

Abstract
Steroids are of profound importance in human biology and medicine. Today, >100 FDA-approved steroidal agents are prescribed daily for indications that include heart failure, inflammation, pain, and cancer, among others. The scientific foundation of this now rich history of clinical success is in chemical synthesis, where advances provided a means to access meaningful quantities of these complex carbocyclic species. While triumphs in organic chemistry have, without question, enabled the establishment and sustained growth of the steroid pharmaceutical industry, production of highly functionalized synthetic steroids of widely varying substitution and stereochemistry (including unnatural ent-steroids) remains challenging, despite the numerous reports of elegant strategies for their de novo synthesis. In other words, broad exploration of >50% of this pharmaceutically privileged area of ‘chemical space’ remains challenging, labor intensive, and costly – potential factors that have contributed to the current reality that 100% of FDA-approved steroidal agents are of the natural antipode (absolute stereochemistry). Here, we describe an advance in chemical synthesis that has established an enantiospecific means to access novel steroidal compositions of matter with unprecedented facility and flexibility through the sequential use of two powerful ring-forming reactions: a modern metallacycle-mediated annulative cross-coupling and a new acid-catalyzed vinylcyclopropane rearrangement cascade. In addition to demonstrating the usefulness of this chemistry for accessing synthetic steroids of either enantiomeric series (nat- or ent-), steroidal products from this synthesis pathway have been selectively functionalized at sites within each of the four carbocyclic rings, a synthetic ent-steroid has been prepared on multi-gram scale, the enantiomer of a selective estrogen has been prepared, and a novel ent-steroid with growth inhibitory properties in three cancer cell lines has been discovered [GI50 = 0.32 – 1.07 μg/mL (1.2 – 4 μM) in MDA-MB-231 (breast), U2OS (osteosarcoma), and AsPC-1 (pancreatic) human cancer cell lines].
Following the first correct structure elucidation of a steroid and the synthesis of equilenin in 1932 and 1939 (Fig. 1a,b), respectively, much attention has been given to developing chemistry capable of producing natural and synthetic steroids1,2. Efforts in this area established a scientific foundation that has delivered >100 FDA-approved steroidal agents as therapeutics, leading to their current status as arguably the most well-studied and successful class of natural product-inspired pharmaceuticals3. While steroids have had a transformative impact on medicine and society, playing vital roles as oral contraceptives and treatments for cancer, heart failure, inflammation, pain, and traumatic brain injury, among others, there exist substantial barriers in preparative organic chemistry that continue to constrain our ability to freely explore the medicinal value of novel synthetic steroidal compositions of matter. Here, we describe a new approach to steroid synthesis that allows for preparation of partially aromatic steroids in as few as five chemical steps from a readily available chiral starting material (epichlorohydrin). The pathway is uniquely capable of accessing structurally diverse steroids in a straightforward fashion (housing modification in each of the four steroidal rings), proceeds in an enantiospecific fashion, and can be used to prepare multigram quantities of nat- or ent-steroidal compositions of matter with ease. This new steroid synthesis has been realized through capitalizing on the power of a modern metallacycle-mediated annulative cross-coupling reaction and a new acid-promoted vinylcyclopropane rearrangement cascade.
Figure 1.

a, The general steroid tetracyclic skeleton. b, Examples of natural steroids. c, Selected approaches to steroid synthesis using semisynthetic and de novo strategies.
Early scientific breakthroughs that established the steroid pharmaceutical industry were based on semisynthesis, where readily available natural steroids were chemically transformed to high-value therapeutic agents. Historical examples include Merck’s bile acid-to-cortisone process, and the Marker degradation (Fig. 1c) – an achievement that cleared the first synthetic pathway from a plant-derived steroid (diosgenin) to testosterone, estrone, estradiol, progesterone, and eventually cortisone4–6. While continuing to play a valuable role in medicinal pursuits7, semisynthesis is often difficult to employ as a strategy to gain access to collections of highly oxygenated/functionalized steroidal targets, and is wholly unsuitable to explore the broad medicinal value of ‘ent-steroids’ (defined by an unnatural absolute stereochemistry of the tetracycle). This latter point deserves further comment, as decades of scientific triumph have positioned steroidal compositions of matter as ‘privileged’ in the pharmaceutical industry – a characterization that is based, in part, on physical properties that are critically important in medicinal chemistry (i.e. ‘drug like’ properties). While pairs of enantiomers share identical physical properties, and in the case of steroids ‘drug-like’ properties, it is surprising that 100% of FDA-approved steroidal drugs are of the natural (‘nat’) enantiomeric series. It is a growing sentiment that ent-steroids define fertile ground for discovery in biomedical science, boasting complementary three dimensional structures of potential relevance for targeting biology that is distinct from the nat-series8–12, and retaining the extremely valuable drug-like properties of the parent class. In other words, ent-steroids are a privileged class of drug-like molecules that remain grossly underexplored in biology and medicine because they are not readily available from natural sources or chemical synthesis.
