

Abstract
Anthracyclines such as doxorubicin are highly effective chemotherapy agents used to treat many common malignancies. However, their use is limited by cardiotoxicity. We previously identified visnagin as protecting against doxorubicin toxicity in cardiac but not tumor cells. In this study, we sought to develop more potent visnagin analogs in order to use these analogs as tools to clarify the mechanisms of visnagin-mediated cardioprotection. Structure-activity relationship studies were performed in a zebrafish model of doxorubicin cardiomyopathy. Movement of the 5-carbonyl to the 7 position and addition of short ester side chains led to development of visnagin analogs with 1,000-fold increased potency in zebrafish and 250-fold increased potency in mice. Using proteomics, we discovered that doxorubicin caused robust induction of Cytochrome P450 family 1 (CYP1) that was mitigated by visnagin and its potent analog 23. Treatment with structurally divergent CYP1 inhibitors, as well as knockdown of CYP1A, prevented doxorubicin cardiomyopathy in zebrafish. The identification of potent cardioprotective agents may facilitate the development of new therapeutic strategies for patients receiving cardiotoxic chemotherapy. Moreover, these studies support the idea that CYP1 is an important contributor to doxorubicin cardiotoxicity and suggest that modulation of this pathway could be beneficial in the clinical setting.
Keywords: Cardiology, Oncology
Keywords: Cardiovascular disease, Drug therapy, Toxins/drugs/xenobiotics
CYP1 inhibitors are identified that protect against chemotherapy-induced cardiotoxicity with high in vivo potency.
Introduction
Anthracyclines such as doxorubicin (DOX) are used to treat a number of common malignancies, including breast cancer, leukemia, lymphoma, and sarcoma. DOX exhibits potent tumoricidal activity but can also cause cardiotoxicity in up to 9% of patients, potentially resulting in congestive heart failure. Although the mechanisms leading to the development of DOX cardiotoxicity have not been clearly defined, oxidative stress and mitochondrial dysfunction have been implicated as key mediators (1–3). Currently, dexrazoxane is the only compound approved by the Food and Drug Administration (FDA) to prevent DOX-induced cardiotoxicity in patients. Dexrazoxane has been proposed to chelate intracellular iron, block iron-assisted oxidative radical production, and inhibit topoisomerase 2β (4–7). Although recent studies have found dexrazoxane to be a safe adjunct to DOX therapy (8, 9), many clinicians choose not to administer dexrazoxane to patients, given concerns that it may interfere with the antitumor efficacy of DOX (10) and/or induce secondary malignancies (11). Thus, new cardioprotective agents are needed for this patient population.
To tackle this problem, our laboratory established an embryonic zebrafish model of DOX cardiomyopathy characterized by pericardial edema, impaired cardiac contractility, and decreased blood flow through the vasculature (12). Through a large chemical screen in this model, we identified visnagin (compound 1 [C1]) as the most potent and least toxic antidote to DOX cardiotoxicity in zebrafish (half maximal effective concentration [EC50] 1 μM; median toxic dose [TD50] > 10 μM; Figure 1). C1 is a furanochromone derived from the plant Ammi visnaga that has previously been used as an herbal remedy for kidney stones and high blood pressure. In cultured cells exposed to DOX, cotreatment with C1 decreased apoptosis in cardiomyocytes but did not affect apoptosis in a number of tumor cell lines. Likewise, C1 preserved cardiac function during acute and chronic DOX administration in adult mice, though it did not affect the tumoricidal activity of DOX in mouse and zebrafish xenograft models. Here we describe structure-activity relationship (SAR) studies of C1, guided primarily by large-scale in vivo phenotypic assessment allowing for rapid determination of efficacy and toxicity in a whole-organism model. The identification of structural modifications that improve the in vivo potency of C1 may facilitate the development of this family of compounds as cardioprotective agents for patients receiving anthracycline chemotherapy.
Figure 1. Optimization of compound 1 (C1, visnagin) in an in vivo model of DOX-induced cardiac toxicity.

EC50 values were calculated based on the percent of zebrafish rescued from the DOX cardiomyopathy phenotype (decreased cardiac contraction, pericardial edema, and decreased tail blood flow) as assessed under light microscopy at 40 hours after treatment. Initial SAR experiments included modification of the tricyclic structure of C1, addition of a methoxy group to the middle phenyl ring, and modification of substituents on the pyrone ring.
Results
Determination of SAR in zebrafish.
Using the zebrafish DOX cardiotoxicity model to explore SAR, we began by assessing the contribution of the tricyclic aromatic structure of C1 to in vivo potency (Figure 1 and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.96753DS1). We discovered that removal of either the pyrone or furan rings (C2 and C3) abolished activity. Although C2 and C3 also lack additional substituents on the pyrone and benzene rings, these substituents were not essential to activity in subsequent studies (e.g., C11), highlighting the importance of maintaining the tricyclic structure as a scaffold for further modifications. We then turned our attention to the ring substituents on the chromone portion of C1. Addition of a methoxy group to the 9 position (C4) modestly improved activity. Movement of the carbonyl group from the 7 position to the 5 position (C5) resulted in a further improvement in potency, as did substitution of a thiocarbonyl in the 5 position. As C5 was commercially available, we chose to use it as a scaffold for subsequent SAR studies.
To further optimize C5, we started with modification of the furan moiety to determine the contribution of this ring to activity in zebrafish (Figure 2A). We first moved the furan ring to the [2,3-h] position and found that the resulting C7 had similar potency to C5. Removal of the furan ring and substitution of an additional methoxy group in the 7 position (C8) resulted in decreased activity relative to C5. Interestingly, removal of both the furan ring and the 4-methoxy group on the chromone core of C5 (C9) resulted in a complete loss of activity. We then studied reduction of the double bond of the furan moiety and discovered that this also resulted in a loss of activity (C10), suggesting that the aromaticity conferred by the furan ring was an important feature contributing to the activity of C5. Finally, we added hydrophobic bulk at various positions on the furan ring (C11–C13). Because these compounds were also lacking the 4-methoxy group on the chromone ring, we compared their activity to C14 and found that potency was further improved with the addition of these simple alkyl substituents. However, C11 and C13 demonstrated significant toxicity at concentrations in the low micromolar range.
Figure 2. Optimization of C5 and C7 through modification of the furan ring and methoxy group.

SAR experiments using C5 (A and B) and C7 (C) as scaffolds highlighted the importance of preserving the furan ring and methoxy group for in vivo activity.
We next focused our attention on the 4-methoxy group on the chromone core of C5 (Figure 2B). Removal of the methoxy group altogether (C14) decreased activity compared with C5, as did moving this group to the 9 position to generate C15. Hydrolysis of the methoxy group to yield C16 completely abolished activity. Given the improved potency we had observed with C4 compared with C1, we added a second methoxy group to the 9 position, though this did not significantly affect activity (C17). Similar maneuvers were performed using C7 as a scaffold, including removal of the methoxy group (C18), movement of the methoxy group from the 5 position to the 6 position (C19), and addition of a methoxy group to the 6 position (C20; Figure 2C). All of these maneuvers decreased activity relative to C7. Thus, the methoxy group and its location on the chromone core of C5–C7 appeared to be critical to these compounds’ potency.
