bontchev RP, Smith JM. Angew Chem, Int Ed. 2009;48:3158. doi: 10.1002/anie.200900381. [DOI] [PubMed] [Google Scholar]
- 24.Wilding MJT, Iovan DA, Betley TA. J Am Chem Soc. 2017;139:12043. doi: 10.1021/jacs.7b06682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iovan DA, Betley TA. J Am Chem Soc. 2016;138:1983. doi: 10.1021/jacs.5b12582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.King ER, Hennessy ET, Betley TA. J Am Chem Soc. 2011;133:4917. doi: 10.1021/ja110066j. [DOI] [PubMed] [Google Scholar]
- 27.Nieto I, Ding R, Bontchev RP, Wang H, Smith JM. J Am Chem Soc. 2008;130:2716. doi: 10.1021/ja0776834. [DOI] [PubMed] [Google Scholar]
- 28.Searles K, Fortier S, Khusniyarov MM, Carroll PJ, Sutter J, Meyer K, Mindiola DJ, Caulton KG. Angew Chem, Int Ed. 2014;53:14139. doi: 10.1002/anie.201407156. [DOI] [PubMed] [Google Scholar]
- 29.Thomas CM, Mankad NP, Peters JC. J Am Chem Soc. 2006;128:4956. doi: 10.1021/ja0604358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verma AK, Nazif TN, Achim C, Lee SC. J Am Chem Soc. 2000;122:11013. [Google Scholar]
- 31.Wang L, Hu L, Zhang H, Chen H, Deng L. J Am Chem Soc. 2015;137:14196. doi: 10.1021/jacs.5b09579. [DOI] [PubMed] [Google Scholar]
- 32.Ni C, Fettinger JC, Long GJ, Brynda M, Power PP. Chem Commun. 2008:6045. doi: 10.1039/b810941a. [DOI] [PubMed] [Google Scholar]
- 33.Hong S, Sutherlin KD, Vardhaman AK, Yan JJ, Park S, Lee YM, Jang S, Lu X, Ohta T, Ogura T, Solomon EI, Nam W. J Am Chem Soc. 2017;139:8800. doi: 10.1021/jacs.7b04695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Badiei YM, Dinescu A, Dai X, Palomino RM, Heinemann FW, Cundari TR, Warren TH. Angew Chem, Int Ed. 2008;47:9961. doi: 10.1002/anie.200804304. [DOI] [PubMed] [Google Scholar]
- 35.Kogut E, Wiencko HL, Zhang L, Cordeau DE, Warren TH. J Am Chem Soc. 2005;127:11248. doi: 10.1021/ja0533186. [DOI] [PubMed] [Google Scholar]
- 36.Lu CC, De Beer George S, Weyhermüller T, Bill E, Bothe E, Wieghardt K. Angew Chem, Int Ed. 2008;47:6384. doi: 10.1002/anie.200800669. [DOI] [PubMed] [Google Scholar]
- 37.Shay DT, Yap GPA, Zakharov LN, Rheingold AL, Theopold KH. Angew Chem, Int Ed. 2005;44:1508. doi: 10.1002/anie.200462529. [DOI] [PubMed] [Google Scholar]
- 38.Hennessy ET, Betley TA. Science. 2013;340:591. doi: 10.1126/science.1233701. [DOI] [PubMed] [Google Scholar]
- 39.Hennessy ET, Liu RY, Iovan DA, Duncan RA, Betley TA. Chem Sci. 2014;5:1526. [Google Scholar]
- 40.Mehn MP, Peters JC. J Inorg Biochem. 2006;100:634. doi: 10.1016/j.jinorgbio.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 41.Ortiz de Montellano PR. Cytochrome P450: Structure, Mechanism, and Biochemistry. 4. Kluwer Academic/Plenum Publishers; New York: 2005. [Google Scholar]
- 42.Wilding MJT. Electronic structure/function relationship in metal ligand multiple bonds for C–H functionalization chemistry. Harvard University; 2016. [Google Scholar]
- 43.Hagen WR. Mol Phys. 2007;105:2031. [Google Scholar]
- 44.Chilton NF, Anderson RP, Turner LD, Soncini A, Murray KS. J Comput Chem. 2013;34:1164. doi: 10.1002/jcc.23234. [DOI] [PubMed] [Google Scholar]
- 45.Neese F. ORCA - An ab initio, Density Functional and Semi-empirical Electronic Structure Package. 7. Vol. 2. Universität Bonn; Bonn, Germany: 2009. [Google Scholar]
- 46.Lukens JT, Kurogi T, Mindiola DJ, Lancaster KM. Unpublished data. [Google Scholar]
- 47.Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]
- 48.Lee CT, Yang WT, Parr RG. Phys Rev B: Condens Matter Mater Phys. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 49.van Lenthe E, van der Avoird A, Wormer PES. J Chem Phys. 1998;108:4783. [Google Scholar]
- 50.van Wüllen C. J Chem Phys. 1998;109:392–399. [Google Scholar]
- 51.Neese F. Inorg Chim Acta. 2002;337:181. [Google Scholar]
- 52.De Beer George S, Petrenko T, Neese F. J Phys Chem A. 2008;112:12936. doi: 10.1021/jp803174m. [DOI] [PubMed] [Google Scholar]
- 53.Devlin FJ, Finley JW, Stephens PJ, Frisch MJ. J Phys Chem. 1995;99:16883. [Google Scholar]
- 54.Schäfer A, Horn H, Ahlrichs R. J Chem Phys. 1992;97:2571. [Google Scholar]
- 55.Schäfer A, Huber C, Ahlrichs R. J Chem Phys. 1994;100:5829. [Google Scholar]
- 56.Weigend F, Ahlrichs R. Phys Chem Chem Phys. 2005;7:3297. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 57.Soda T, Kitagawa Y, Onishi T, Takano Y, Shigeta Y, Nagao H, Yoshioka Y, Yamaguchi K. Chem Phys Lett. 2000;319:223. [Google Scholar]
- 58.Yamaguchi K, Takahara Y, Fueno T. Applied Quantum Chemistry. Springer; Dordrecht: 1986. [Google Scholar]
- 59.Mayer JM. Acc Chem Res. 2011;44:36. doi: 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thacker NC, Lin Z, Zhang T, Gilhula JC, Abney CW, Lin W. J Am Chem Soc. 2016;138:3501. doi: 10.1021/jacs.5b13394. [DOI] [PubMed] [Google Scholar]
- 61.Broere DL, de Bruin B, Reek JNH, Lutz M, Dechert S, van der Vlugt JI. J Am Chem Soc. 2014;136:11574. doi: 10.1021/ja502164f. [DOI] [PubMed] [Google Scholar]
- 62.Warren JJ, Tronic TA, Mayer JM. Chem Rev. 2010;110:6961. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo Y-R. Handbook of Bond Dissociation Energies in Organic Compounds. CRC Press LLC; Boca Raton, FL: 2003. [Google Scholar]
- 64.Parsell TH, Yang MY, Borovik AS. J Am Chem Soc. 2009;131:2762. doi: 10.1021/ja8100825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rosenthal J, Pistorio BJ, Chng LL, Nocera DG. J Org Chem. 2005;70:1885. doi: 10.1021/jo048570v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.