While important contributions in organic chemistry have established de novo synthesis pathways to synthetic steroids from non-steroidal starting materials, notably the Smith–Torgov synthesis of estranes13 and biomimetic cation–olefin cyclization14 processes (Fig. 1c), among others15–21, semisynthesis remains the primary means by which pharmaceutically relevant steroids are prepared7,22. This fact is humbling and reinforces the reality that substantial challenges persist with the de novo synthesis of complex carbocyclic structures, and that barriers continue to obstruct a more free exploration of this pharmaceutically privileged area of chemical space.
To address this gap at the interface of chemistry and medicine, efforts were focused on developing a conceptually unique approach to de novo steroid synthesis that would be: (1) convergent, to allow for ease in diversifying structure by changing the nature of coupling partners, (2) stereochemically flexible, (3) step economical23 (proceeding in a small number of chemical steps), (4) enantiospecific, allowing access to either nat- or ent-steroidal systems with equal facility, and (5) capable of providing steroidal tetracycles that are well suited for a variety of subsequent functionalization processes (i.e. manipulation of functionality in each ring of tetracyclic nucleus).
As illustrated in Figure 2, it was thought that late-stage establishment of a steroidal tetracycle (1) may be possible through strategic formation of the C5–C6 bond by functionalization and activation of a tricyclic ACD-ring-containing system (2). Compound 2 would then surface as an intermediate of high value, and one that was recognized to be accessible from metallacycle-mediated annulative cross-coupling between a suitably functionalized alkyne (3) and a chiral enyne (4)24. While not yet widely adopted in organic synthesis, metallacycle-mediated cross-coupling is an area of great recent growth25–27. A variety of new convergent C–C bond-forming reactions that realize unique retrosynthetic relationships in organic chemistry have been described within this broad class of chemical reactivity, and applications in the context of complex molecule synthesis continue to surface28. Here, a metallacycle-mediated annulative cross-coupling reaction was viewed as being particularly powerful, as it could provide a functionalized hydrindane (2) of great value for steroid synthesis from the stepwise coupling of simple starting materials.
Figure 2.

Retrosynthetic strategy for a convergent, flexible, and enantiospecific synthesis of partially aromatic (C19-nor) steroids by metallacycle-mediated annulative cross-coupling and late-stage formation of the steroidal B-ring.
The reduction of this chemical strategy to practice is depicted in Figure 3a. With initial focus on the nat-steroid series, conversion of epichlorohydrin (5) to the functionalized enyne 6, followed by metallacycle-mediated annulative cross-coupling with TMS-phenylacetylene 7 delivered hydrindane 8. This coupling process is unique in organic chemistry and particularly powerful here, delivering an angularly substituted trans-fused hydrindane in a convergent manner from acyclic precursors, and establishing three σC–C bonds and two stereocenters (one of which is quaternary). Our working mechanistic hypothesis for this annulation reaction is summarized in Fig. 3b, and includes: i) alkoxide-directed metallacycle-mediated cross-coupling to generate (A) (rs ≥ 20:1)29, ii) stereoselective intramolecular [4+2] cycloaddition to furnish a bridged polycyclic metallacyclopentene (C) (ds ≥ 20:1), iii) elimination to provide a tertiary allylic metal species (D), iv) isomerization to the primary allylic organometallic (E), and v) regio- and stereoselective protonation to deliver hydrindane 8.
Figure 3.

Establishment of a new chemical pathway for the enantiospecific synthesis of steroids. a, Establishment of a synthesis pathway from optically active epichlorohydrin to an enantiodefined estrane by way of a metallacycle-mediated annulative cross-coupling and an acid-mediated vinylcyclopropane rearrangement cascade. b, Proposed mechanism for the metallacycle-mediated annulative cross-coupling reaction.
Moving forward, a “functionalization” protocol was sought that could install the steroidal C6 carbon in concert with revealing a reactive intermediate for B-ring formation. Cyclopropanation was selected as the means to accomplish such a process and, as depicted in Fig. 3a, can be employed to selectively engage the 1,1-disubstituted alkene of 8 en route to the vinylcyclopropane intermediate 1030,31. While efficiency in this initial attempt at cyclopropanation was rather low (41% isolated yield), subsequent studies have concluded that higher yields are routinely encountered if protection of the secondary alcohol is conducted prior to cyclopropanation (vide infra). B-ring formation by way of the vinylcyclopropane 10 was initially thought possible from ionization and electrocyclic ring opening of the resulting cyclopropyl cation, followed by intramolecular Friedel–Crafts alkylation32–35. Unfortuantely, all attempts to accomplish such a transformation were unsuccessful, ultimately leading to the triene 11, presumably through pentadienyl cation intermediate I. In an attempt to achieve cyclization without proceeding through this unstable cationic intermediate, it was found that treatment of 10 with TiCl4 in nitromethane resulted in the formation of steroidal product 12 in 68% yield. Here, reaction of TiCl4 with the D-ring hydroxy group (or with adventitious water) is thought to produce protic acid in situ and convert 10 to the reactive homoallylic cation intermediate II, presumably through a process that includes protodesilylation, protonation of the resulting styrenyl alkene, and regioselective cyclopropane fragmentation36–39. Ring closure would then be possible by engaging the phenyl ring in an intramolecular Friedel–Crafts alkylation to deliver an intermediate that is transformed to 12 through loss of HBr40,41.