Given the importance of the 4-methoxy group for in vivo activity, we subsequently explored whether modification of this area could alter the potency of C5 by attaching a variety of substituents to the pendant ether group (Figure 3). We began by attaching an isopropyl group (C21), resulting in a roughly 2-fold increase in potency compared with C5. Attachment of an epoxypropyl group (C22) further increased activity, resulting in protection against DOX cardiomyopathy at concentrations less than 100 nM. Notably, attachment of a bulky tert-butyl carbamate to yield C23 increased potency dramatically, resulting in EC50 values below 10 nM in the zebrafish model.
Figure 3. Optimization of C5 through a series of modifications of the pendant ether group.

The addition of short ester side chains led to development of cardioprotective compounds with 1,000-fold increased potency in zebrafish.
We subsequently used C23 as a scaffold to investigate SAR of the pendant group, beginning at the proximal end with modification of the alkyl chain. Removal of 1 carbon from the chain did not significantly change potency (C24). Incorporation of a carbonyl group into this area resulted in a complete loss of activity (C25), perhaps due to in vivo reduction to an alcohol resulting in C16. A similar loss of activity was observed with substitution of the alkyl chain with an acetyl group (C26), a carboxylic acid (C27), or an ethyl ester (C28). However, the addition of a tert-butyl ester (C29) restored activity at low nanomolar concentrations, highlighting the importance of incorporating bulk distal to the ester group. Addition of carbons to the alkyl chain of 29 to generate 30 and 31 further improved potency but also resulted in significant toxicity at micromolar concentrations.
We then examined the individual components of the carbamate group shared by C23 and C24. Use of an azetidine instead of an amine (C32) did not significantly affect potency, nor did the use of a carbamothioate group in place of the original carbamate (C33). We hypothesized that the presence of a hydrolyzable ester or thioester was important to the increased potency of C23 and C24, given the decrease in activity observed with compounds lacking the ester moiety (C34 and C35). To test this hypothesis, we synthesized C36 and found that replacement of the carbamate group with an ethyl amide resulted in a 100-fold loss of activity compared with C24. Replacement of the tert-butyl group at the distal end of the pendant chain with a bulky phenyl group (C37) did not significantly affect activity.
Finally, given the improved potency observed with substitution of a thiocarbonyl for the carbonyl group in the 5 position of the chromone (C6), we synthesized C38 using C6 as a scaffold attached to the pendant group of C24. We found that incorporation of the thiocarbonyl group did not improve potency compared with C24 (Supplemental Figure 2).
Assessment of efficacy of visnagin analogs in a mouse model of DOX cardiotoxicity.
We selected one of our most potent and least toxic visnagin analogs in zebrafish (C23; EC50 7 nM, TD50 > 10 μM) and tested this compound in a mouse model of acute DOX-induced cardiotoxicity. DOX fluorescence was assessed directly by fluorescence reflectance imaging. Inherent DOX fluorescence intensity increased linearly with increasing DOX concentration (Figure 4A). DOX uptake in the heart was, thus, quantifiable and allowed a threshold to be applied so that therapeutic impact was examined only in the hearts in which DOX retention, and hence the potential for toxicity, was confirmed (Figure 4B). To assess for cardiac apoptosis in mice, we performed ex vivo imaging of the myocardium using an annexin V conjugate with a near-infrared fluorochrome, Annexin-Vivo 750 (13), a technique with which we had previously shown suppression of DOX-induced apoptosis with C1 at a dose of 25 mg/kg (12). C23 significantly reduced Annexin-Vivo fluorescence in the mouse heart at concentrations as low as 0.1 mg/kg by i.p. injection (Figure 4, C and D). DOX retention in the heart was consistently high in all the cohorts analyzed (Figure 4E). The potency observed with C23 was, thus, 250-fold higher than previously reported with C1 in reducing DOX-induced cardiomyocyte apoptosis, consistent with the increased potency observed in our zebrafish assay.
Figure 4. C23 attenuates DOX-induced cardiac apoptosis in mice.

DOX uptake in the heart was assessed by reflectance fluorescence imaging, and the resultant apoptosis was simultaneously imaged with Anx-750, a near-infrared fluorescent annexin V probe. (A) DOX fluorescence in a phantom calibration study increased linearly with increasing concentration. (B) Representative ex vivo short-axis heart slices of mouse hearts showing differential DOX uptake 24 hours after a 15 mg/kg DOX injection. (C) Representative ex vivo images of Anx-750 accumulation showed marked reduction in Anx fluorescence after cotreatment with C23 at a dose of 2 mg/kg. A significant reduction in apoptosis was seen with all 3 doses of C23 tested. (D) Cardiac annexin uptake increased significantly in the DOX-only group (DOX+carrier) compared with the control mice injected with saline. Annexin uptake was reduced significantly with treatment with C23. C23 administered at doses of 0.1, 1, and 2 mg/kg all conferred a significant cardioprotective effect. Positive DOX uptake in the heart was confirmed by direct imaging. A total of 45 mice were included in the study as follows: 0.9% saline, i.p. injection (n = 7); 15 mg/kg DOX in saline, and carrier only, i.p. (n = 17); 15 mg/kg DOX in saline, and 0.1 mg/kg 23, i.p. (n = 7); 15 mg/kg DOX in saline, and 1 mg/kg 23, i.p. (n = 7); and 15 mg/kg DOX in saline, and 2 mg/kg 23, i.p. (n = 7). Data are represented as mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, 1-way ANOVA followed by the Tukey’s test. Carrier, 10% DMSO + 90% olive oil. (E) DOX retention in the heart was assessed by direct reflectance fluorescence imaging, and quantified as an increase in signal/noise ratio (SNR) compared with the control mice without DOX injection. No significant difference in DOX fluorescence increase was seen between the DOX only and any of the C23-treated mice. P > 0.05, 1-way ANOVA. Veh., 10% DMSO.
Determination of in vitro potency of visnagin analogs.
We also assessed the ability of select visnagin analogs to prevent DOX-induced cell death in cultured HL-1 cardiomyocytes (Supplemental Table 1). Interestingly, many compounds with increased potency in the zebrafish model of DOX cardiotoxicity were ineffective at doses up to 50 μM in cultured HL-1 cardiomyocytes. For instance, C23 had an EC50 of 7 nM in the zebrafish model and was cardioprotective at a dose of 0.1 mg/kg in the mouse model, but it was ineffective in the HL-1 cell culture model. This suggests that these analogs may be prodrugs that require in vivo metabolism in order to confer cardioprotection or that they are cardioprotective by exerting action at a location other than the cardiomyocyte itself.
Visnagin analogs inhibit DOX-mediated induction of CYP1 enzymes.