As illustrated in Table 1, this chemical pathway is useful for generating a wide range of steroidal systems in an exceptionally concise and enantiospecific fashion. First, to demonstrate the facility with which this chemistry delivers ent-steroids, ent-6 (derived from (+)-epichlorohydrin) was smoothly converted to ent-12 (entry 1). As experienced with our first attempt, depicted earlier in Figure 3A, the efficiency of the two-step ring-closing process (ent-8 → ent-12) was fairly low due to the challenge of accomplishing cyclopropanation in the presence of the C16-OH. Entry 2 reveals that the efficiency of the overall process is substantially greater if the C16 hydroxy group is silylated prior to cyclopropanation and, in combination with entry 3, demonstrates the ease with which A-ring aromatic steroids containing varied oxygenation patterns can be preapared (15 and 18). Interestingly, while proceeding uneventfully in these two examples, it was later found that the acid-mediated vinylcyclopropane rearrangement can be more complex.
Table 1.

|
yield reported is for the combination of diene iosmers (trans + endo).
stereoselectivity for the annulation process (trans:cis) is typically ~6:1
yield reported is for the 2–3 step sequence.
cyclization sequence was conducted without protection of the C16 hdyroxy group.
regioselectivity based on 1H NMR of the crude reaction product.
yield for this unselective process was not determined.
structure confirmed by X-ray diffraction - See Supporting Information.
As illustrated in entry 4, use of the p-chloro-substituted phenylacetylene 19 led ultimately to the discovery that the final ring closure can proceed with additional rearrangement. Here, steroidal products 21a and 21b were produced in roughly equal proportions – an observation that is consistent with the mechanistic proposal depicted in Figure 4, where cyclization may proceed either through direct Friedel–Crafts alkylation (II → III → F), or by initial formation of a spirocyclic intermediate and subsequent rearrangement with selective migration of C9 (II → IV → G).
Figure 4.
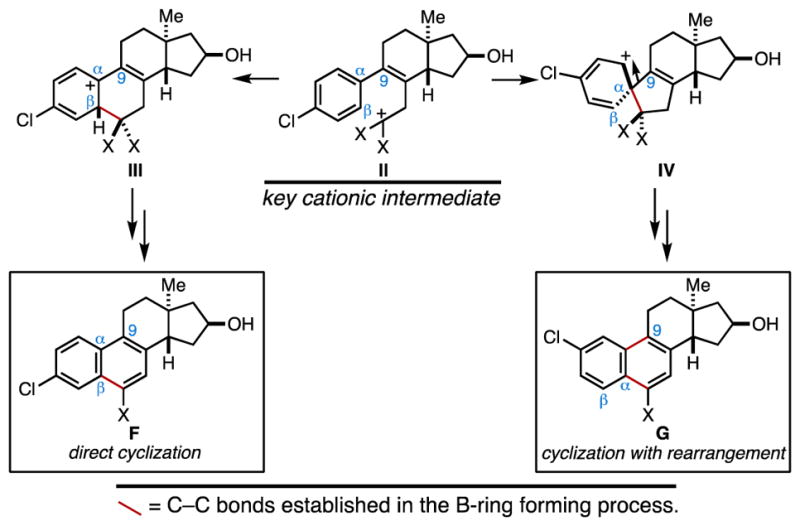
Mechanistic divergence in the vinylcyclopropane rearrangement and intramolecular Friedel–Crafts alkylation process.
As illustrated in entry 5, it is possible to achieve high levels of selectivity in favor of ring closure by rearrangement. Here, the p-methoxy-substituent of 23 plays a signficant role in biasing the course of reaction, presumably due to its electronic contribution to favoring the formation of a spirocyclic intermediate akin to IV (Figure 4). Here, production of the C2-methoxy-substituted steroid 24 was found to proceed with very high levels of seletivity (rs ≥ 20:1). Interestingly, as depicted in entry 6, further modification of A-ring substitution (26) was found to restore a preference for cyclization without rearrangement. Here, cyclization delivered the C3-methoxy-substituted steroid 27 as a single regioisomer in a modest 31% yield (over three steps) – an observation that is consistent with steric effects stemming from the chloride substituents that may dissuade oxonium ion formation in the spirocyclic intermediate (i.e. an oxonium ion intermediate would be destabilized by significant 1,5-interactions between the methyl group of the oxonium ion and one of the ortho chlorine substitutents). Additional examples that illustrate the facility with which this steroid synthesis provides access to products of varying A-ring structure and substitution are depicted in entries 7 and 8.