Using an unbiased proteomics approach, we measured 6,085 proteins in zebrafish treated with DOX with or without C1 (Supplemental Data). Fish treated with DOX alone demonstrated significant upregulation of the CYP1 family of enzymes (CYP1A, CYP1B1, and CYP1C1), whereas fish cotreated with DOX and C1 showed significantly less CYP1 induction (Table 1). This was in contrast with other proteins that were modulated to a similar extent in fish treated with DOX and were not changed by cotreatment with C1. For instance, levels of myosin heavy chain B (MYHB) were decreased in fish treated with DOX alone and in fish treated with DOX + C1. Downregulation of MYHB has been previously described with DOX treatment (14). Likewise, the inflammatory marker C-reactive protein 3 (CRP3) was increased in both DOX and DOX + C1–treated fish. These observations support the specificity of CYP1 downregulation by C1 and suggest that C1 does not confer cardioprotection by simply preventing DOX entry into the cell.
Table 1. Proteomics analysis of zebrafish treated with DOX, DOX + C1, or C1.
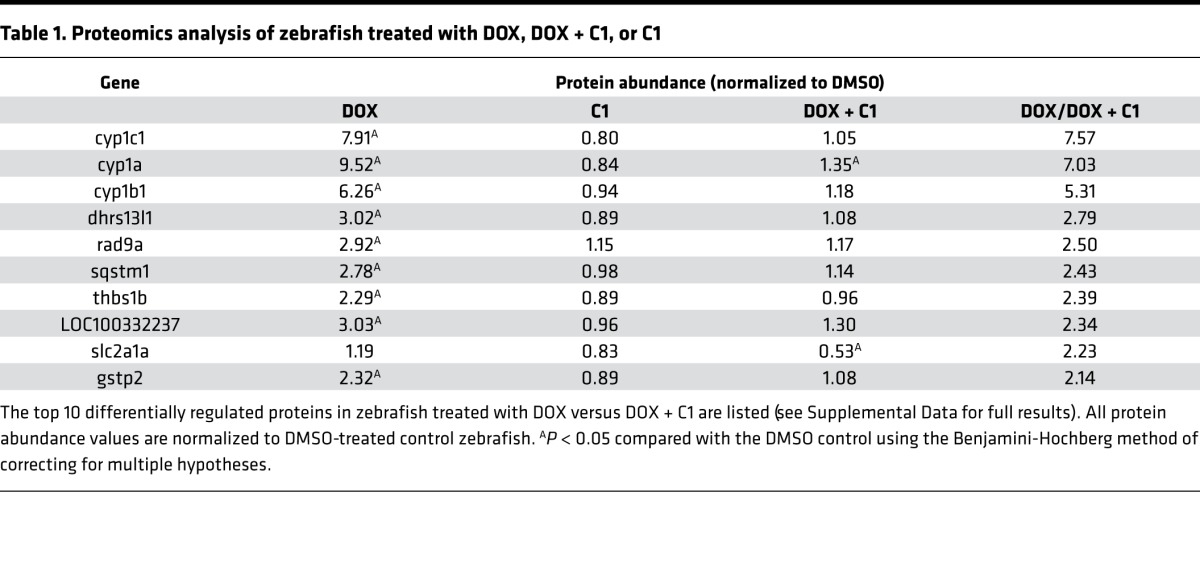
These findings were confirmed by assessing RNA and protein levels using quantitative PCR (qPCR) (Figure 5A) and Western blot with a zebrafish-specific antibody against CYP1A (Figure 5B). Notably, cotreatment with both C1 and C23 mitigated induction of CYP1A as assessed by Western blot as early as 18 hours after treatment (hpt), prior to the development of the cardiomyopathy phenotype at 42 hpt.
Figure 5. C1 and C23 inhibit induction of CYP1 enzymes by DOX.

(A) RNA levels of Cyp1a, Cyp1b1, and Cyp1c1 were increased in zebrafish treated with DOX. This effect was attenuated in zebrafish cotreated with C1 (10 μM) and C23 (10 μM), as measured by qPCR 18 hours after treatment (prior to the development of the cardiomyopathy phenotype). Expression levels were normalized to a housekeeping gene (β-actin or ornithine decarboxylase 1) and to control fish treated with DMSO. Data are depicted as mean ± SEM. (B) Western blotting of fish tissue lysate using a zebrafish-specific CYP1A antibody demonstrated induction of CYP1A by DOX and normalization by C1 (10 μM) and C23 (10 μM). RNA levels of Cyp1a (C) and Cyp1b1 (D) were normalized in zebrafish cotreated with DOX and either C1, C23, CYP1 inhibitors (CYP1i; α-NF and Pyr), or an AHR inhibitor (AHRi; CH) when compared with zebrafish treated with DOX alone. All cotreatments were administered at a concentration of 10 μM. α-NF, α-naphthoflavone; Pyr, pyrene; TM, 2,4,3’,5’-tetramethoxystilbene; CH, CH-233191. (E) Zebrafish cotreated with inhibitors of CYP1 and the AHR demonstrated increased percent rescue from the DOX cardiomyopathy phenotype (decreased cardiac contraction, pericardial edema, and decreased tail blood flow) as assessed under light microscopy at 40 hours after treatment. All inhibitors were administered at a dose of 10 μM. Student’s t test was performed, followed by Bonferroni correction for multiple hypotheses. (F) Percent rescue from the DOX cardiomyopathy phenotype in zebrafish cotreated with C1 (20 μM) and the AHR agonist methyl 2-(1H-indole-3-carbonyl)-1,3-thiazole-4-carboxylate (ITE). One-way ANOVA was performed, followed by the Tukey’s test. (G) Using CRISPR/Cas9-mediated mutagenesis, single-guide RNAs (sgRNAs) targeting Cyp1a were injected into zebrafish embryos to generate potential founder (F0) Cyp1-KO fish. Across this mosaic population, injected zebrafish demonstrated 40% knockdown of Cyp1a RNA levels as assessed by qPCR compared with uninjected zebrafish from the same clutch. (H) Zebrafish injected with sgRNAs targeting Cyp1a demonstrated nearly 50% rescue from DOX-induced cardiomyopathy. Data represent mean ± SEM. One-way ANOVA was performed, followed by the Tukey’s test. For all chemical treatments in zebrafish, data represent 12 fish treated per condition per experiment, and each experiment was repeated 3 times. *P < 0.05. **P < 0.01. ***P < 0.001.
As with other polycyclic aromatic hydrocarbons, treatment with DOX has been shown to activate the aryl hydrocarbon receptor (AHR), a transcriptional regulator that translocates to the nucleus to induce expression of the CYP1 family of enzymes (15). In line with these observations, we noted that zebrafish treated with DOX also demonstrated induction of DHRS13L1, THBS1B, and GSTP2, expression levels that have been previously reported to correlate with AHR activity (16–18). Thus, we hypothesized that potent analogs of C1 may serve as AHR antagonists. Using a cell line stably transfected with human AHR and the corresponding response elements (Puracyp), we found that both C1 and C23 inhibited induction of the AHR mediated by 2,3,7,8-tetrachlorodibenzodioxin (TCDD) at low micromolar concentrations (Table 2). Although these data suggest that C1 and C23 are AHR inhibitors, the increased potency of C23 does not appear to be related to more potent antagonism of the AHR as measured in vitro.
Table 2. Inhibition of human AHR activity in vitro.
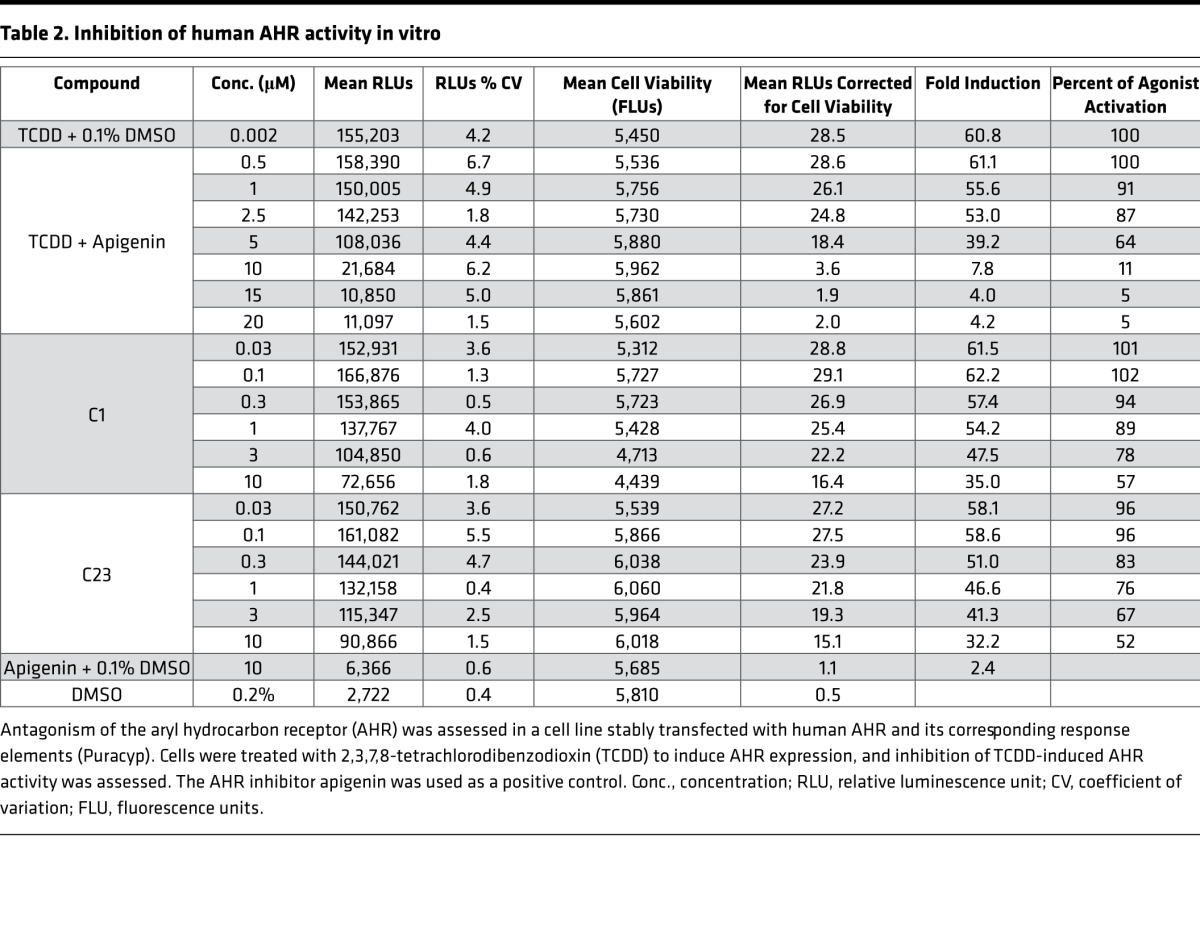
To assess whether inhibition of this pathway was responsible for cardioprotection, we treated fish with DOX and CYP1 family inhibitors α-naphthoflavone, pyrene, and 2,4,3’,5’-tetramethoxystilbene (TM) (19) or the AHR inhibitor CH-233191. As with cotreatment with C1 and C23, each inhibitor decreased DOX-induced CYP1 expression (Figure 5, C and D) and resulted in significant rescue from the cardiomyopathy phenotype (Figure 5E). Finally, rescue from DOX-induced cardiomyopathy mediated by C1 was abrogated when fish were also treated with the endogenous AHR agonist methyl 2-(1H-indole-3-carbonyl)-1,3-thiazole-4-carboxylate (ITE; Figure 5F) at nanomolar doses. ITE has been shown to serve as an agonist for the AHR across a range of species, from humans to zebrafish (20). Notably, ITE itself was not toxic when administered to zebrafish embryos without DOX at this range of concentrations, although toxicity was observed at micromolar doses.
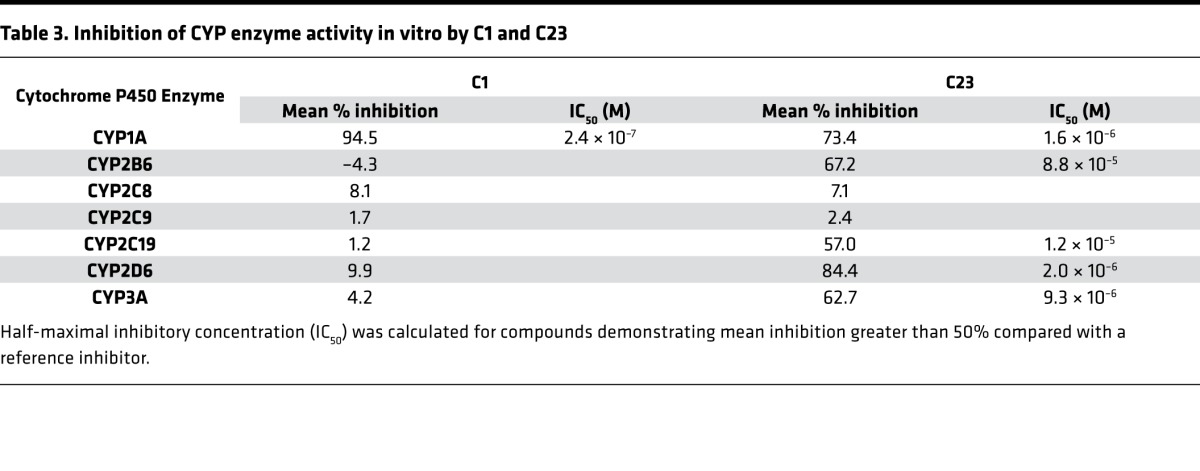
Given that furanocoumarins such as C1 and C23 have also been described to directly modulate CYP enzymatic activity, we assessed the ability of these compounds to inhibit a panel of CYP enzymes in vitro using a series of human liver microsome assays. C23 inhibited a broad range of CYP enzymes at micromolar concentrations, although C1 was a more potent inhibitor of CYP1A (Table 3). This shared inhibitory activity against CYP1A supports the potential role of CYP1 enzymes in cardioprotection mediated by C1 and C23. However, these data also suggest that the increased potency of C23 as a cardioprotectant does not arise solely from direct inhibition of enzyme activity. In line with this observation, C23 was significantly more potent than other previously published CYP1 inhibitors in the zebrafish DOX model, including α-naphthoflavone (EC50 293 nM), pyrene (EC50 3.7 μM), and TM (EC50 8.6 μM).