This enantiospecific entry to steroids can be used to gain access to a range of systems that boast additional substitution and varying stereochemistry (Figure 5a). In products 34 and 35, the C13 quaternary center at the junction of rings C and D was altered simply by changing the Grignard reagent used to prepare the initial enyne for annulation (R1 = Et or Bn). In product 36, the stereochemistry at C14 was altered by using a variant of the metallacycle-mediated annulation process that furnishes the cis-fused isomer42. Finally, simple functional group manipulations can be used to gain access to steroidal compositions of matter possessing varied structure within each of the four rings of the tetracycle (37–41) – including a remarkably stable naphthoquinone methide (39)43–50. Finally, as depicted in Figure 5b, this synthesis pathway is capable of producing multigram quantities of steroidal products with ease. Here, 4.0 g of ent-12 was prepared in just five steps from epoxide 42 with an overall isolated yield of 20%.
Figure 5.

a. Access to steroids with varying substitution and stereochemistry (see Supporting Information for experimental details associated with the syntheses of 34–41). b. Multigram-scale preparation of a synthetic ent-steroid. c. Preparation of ent-estra-1,3,5(10),6,8-pentaene-3,16α-diol. d. Discovery of an ent-steroid with cytotoxic properties: Cells were plated at 1000 cells/well of a 96 well plate. The following day, compound 39 was added in 2-fold dilutions (8 wells/concentration). After 7 days growth, cells were lyzed and analyzed for total DNA content as previously described52.
The synthetic pathway to ent-steroids that has been established here is useful for the preparation of antipodes medicinally relevant agents. For example, 16-hydroxyestratrienes have been identified as synthetic estrogens that have a dissociation in favor of their estrogenic action on bone rather than the uterus (Figure 5c)51. While estra-1,3,5(10),6,8-pentaene-3,16α-diol is a representative member of this class, its enantiomer has never been described. Because the eanantiomer of estradiol is known to have neurological activity of potential value for the treatment of traumatic brain injury, and lacks activity as an estrogenic compound, ent-estranes are beginning to emerge as a broader class with potentially useful “non-steroidal” pharmaceutical properties. Here, it is demonstrated that one of the products from this synthesis pathway (27) can be easily transformed to ent-estra-1,3,5(10),6,8-pentaene-3,16α-diol (43). We look forward to future efforts that explore the neurological and estrogenic activities of this and related synthetic ent-16-hydroxyestranes.
Finally, a novel synthetic ent-steroid that was prepared in these studies (39) induced growth inhibition in three cancer cell lines with 50% inhibitory concentrations of 1.2 – 4 μM (0.32 – 1.07 μg/mL) (Figure 5d).
In conclusion, steroids are unquestionably the most appreciated and well-developed class of natural product-inspired pharmaceutical agents. While originally pursued as treatments for inflammation and fertility, they continue to define a rich platform for drug discovery and development across a diverse therapeutic landscape. However, the greater potential of steroids as privileged drug-like skeletons has been, and will continue to be, limited by the chemistry that is available to access their fused polycyclic skeletons. While accomplishments in drug development have been primarily fueled by semisynthesis, a dependence on this chemical strategy will continue to greatly constrain our ability to take advantage of the favorable drug-like properties of diverse steroidal compositions across a broad therapeutic landscape. Chemical strategies based on semisynthesis are inherently limited in scope, proceeding from a subset of abundant natural steroidal starting materials of only a single antipode (chirality) and typically requiring significant effort to install or remove functionality through uneconomical means. De novo synthesis of steroids has a rich history in organic chemistry, yet crowning achievements in this area do not routinely surface as enabling technology in discovery or development. Such a reality is not uncommon in natural product-inspired drug discovery, and attention to this gap between accomplishments in total synthesis and the persistent difficulties associated with discovery and development of natural product-inspired drugs, will continue to be an important consideration in the ongoing evolution of synthetic organic chemistry53. Here, a step-economical (concise), flexible, convergent, and enantiospecific synthesis of partially aromatic synthetic steroids has been established from a readily available chiral starting material (epichlorohydrin). The route proceeds by way of a complex metallacycle-mediated annulative cross-coupling reaction and a new acid-catalyzed vinylcyclopropane rearrangement cascade, and can be employed to gain access to novel steroidal compositions of varying stereochemistry and substitution. Given the value of steroidal structures as platforms for drug discovery, and the growing appreciation of the potential therapeutic value of ent-steroids, this chemical advance can play an important, even enabling, role in the future identification of novel and biomedically relevant steroidal compositions of matter.
Methods
A representative set of experimental procedures is provided below for the multigram synthesis of ent-12.
To a stirring solution of epoxide 42 (14.8 g, 0.079 mol, 1 equiv) in 300 mL THF under N2 atmosphere at −78 °C was added CuI (3.0 g, 0.016 mol, 0.2 equiv) followed by 2-propenylmagnesium bromide (189 mL, 0.094 mol, 0.5 M, 1.2 equiv). The resulting yellow suspension was stirred for 1 h at −78 °C, warmed to rt, and then stirred until the reaction was judged to be complete by TLC analysis. The reaction was quenched by adding 200 mL sat. NH4Cl (aq) to the reaction mixture. The organic layer was separated, and the aqueous layer was extracted with EtOAc (200 mL × 3). The combined organic layers were dried over anhydrous MgSO4, filtered, and the solvents were removed in vacuo. Purification of the crude product by SiO2 flash column chromatography (20% EtOAc/80% Hexane) afforded 14.8 g of the enyne ent-6 as yellow oil (81%).
To a stirring solution of alkyne 7 (30 g 0.17 mol, 2.7 equiv) in 1.2 L of dry toluene at rt under N2 atmosphere was added Ti(Oi-Pr)4 (49.1 g, 0.17 mmol 2.7 equiv). The resulting mixture was cooled to −78 °C, and n-BuLi (2.7 M, 135 mL, 0.364 mol, 5.7 equiv) was added dropwise. The resulting black mixture was warmed first to rt, then heated to 50 °C and stirred at that temperature for 1 h (no reflux condenser required). In a separate flask under N2 atmosphere, enyne ent-6 (14.7 g, 0.064 mol, 1.0 equiv) was dissolved in 300 mL of dry toluene, cooled to −78 °C, and treated with n-BuLi (2.5 M, 25 mL, 0.064 mmol, 1.0 equiv); added dropwise at −78 °C. The resulting mixture was warmed to rt, and then transferred by cannula to the black Ti-alkyne complex at −78 °C. The mixture was slowly warmed to rt overnight (approx. 17 h). After this period, 1.5 L of dry MeOH was cooled to −78 °C in a separate flask under N2 atmosphere, and the reaction mixture was transferred by cannula to the pre-cooled MeOH. Once the addition was complete, the reaction mixture was warmed to rt, and 500 mL of DI H2O was added. 500 mL of EtOAc was added, the organic phase was separated, and the aqueous layer was extracted with EtOAc (500 mL × 3). The combined organic layers were dried over MgSO4, the drying agent was removed by filtration, and the resulting solution was concentrated in vacuo. Purification of the crude product by flash column chromatography afforded 12.4 g of the title hydrindane ent-8 as yellow film (60%, isolated as 4:1 mixture of ent-8: ent-9).
The following three-step procedure was used to convert 9.9 g of the trans-fused hydrindane ent-8 to the steroidal product ent-12 – a process that proceeded with an overall 40% isolated yield. This yield is based on the amount of hydrindane ent-8 present in a 4:1 mixture with the unreactive “endo” diene isomer: To a solution of 12.4 g of the 4:1 mixture [containing 9.9 g of hydrindane ent-8 (0.030 mol, 1.0 equiv) and its corresponding endo diene isomer ent-9 (2.5 g, 0.008 mol)] in 500 mL THF was added TBSCl (11.4 g, 0.076 mol, 2.5 equiv) and imidazole (5.2 g, 0.076 mmol, 2.5 equiv). The reaction mixture was stirred at rt under N2 atmosphere overnight (approx. 17 h), then partitioned between a solution of 200 mL sat. NaHCO3(aq) and 200 mL ethyl acetate. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (200 mL × 3). The combined organic layers were washed with brine and dried over anhydrous MgSO4. The resulting suspension was filtered through a course glass fritted funnel to remove solid material, and the filtrate was concentrated in vacuo. The resulting residue was filtered through a pad of silica gel using a hexanes and ethyl acetate solvent mixture as eluent (hexanes/EtOAc = 80/1). Concentration of the resulting dilute solution afforded 15.8 g of a yellow oil. This crude product was used in the subsequent step without further purification.
To a solution of the above crude product (15.8 g) and triethylbenzylammonium chloride (1.7 g, 7.3 mmol) in 73 mL CHBr3 at rt was added KOH (24.6 g, 0.44 mol) in water (25 mL). The reaction mixture was stirred at 45 °C overnight (approx. 17 h), cooled to rt, then partitioned between a solution of 100 mL water and 200 mL CH2Cl2. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (200 mL × 3). The combined organic layers were washed with brine and dried over anhydrous MgSO4. The resulting suspension was filtered through a course glass fritted funnel to remove solid material, and the filtrate was concentrated in vacuo to afford 19.6 g of a crude light brown oil that was used in the subsequent step without further purification.