Table 3. Inhibition of CYP enzyme activity in vitro by C1 and C23.
Because chemical modulators of CYP1 and AHR activity can have nonspecific or off-target effects, we began to investigate the impact of genetic knockdown of this pathway on the development of DOX cardiomyopathy. Using CRISPR/Cas9-mediated mutagenesis, single-guide RNAs (sgRNAs) targeting cyp1a were injected into zebrafish embryos to generate potential founder (F0) Cyp1-KO fish. Across this mosaic population, injected zebrafish demonstrated 40% knockdown of cyp1a RNA levels and nearly 50% rescue from DOX-induced cardiomyopathy (Figure 5, G and H), confirming that Cyp1a expression is necessary for the development of cardiomyopathy in this model.
As oxidative stress has been reported as a key mechanism of DOX-induced cardiotoxicity, we previously assessed hydrogen peroxide (H2O2) levels in cultured cardiomyocytes (12). DOX treatment was associated with an increase in H2O2 levels that was not affected by the addition of C1. To assess whether C1 and its analogs modulate other pathways associated with DOX cardiotoxicity, we measured RNA levels of topoisomerase 2β (Top2B), the mitochondrial iron transporters solute carrier family 25 member 28 (Slc25a28, also known as mitoferrin-2), and ATP-binding cassette sub-family B (MDR/TAP) member 8 (Abcb8). Neither C1 nor C23 significantly modulated expression levels of these genes in zebrafish treated with DOX (Supplemental Figure 3). Finally, despite prior literature implicating mitochondrial dysfunction as a mediator of DOX cardiotoxicity, we did not observe any significant change in metabolites involved in mitochondrial function, such as the TCA cycle metabolites, in zebrafish cotreated with DOX and analogs of C1 (data not shown).
Discussion
In this study, we pursued serial optimization of the plant derivative visnagin (C1), using the effects of structural modifications on cardioprotective activity in zebrafish to guide our synthetic strategy. SAR studies have traditionally been performed in vitro, given the time and cost associated with testing a large number of compounds at a wide range of doses. Binding assays are typically employed to determine the affinity of a small molecule for a specific target. However, it can be challenging to replicate the complexity of the in vivo milieu in in vitro systems, limiting the translation of compounds identified in SAR studies to clinically meaningful therapeutics. The zebrafish model allows for rapid phenotypic screening to simultaneously assess drug potency, metabolism, and toxicity at the level of the whole organism. By using the end phenotype rather than binding affinity to determine efficacy, highly potent compounds can be identified in a target-agnostic fashion. Accordingly, in vivo determination of SAR has the potential to increase the biological relevance of compounds discovered through these assays.
Using the zebrafish model of DOX cardiotoxicity, we demonstrated that the addition of a side chain containing a hydrolyzable carbamate group increased the potency of visnagin up to 1,000-fold without evidence of toxicity. Carbamates are frequently used in the synthesis of prodrugs due to their stability during systemic absorption and first-pass metabolism, thereby increasing permeability across cell membranes (21). We hypothesize that the incorporation of a carbamate group increases delivery of visnagin analogs to their site of action, where esterases then participate in bioconversion and release of the active metabolite. Future studies will seek to characterize the metabolism of potent visnagin analogs in detail to enable further optimization of these compounds.
Importantly, one member of this family of analogs (C23) protected against cardiotoxicity at a concentration of 0.1 mg/kg in adult mice treated with DOX 15 mg/kg. The currently available cardioprotectant, dexrazoxane, is typically prescribed to patients undergoing treatment with DOX at a ratio of 10:1 (dexrazoxane/DOX) based on the dose required to significantly reduce the incidence of DOX-induced cardiomyopathy in mice (22, 23). Currently, dexrazoxane is approved in the US only for adult patients with advanced breast cancer who require more than 300 mg/m2 of DOX, although patients can develop cardiotoxicity at lower doses (24). Based on the concern for precipitating cardiotoxicity, patients may not receive the anthracycline dose necessary for optimal treatment of their cancer. Therefore, there is a clinical need for potent, selective cardioprotective agents that can be administered to a broader population of patients at the time of DOX therapy.
We previously reported that C1 binds to mitochondrial malate dehydrogenase (MDH2) and postulated that modulation of MDH2 may be responsible for the cardioprotective effect of C1 (12). However, we have found manipulation of this pathway for therapeutic purposes to be challenging, given the importance of MDH2 in maintaining normal mitochondrial metabolism and bioenergetics. As such, subsequent work has focused on identifying additional and potentially complementary mechanisms that may explain the cardioprotection conferred by C1 and its analogs.
Through serial modifications of the chemical structure of C1, we identified the furanocoumarin core as being essential to its cardioprotective activity. The furanocoumarin class of compounds are well-characterized inhibitors of cytochrome P450 and, in particular, CYP family 1 (19). In zebrafish, DOX treatment significantly increased levels of CYP1 enzymes, an effect that was reduced by C1 and completely abolished by C23. Moreover, 3 structurally different CYP1 inhibitors, an AHR inhibitor, and genetic knockdown of Cyp1a all protected against DOX cardiotoxicity in our zebrafish model. Of note, inhibitors of other CYP enzymes (e.g., CYP3A4) did not confer cardioprotection in our model (data not shown). These observations suggest that C1 and its potent structural analogs protect against cardiotoxicity by suppressing DOX-mediated CYP1 induction, in addition to direct inhibition of CYP1 enzyme activity. This effect may be related to inhibition of the AHR but does not seem to explain C23’s increased potency in our initial in vitro studies using the well-characterized AHR agonist TCDD. The functional activity of the AHR has been reported to vary based on the specific ligand to which it is bound (25), and it is possible that C23 is a more potent inhibitor of the AHR when activated by DOX rather than by TCDD. The effect of C23 on inhibition of DOX-mediated CYP1 induction may also occur through a mechanism that is independent of direct AHR antagonism, for instance through degradation of the AHR, inhibition of the AHR nuclear translocator, or direct effects on CYP1 transcription. Finally, local accumulation or bioconversion of C23 may also contribute to its increased potency in vivo. Future studies will focus on clarifying these mechanisms.
CYP1 is a family of highly conserved monooxygenases responsible for the metabolism of environmental toxicants, including polycyclic aromatic hydrocarbons similar in structure to DOX (26). One possibility is that C1 and its analogs modulate the metabolism of DOX itself through previously uncharacterized pathways. CYP1 enzymes also play an important role in the metabolism of endogenous bioactive polyunsaturated fatty acids (27), such as the hydroxyeicosatetraenoic acids (HETEs). HETEs are arachidonic acid metabolites that have been previously implicated in the pathogenesis of DOX cardiotoxicity (28) and other cardiovascular diseases such as hypertension (29). It is possible that CYP1 inhibitors such as C1 and its analogs prevent the generation of cardiotoxic metabolites such as those downstream of DOX or the HETEs in order to protect against DOX cardiomyopathy. Human and mouse CYP1 enzymes are divided into 2 subfamilies: 1A1/1A2 and 1B1. Zebrafish CYP1 enzymes are divided into 4 subfamilies, 2 of which are homologous to those found in mammalian species: 1A, 1B1, 1C1/1C2 (may complement 1B1 activity in fish), and 1D1 (pseudogene in humans) (30). Although our initial genetic knockdown model suggests that Cyp1a is necessary for the development of cardiotoxicity, future studies will focus on the development of stable transgenic lines in zebrafish to address the role of each CYP1 family member and identify potential downstream metabolites that may contribute to the pathogenesis of DOX-induced cardiotoxicity.