To a solution of the above crude product (19.6 g) in 600 mL nitromethane was added i-PrOH (20.4 g, 0.34 mol) and TiCl4 (15.9 g, 0.084 mol). The resulting mixture was stirred at rt for 1 h under N2 atmosphere and a second aliquot of i-PrOH (20.4 g, 0.34 mol) and TiCl4 (15.9 g, 0.084 mol) was added. The mixture was stirred for another 1 h at rt under N2 atmosphere, then partitioned between a solution of 200 mL sat. NaHCO3(aq) and 200 mL CH2Cl2. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (200 mL × 3). The combined organic layers were washed with brine and dried over anhydrous MgSO4. The resulting suspension was filtered through a course glass fritted funnel to remove solid material, and the filtrate was concentrated in vacuo. Purification of the crude product by flash column chromatography afforded 4.0 g of the title compound as yellow solid (40% over 3 steps).
Spectral data for ent-12: 1H NMR (600 MHz, CD2Cl2) δ 8.22–8.18 (m, 1H), 8.00–7.96 (m, 1H), 7.57–7.52 (m, 3H), 4.72–4.63 (app. m, 1H), 3.27 (app. dd, J = 18.5, 8.0 Hz, 1H), 3.22–3.14 (m, 2H), 2.29 (dd, J = 12.4, 7.3 Hz, 1H), 2.19 – 2.15 (app. m, 2H), 2.13 (app. dd, J = 12.7, 7.8 Hz, 1H), 1.91 (td, J = 11.8, 7.8 Hz, 1H), 1.68 (s, 1H), 1.40 (dd, J = 12.4, 5.7 Hz, 1H), 0.64 (s, 3H); 13C NMR (150 MHz, CD2Cl2) δ 138.3, 134.0, 131.7, 130.8, 129.6, 128.0, 127.1, 126.5, 124.2, 120.9, 72.5, 51.1, 48.3, 41.9, 37.8, 36.3, 25.0, 18.0; IR (thin film): 3335, 2930, 2856, 1584, 1266, 1200, 1044, 753, 666 cm–1; HRMS (EI+) Calculated for C18H19OBr [M+]330.0619, found 330.0610; −21 (c 6.8, CHCl3)
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support of this work by the National Institutes of Health–NIGMS (GM80266). The authors also thank Professor Gordon Gribble, Professor Peter Jacobi, Professor Jimmy Wu, and Dr. Brian Heasley for helpful discussions.
Footnotes
Author Contributions W.S.K., K.D., R.P.H. and G.C.M. contributed to the chemical experiments, R.P.H performed in silico experiments to explore the mechanism of the vinylcyclopropane rearrangement, W.S.K. and K.D. performed all chemical reactions reported, A.E. performed the in vitro evaluation of ent-steroid 39, and G.C.M. wrote the manuscript with contributions from all authors.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
Supporting Information is available in the online version of the paper.
References
- 1.Windaus A. Concerning the constitution of cholesterol and biliary acid. Z Physiol Chem. 1932;213:147–187. [Google Scholar]
- 2.Bachmann WE, Cole W, Wilds AL. The total synthesis of the sex hormone equilenin. J Am Chem Soc. 1939;61:974–975. [Google Scholar]
- 3.Lednicer D. Steroid chemistry at a glance. Wiley; New York: 2011. [Google Scholar]
- 4.Pines SH. The Merck bile acid cortisone process: The next-to-last word. Org Proc Res Dev. 2004;8:708–724. [Google Scholar]
- 5.Marker RE, Tsukamoto T, Turner DL. Sterols. C Diosgenin. J Am Chem Soc. 1940;62:2525–2532. [Google Scholar]
- 6.Renneberg R. Mexico, the father of the pill and the race for cortisone. Biotechnol J. 2008;3:449–451. doi: 10.1002/biot.200890042. [DOI] [PubMed] [Google Scholar]
- 7.Hanson JR. Steroids: Partial synthesis in medicinal chemistry. Nat Prod Rep. 2010;27:887–899. doi: 10.1039/c001262a. [DOI] [PubMed] [Google Scholar]
- 8.Biellmann JF. Enantiomeric steroids: Synthesis, physical, and biological properties. Chem Rev. 2003;103:2019–2033. doi: 10.1021/cr020071b. [DOI] [PubMed] [Google Scholar]
- 9.Covey DF. ent-Steroids: novel tools for studies of signaling pathways. Steroids. 2009;74:577–585. doi: 10.1016/j.steroids.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green PS, et al. The nonfeminizing enantiomer of 17beta-estradiol exerts protective effects in neuronal cultures and a rat model of cerebral ischemia. Endocrinology. 2001;142:400–406. doi: 10.1210/endo.142.1.7888. [DOI] [PubMed] [Google Scholar]
- 11.Akwa Y, Ladurelle N, Covey DF, Baulieu EE. The synthetic enantiomer of pregnenolone sulfate is very active on memory in rats and mice, even more so than its physiological neurosteroid counterpart: distinct mechanisms? Proc Natl Acad Sci USA. 2001;98:14033–14037. doi: 10.1073/pnas.241503698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petit GH, et al. Pregnenolone sulfate and its enantiomer: differential modulation of memory in a spatial discrimination task using forebrain NMDA receptor deficient mice. Eur Neuropsychopharmacol. 2011;21:211–215. doi: 10.1016/j.euroneuro.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeung Y-Y, Chein R-J, Corey EJ. Conversion of Torgov’s synthesis of estrone into a highly enantioselective and efficient process. J Am Chem Soc. 2007;129:10346–10347. doi: 10.1021/ja0742434. [DOI] [PubMed] [Google Scholar]
- 14.Yoder RA, Johnston JN. A case study in biomimetic total synthesis: polyolefin carbocyclizations to terpenes and sterols. Chem Rev. 2005;105:4730–4756. doi: 10.1021/cr040623l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeelen FJ. Steroid total synthesis. Nat Prod Rep. 1994;11:607–612. doi: 10.1039/np9941100607. [DOI] [PubMed] [Google Scholar]
- 16.Chapelon A, Moraléda D, Rodriguez R, Ollivier C, Santelli M. Enantioselective synthesis of steroids. Tetrahedron. 2007;63:11511–11616. [Google Scholar]
- 17.Mackay EG, Sherburn MS. The Diels–Alder reaction in steroid synthesis. Synthesis. 2015;47:1–21. [Google Scholar]
- 18.Funk RL, Vollhardt KPC. A cobalt-catalyzed steroid synthesis. J Am Chem Soc. 1977;99:5483–5484. doi: 10.1021/ja00458a044. [DOI] [PubMed] [Google Scholar]
- 19.Funk RL, Vollhardt KPC. The cobalt way to dl-estrone. A highly regiospecific functionalization of 2,3-bis(trimethylsilyl)extratrien-17-one. J Am Chem Soc. 1979;101:215–217. [Google Scholar]
- 20.Vollhardt KPC. Cobalt-mediated steroid synthesis. Pure & Appl Chem. 1985;57:1819–1826. [Google Scholar]
- 21.Kaplan W, Khatri HR, Nagorny P. Concise enantioselective total synthesis of cardiotonic steroids 19-hydroxysarmentogenin and trewianin aglycone. J Am Chem Soc. 2016;138:7194–7198. doi: 10.1021/jacs.6b04029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nising CF, Bräse S. Highlights in steroid chemistry: total synthesis versus semisynthesis. Angew Chem Int Ed. 2008;47:9389–9391. doi: 10.1002/anie.200803720. [DOI] [PubMed] [Google Scholar]
- 23.Wender PA, et al. Function-Oriented Synthesis, Step Economy, and Drug Design. Accounts Chem Res. 2008;41:40–49. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- 24.Jeso V, et al. Synthesis of angularly substituted trans-fused hydroindanes by convergent coupling of acyclic precursors. J Am Chem Soc. 2014;136:8209–8212. doi: 10.1021/ja504374j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reichard HA, McLaughlin M, Chen MZ, Micalizio GC. Regioselective reductive cross-coupling reations of unsymmetrical alkynes. Eur J Org Chem. 2010:391–409. doi: 10.1002/ejoc.200901094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reichard HA, Micalizio GC. Metallacycle-mediated cross-coupling with substituted and electronically unactivated alkenes. Chem Sci. 2011;2:573–589. doi: 10.1039/C0SC00394H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Micalizio GC, Mizoguchi H. The development of alkoxide-directed metallacycle-mediated annulative cross-coupling chemistry. Isr J Chem. 2017;57:228–238. doi: 10.1002/ijch.201600098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Micalizio GC, O’Rourke NF, Kier MJ. Metallacycle-mediated cross-coupling in natural product synthesis. Tetrahedron. 2016;72:7093–7123. doi: 10.1016/j.tet.2016.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan J, Micalizio GC. An alkoxide-directed carbometalation of internal alkynes. J Am Chem Soc. 2006;128:2764–2765. doi: 10.1021/ja057352w. [DOI] [PubMed] [Google Scholar]
- 30.Makosza M, Wawrzyniewicz M. Reactions of organic anions. XXIV Catalytic method for preparation of dichlorocyclopropane derivatives in aqueous medium. Tetrahedron Lett. 1969;10:4659–4662. [Google Scholar]
- 31.Fedorynski M. Synthesis of gem-dihalocyclopropanes and their use in organic synthesis. Chem Rev. 2003;103:1099–1132. doi: 10.1021/cr0100087. [DOI] [PubMed] [Google Scholar]
- 32.Sasaki T, Eguchi S, Kiriyama T. Studies on heterocage compounds. V Reaction of 5-hydroxymethyl-2-norbornene with dihalocarbene Novel synthesis of some oxa-modified adamantane analogs. J Org Chem. 1973;38:2230–2234. [Google Scholar]