Interestingly, CYP1 may also play a role in estrogen-mediated tumor formation (31). Although all CYP1 family members are expressed in extrahepatic tissues, CYP1B1 in particular is overexpressed in a wide range of tumors including breast cancer (32). Notably, CYP1B1 catalyzes C-4 hydroxylation of estradiol, a process that has been implicated in estrogen-related tumorigenesis (33). These observations suggest that CYP1 inhibition may simultaneously confer cardioprotection while enhancing the antitumor effect of DOX, increasing its appeal as a therapeutic strategy.
To further assess whether our findings can be translated to patients receiving DOX, future studies will aim to determine the efficacy of potent analogs of C1 in the setting of chronic anthracycline exposure in mammalian models, which more accurately simulates chemotherapy regimens administered to patients. In parallel, ongoing efforts seek to define downstream mediators of cardioprotection conferred by CYP1 inhibition and to identify ways in which the CYP1 pathway could be modulated to protect against cardiotoxicity in the clinical setting.
Methods
Chemistry
Procedures for small molecule synthesis are described in the Supplemental Methods.
Reagents
Commercially available compounds were purchased from MilliporeSigma unless otherwise specified.
Zebrafish DOX model
TuAB zebrafish embryos at 30 hours after fertilization were treated with DOX 100 μM in 96-well plates as previously described (12). Visnagin analogs were dissolved in DMSO and added at concentrations ranging from 3 nM–10 μM (n = 6 fish per dose; < 1% DMSO v/v). The cardiomyopathy phenotype (decreased cardiac contraction, pericardial edema, and decreased tail blood flow) was assessed under light microscopy (5× magnification) at 40 hpt. Percent rescue from the cardiomyopathy phenotype was calculated for each concentration tested. Zebrafish were considered to be rescued from DOX-induced cardiomyopathy if all 3 features of the phenotype were absent. For those compounds demonstrating activity in zebrafish, each dose-response experiment was repeated 3 times.
Mouse annexin model
DOX was dissolved in 0.9% normal saline and administered at a dose of 15 mg/kg to adult male C57/Bl6 mice (8–10 weeks of age) via i.p. injection. We previously showed that this regimen causes acute cardiotoxicity manifest by a decline in fractional shortening and decreased strain rate as assessed by echocardiography within 5 days after treatment, effects that are ameliorated by cotreatment with C1 (12). For the treatment arm, C23 dissolved in 10% DMSO and 90% olive oil (carrier) or carrier alone was administered via i.p. injection on the contralateral side.
A total of 45 mice were included in the study. Mice cohorts imaged were: 0.9% saline, i.p. injection (n = 7); 15 mg/kg DOX in saline, and carrier only, i.p. (n = 17); 15 mg/kg DOX in saline and 0.1 mg/kg C23, i.p. (n = 7); 15 mg/kg DOX in saline, and 1 mg/kg C23, i.p. (n = 7); and 15 mg/kg DOX in saline, and 2 mg/kg C23, i.p. (n = 7).
Twenty-two hours after DOX and treatment injections, Anx-750 (Perkin Elmer) was injected i.v. according to manufacturer’s instructions. After 2 hours of probe circulation, mice were euthanized with isoflurane. Hearts were excised, sectioned into 1-mm slices along the short axis, and imaged on a commercial imaging system (IVIS spectrum, Perkin Elmer) to assess apoptosis as previously described (13).
Fluorescence reflectance imaging.
DOX fluorescence imaging was carried out on IVIS spectrum with the filter setting 500 nm excitation/600 nm emission, 30-second exposure, with 135 μm in-plane resolution. A series of DOX phantoms, ranging from 0–0.03 mM was prepared in saline and imaged on the IVIS spectrum with identical settings. Anx-750 fluorescence was detected with the filter setting 745 nm excitation/800 nm emission, 30-second exposure, and 135 μm resolution.
Image quantification and analysis.
Annexin and DOX fluorescence signal was quantified and analyzed in ImageJ (NIH). For each heart, fluorescence intensity was averaged across 4–6 slices. Annexin uptake was normalized to the saline-injected control animals. DOX fluorescence in the heart was quantified by calculating the signal/noise ratio (SNR; mean fluorescence intensityheart/standard deviationair).
Cultured HL-1 cardiomyocyte model
The cardiomyocyte cell line HL-1 derived from mouse atrial tumor was a gift from W. Claycomb (Louisiana State University School of Medicine, New Orleans, Louisiana, USA). Cells were cultured as previously published (34) and treated with DOX 5 μM with or without visnagin analogs at a concentration up to 50 μM. Viability was assessed after 18 hours of using a CellTiter-Glo Luminescent assay from Promega. Each dose-response experiment was repeated 3 times.
Zebrafish proteomics
Zebrafish embryos were treated with DMSO, DOX, DOX + C1 20 μM, or C1 20 μM as described. One-hundred five embryos were treated per condition and lysed in ultrapure water at 40 hpt. Two sets of biological replicates were assessed. Proteomics analyses were performed by OmicScouts in collaboration with Merck. Following protein digestion, peptides from each treatment condition were labeled using isobaric tandem mass tags (TMT10-plex) followed by fractionation and analysis by liquid chromatography–tandem mass spectrometry (LC-MS/MS) using a Thermo Scientific Exactive hybrid quadrupole-Orbitrap mass spectrometer with a 120-minute gradient. Peptide sequences (55,444 sequences)corresponding to 7,090 unique proteins were identified, and 6,085 proteins were quantified using MaxQuant software (35, 36).