- 33.Danheiser RL, Morin JM, Jr, Yu M, Basak A. Cationic cyclizations initiated by electrocyclic cleavage of cyclopropanes. Synthesis of lactones, tetrahydropyrans, and tetrahydrofurans. Tetrahedron Lett. 1981;22:4205–4208. [Google Scholar]
- 34.Gassman PG, Tan L, Hoye TR. Intramolecular cationic cyclizations initiated by electrocyclic cleavage of cyclopropanes. Synthesis of trienic cyclopentane derivatives. Tetrahedron Lett. 1996;37:439–442. [Google Scholar]
- 35.Banwell MG, Harvey JE, Hockless DCR, Wu AW. Electrocyclic ring-opening/π-allyl cation cyclization reaction sequences involving gem-dihalocyclopropanes as substrates: Application to syntheses of (+/−)-, (+)-, and (−)-γ-lycorane. J Org Chem. 2000;65:4241–4250. doi: 10.1021/jo991791u. [DOI] [PubMed] [Google Scholar]
- 36.Olah GA, Bollinger JM. Stable carbonium ions. LXVIII Protonation and ionization of cyclopropyl halides Measurement of rotational barriers in substituted allyl cations. J Am Chem Soc. 1968;90:6082–6086. [Google Scholar]
- 37.Poulter CD, Winstein S. The cyclopropylcarbinyl–allyl rearrangement of a hexamethylcyclopropylcarbinyl system. J Am Chem Soc. 1969;91:3649–3650. [Google Scholar]
- 38.Poulter CD, Winstein S. Solvolysis and degenerate cyclopropylcarbinyl–cyclopropylcarbinyl rearrangement of a hexamethylcyclopropylcarbinyl system. J Am Chem Soc. 1969;91:3650–3652. [Google Scholar]
- 39.Sorensen TS, Ranganayakulu K. Cyclopropylcarbinyl–allylcarbinyl–allyl cation rearrangements. Tetrahedron Lett. 1970;11:659–662. [Google Scholar]
- 40.Seko S, Tanabe Y, Suzukamo G. A novel synthesis of α- and β-halonaphthalenes via regioselective ring cleavage of aryl(gem-dihalocyclopropyl)methanols and its application to total synthesis of lignan lactones, Justicidin E and Taiwanin C. Tetrahedron Lett. 1990;31:6883–6886. [Google Scholar]
- 41.Tanabe Y, et al. Novel method for the synthesis of α- and β-halogenonaphthalenes by regioselective benzannulation of aryl(gem-dihalogenocyclopropyl)methanols: application to the total synthesis of the lignan lactones, justicidin E and taiwanin C. J Chem Soc Perkin Trans. 1996;1:2157–2165. [Google Scholar]
- 42.Kim WS, Aquino C, Mizoguchi H, Micalizio GC. LiOOt-Bu as a terminal oxidant in a titanium alkoxide-mediated [2+2+2] reaction cascade. Tetrahedron Lett. 2015;56:3557–3559. doi: 10.1016/j.tetlet.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rabideau PW. The metal–ammonia reduction of aromatic compounds. Tetrahedron. 1989;45:1579–1603. [Google Scholar]
- 44.Rabideau PW, Marcinow Z. The Birch reduction of aromatic compounds. Org React. 1992;42:1–334. [Google Scholar]
- 45.Pellissier H, Santelli M. The Birch Reduction of Steroids. A Review. Org Prep Proc Int. 2002;34:609–642. [Google Scholar]
- 46.Subba Rao GSR. Birch reduction and its application in the total synthesis of natural products. Pure Appl Chem. 2003;75:1443–1451. [Google Scholar]
- 47.Fajkoš J, Joska J. On steroids. LV Bromination of 3β-acetoxy-5α-androstan-16-one. Collect Czech Chem Commun. 1960;25:2863–2877. [Google Scholar]
- 48.Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem Rev. 1995;95:2457–2483. [Google Scholar]
- 49.Sawyer JS. Recent advances in diaryl ether synthesis. Tetrahedron. 2000;56:5045–5065. [Google Scholar]
- 50.Syper L. Partial oxidation of aliphatic side chains with cerium (IV) Tetrahedron Lett. 1966;7:4493–4498. [Google Scholar]
- 51.Kuenzer H, et al. 16-Hydroxyestratrienes as selectively active estrogens. 7,109,360 B1. US. 2016 Sep 19;
- 52.Montano R, Chung I, Garner KM, Parry D, Eastman A. Preclinical development of the novel Chk1 inhibitor SCH900776 in combination with DNA damaging agents and antimetabolites. Mol Cancer Therap. 2012;11:427–438. doi: 10.1158/1535-7163.MCT-11-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eastgate MD, Schmidt MA, Fandrick KR. On the design of complex drug candidate syntheses in the pharmaceutical industry. Nat Rev Chem. 2017;1 doi: 10.1038/s41570-017-0016. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.