Zebrafish cytochrome P450 family 1 (CYP1) experiments
For qPCR, zebrafish embryos were treated as described and lysed in RNAzol RT at 40 hpt for isolation of RNA. cDNA was synthesized using the QuantiTect Reverse Transcription Kit from Qiagen. PCR primers for zebrafish Cyp1a and Cyp1b1 were used as previously published (37). PCR primers for Top2B, Slc25a28, and Abcb8 were as follows: (Top2B forward) 5′ - GGGTGAGATAGATGCGGCTGTG - 3′, (Top2B reverse) 5′ - TTTGGGTTTGGCTCCTGGGT - 3′, (Slc25a28 forward) 5′ - TGAGGGCGTTTCCGTGTTCC - 3′, (Slc25a28 reverse) 5′ - TCCCACGCCCGATGTTTACG - 3′, (Abcb8 forward) 5′ - TTGGTCGCCTCGCAAACAGT - 3′, and (Abcb8 reverse) 5′ - AGTGAGCGGAACAGAGGGGT - 3′. All results were normalized to a housekeeping gene (β-actin or ornithine decarboxylase 1). For Western blotting, zebrafish were treated as described and homogenized in lysis buffer (10 mM Tris [pH 7.4]; 150 mM NaCl; 1 mM CaCl2; 1% Triton X-100; supplemented with protease inhibitor cocktail and phosphatase inhibitor; MilliporeSigma) at 40 hpt using a motorized pestle. The homogenate was centrifuged for 10 minutes at 14,000 g at 4°C, and protein concentration of the supernatant was assessed using a Pierce BCA Protein Assay Kit from ThermoFisher Scientific. Following SDS-PAGE, proteins were transferred to a polyvinylidene difluoride membrane (Bio-Rad), blocked with 5% dry milk, and incubated with zebrafish-specific anti-CYP1A1 antibody (Abcam, catalog ab209824) at 4°C overnight. Immunodetection was performed using a mouse anti-rabbit IgG light-chain specific secondary antibody (Jackson ImmunoResearch) and Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare Life Sciences). To generate potential founder (F0) cyp1-KO fish, CRISPR/Cas9-mediated mutagenesis was performed as previously published (38). Five sgRNAs were multiplexed to target the following sites in the zebrafish Cyp1a gene: 5′ - CGATGAGTTCGGGAAGATCG; 5′ - GCGGGATCTGTTTCGGACGC - 3′; 5′ - GAATGGATCAAAGCTTCCAT - 3′; 5′ - GCCCGTCTGGGATTTTCGTG - 3′; 5′ - CGACAGGCGCTCCTAAAACA - 3′. The overall percent of gene knockdown across the injected fish was assessed using qPCR as described above.
AHR inhibition assays
AHR inhibition experiments were performed by Puracyp Services using 1A2-DRE cells, a cell line stably transfected with human AHR and its response elements. Twenty-four hours after seeding the cells on a 96-well plate, they were pretreated with 6 concentrations of C1 and C23, as well as 6 concentrations of the positive control apigenin. Six hours after pretreatment, all cells were treated with 2 nM TCDD (AHR agonist). Negative controls were treated with DMSO alone. Cells were incubated for 24 hours, after which the Cell Titer-Fluor assay (Promega) was used to assess cell viability. The ONE-Glo luciferase assay (Promega) was then performed to assess reporter gene activity.
CYP inhibition assays
CYP inhibition experiments were performed by Cerep Panlabs (Eurofins) using human liver microsomes and the following substrates: phenacetin, 10 μM (CYP1A); buproprion, 100 μM (CYP2B6); paclitaxel, 10 μM (CYP2C8); diclofenac, 10 μM (CYP 2C9); omeprazole, 0.5 μM (CYP2C19); dextromethorphan, 5 μM (CYP2D6); and testosterone, 50 μM (CYP3A). Metabolites of each substrate were measured using LC-MS/MS. Mean percent inhibition was calculated using the following reference inhibitors: furafylline (CYP1A), clopidogrel (CYP2B6), montelukast (CYP2C8), sulfaphenazole (CYP2C9), oxybutynin (CYP2C19), quinidine (CYP2D6), and ketoconazole (CYP3A). For compounds demonstrating mean inhibition greater than 50%, a full dose-response experiment was performed to calculate the half-maximal inhibitory concentration (IC50). All concentrations were tested in duplicate to obtain the final results.
Statistics
For small molecules tested in the zebrafish DOX model, dose-response curves were constructed using Prism (GraphPad). Raw data representing percent rescue at each dose were log-transformed, and EC50 values were calculated using nonlinear regression analysis. The TD50 was defined for each compound by assessing for pericardial edema, nonspecific deformities, and death in the absence of DOX treatment (data not shown). Concentrations at and above the TD50 were excluded from dose-response curves. For the mouse annexin model, a threshold of 2 SDs above the saline injected control animals was applied to all DOX-injected animals, with or without therapeutics. Only animals that met the threshold (thus, DOX-positive hearts) were included in the analysis. For mouse Annexin experiments and for zebrafish experiments testing 3 chemical treatments simultaneously, 1-way ANOVA was performed followed by the Tukey’s test. For zebrafish experiments testing more than 3 chemical treatments, the Bonferroni method was used to correct for multiple hypotheses. For zebrafish proteomics, the Benjamini-Hochberg method was used to correct for multiple hypotheses. For all experiments, P < 0.05 was considered statistically significant. All statistical analysis was performed in either Prism (Graphpad) or in R.
Study approval
All zebrafish and mouse experiments were reviewed and approved by the Massachusetts General Hospital (MGH) IACUC.
Author contributions
AA, YW, YL, BZ, HHC, DES, and RTP designed the experiments, interpreted the data, and wrote the paper. AA and YL performed zebrafish, cultured cell, and mouse experiments. HHC and DES performed mouse experiments, including imaging acquisition and analysis. YW and BZ synthesized the compounds. AC, ICT, HW, and DGJ performed proteomics analysis. AV contributed to zebrafish CYP1 experiments. All authors reviewed and revised the manuscript for intellectual content.
Supplementary Material
Acknowledgments
This work was supported in part by grants from Merck and the American Heart Association. AA received funding from the John S. LaDue Memorial Fellowship (Harvard Medical School), the Eleanor and Miles Shore 50th Anniversary Fellowship (Massachuetts General Hospital and Harvard Medical School), and the Sarnoff Scholar Award (Sarnoff Cardiovascular Research Foundation).
Version 1. 01/11/2018
Electronic publication
Footnotes
Conflict of interest: AA, YW, YL, BZ, HHC, DES, and RTP have applied for a patent on the compounds described in the manuscript.
Reference information: JCI Insight. 2018;3(1):e96753. https://doi.org/10.1172/jci.insight.96753.
Contributor Information
Aarti Asnani, Email: aasnani@bidmc.harvard.edu.
Yan Liu, Email: yanliu2001@gmail.com.
You Wang, Email: ywangcpu@hotmail.com.
Anita Vohra, Email: avohra@bidmc.harvard.edu.
An Chi, Email: an_chi@merck.com.
Ivan Cornella-Taracido, Email: ivan.cornella.taracido@merck.com.
Huijun Wang, Email: huijun.wang@merck.com.
Douglas G. Johns, Email: douglas_johns@merck.com.
References
- 1.Suliman HB, Carraway MS, Ali AS, Reynolds CM, Welty-Wolf KE, Piantadosi CA. The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J Clin Invest. 2007;117(12):3730–3741. doi: 10.1172/JCI32967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace KB. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol Toxicol. 2003;93(3):105–115. doi: 10.1034/j.1600-0773.2003.930301.x. [DOI] [PubMed] [Google Scholar]
- 3.Zhou S, Starkov A, Froberg MK, Leino RL, Wallace KB. Cumulative and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res. 2001;61(2):771–777. [PubMed] [Google Scholar]
- 4.Hasinoff BB, Herman EH. Dexrazoxane: how it works in cardiac and tumor cells. Is it a prodrug or is it a drug? Cardiovasc Toxicol. 2007;7(2):140–144. doi: 10.1007/s12012-007-0023-3. [DOI] [PubMed] [Google Scholar]
- 5.Ichikawa Y, et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. 2014;124(2):617–630. doi: 10.1172/JCI72931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyu YL, et al. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007;67(18):8839–8846. doi: 10.1158/0008-5472.CAN-07-1649. [DOI] [PubMed] [Google Scholar]
- 7.Zhang S, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18(11):1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 8.Asselin BL, et al. Cardioprotection and Safety of Dexrazoxane in Patients Treated for Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia or Advanced-Stage Lymphoblastic Non-Hodgkin Lymphoma: A Report of the Children’s Oncology Group Randomized Trial Pediatric Oncology Group 9404. J Clin Oncol. 2016;34(8):854–862. doi: 10.1200/JCO.2015.60.8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seif AE, et al. Dexrazoxane exposure and risk of secondary acute myeloid leukemia in pediatric oncology patients. Pediatr Blood Cancer. 2015;62(4):704–709. doi: 10.1002/pbc.25043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swain SM, et al. Cardioprotection with dexrazoxane for doxorubicin-containing therapy in advanced breast cancer. J Clin Oncol. 1997;15(4):1318–1332. doi: 10.1200/JCO.1997.15.4.1318. [DOI] [PubMed] [Google Scholar]
- 11.Tebbi CK, et al. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin’s disease. J Clin Oncol. 2007;25(5):493–500. doi: 10.1200/JCO.2005.02.3879. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, et al. Visnagin protects against doxorubicin-induced cardiomyopathy through modulation of mitochondrial malate dehydrogenase. Sci Transl Med. 2014;6(266):266ra170. doi: 10.1126/scitranslmed.3010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen HH, Josephson L, Sosnovik DE. Imaging of apoptosis in the heart with nanoparticle technology. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2011;3(1):86–99. doi: 10.1002/wnan.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ito H, et al. Doxorubicin selectively inhibits muscle gene expression in cardiac muscle cells in vivo and in vitro. Proc Natl Acad Sci USA. 1990;87(11):4275–4279. doi: 10.1073/pnas.87.11.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volkova M, Palmeri M, Russell KS, Russell RR. Activation of the aryl hydrocarbon receptor by doxorubicin mediates cytoprotective effects in the heart. Cardiovasc Res. 2011;90(2):305–314. doi: 10.1093/cvr/cvr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dabir P, Marinic TE, Krukovets I, Stenina OI. Aryl hydrocarbon receptor is activated by glucose and regulates the thrombospondin-1 gene promoter in endothelial cells. Circ Res. 2008;102(12):1558–1565. doi: 10.1161/CIRCRESAHA.108.176990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodale BC, et al. Ligand-Specific Transcriptional Mechanisms Underlie Aryl Hydrocarbon Receptor-Mediated Developmental Toxicity of Oxygenated PAHs. Toxicol Sci. 2015;147(2):397–411. doi: 10.1093/toxsci/kfv139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Tiem LA, Di Giulio RT. AHR2 knockdown prevents PAH-mediated cardiac toxicity and XRE- and ARE-associated gene induction in zebrafish (Danio rerio) Toxicol Appl Pharmacol. 2011;254(3):280–287. doi: 10.1016/j.taap.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Sridhar J, Foroozesh M. Cytochrome P450 family 1 inhibitors and structure-activity relationships. Molecules. 2013;18(12):14470–14495. doi: 10.3390/molecules181214470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song J, et al. A ligand for the aryl hydrocarbon receptor isolated from lung. Proc Natl Acad Sci USA. 2002;99(23):14694–14699. doi: 10.1073/pnas.232562899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosh AK, Brindisi M. Organic carbamates in drug design and medicinal chemistry. J Med Chem. 2015;58(7):2895–2940. doi: 10.1021/jm501371s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imondi AR, et al. Dose-response relationship of dexrazoxane for prevention of doxorubicin-induced cardiotoxicity in mice, rats, and dogs. Cancer Res. 1996;56(18):4200–4204. [PubMed] [Google Scholar]
- 23.Imondi AR. Preclinical models of cardiac protection and testing for effects of dexrazoxane on doxorubicin antitumor effects. Semin Oncol. 1998;25(4 Suppl 10):22–30. [PubMed] [Google Scholar]
- 24.Mitani I, Jain D, Joska TM, Burtness B, Zaret BL. Doxorubicin cardiotoxicity: prevention of congestive heart failure with serial cardiac function monitoring with equilibrium radionuclide angiocardiography in the current era. J Nucl Cardiol. 2003;10(2):132–139. doi: 10.1067/mnc.2003.7. [DOI] [PubMed] [Google Scholar]
- 25.Soshilov AA, Denison MS. Ligand promiscuity of aryl hydrocarbon receptor agonists and antagonists revealed by site-directed mutagenesis. Mol Cell Biol. 2014;34(9):1707–1719. doi: 10.1128/MCB.01183-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimada T, Fujii-Kuriyama Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci. 2004;95(1):1–6. doi: 10.1111/j.1349-7006.2004.tb03162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Divanovic S, et al. Contributions of the three CYP1 monooxygenases to pro-inflammatory and inflammation-resolution lipid mediator pathways. J Immunol. 2013;191(6):3347–3357. doi: 10.4049/jimmunol.1300699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maayah ZH, Althurwi HN, Abdelhamid G, Lesyk G, Jurasz P, El-Kadi AO. CYP1B1 inhibition attenuates doxorubicin-induced cardiotoxicity through a mid-chain HETEs-dependent mechanism. Pharmacol Res. 2016;105:28–43. doi: 10.1016/j.phrs.2015.12.016. [DOI] [PubMed] [Google Scholar]
- 29.Jenkins CM, Cedars A, Gross RW. Eicosanoid signalling pathways in the heart. Cardiovasc Res. 2009;82(2):240–249. doi: 10.1093/cvr/cvn346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldstone JV, et al. Identification and developmental expression of the full complement of Cytochrome P450 genes in Zebrafish. BMC Genomics. 2010;11:643. doi: 10.1186/1471-2164-11-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruno RD, Njar VC. Targeting cytochrome P450 enzymes: a new approach in anti-cancer drug development. Bioorg Med Chem. 2007;15(15):5047–5060. doi: 10.1016/j.bmc.2007.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murray GI, et al. Tumor-specific expression of cytochrome P450 CYP1B1. Cancer Res. 1997;57(14):3026–3031. [PubMed] [Google Scholar]
- 33.Liehr JG, Ricci MJ. 4-Hydroxylation of estrogens as marker of human mammary tumors. Proc Natl Acad Sci USA. 1996;93(8):3294–3296. doi: 10.1073/pnas.93.8.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Claycomb WC, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA. 1998;95(6):2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alli Shaik A, et al. Functional mapping of the zebrafish early embryo proteome and transcriptome. J Proteome Res. 2014;13(12):5536–5550. doi: 10.1021/pr5005136. [DOI] [PubMed] [Google Scholar]
- 36.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 37.Jönsson ME, Orrego R, Woodin BR, Goldstone JV, Stegeman JJ. Basal and 3,3’,4,4’,5-pentachlorobiphenyl-induced expression of cytochrome P450 1A, 1B and 1C genes in zebrafish. Toxicol Appl Pharmacol. 2007;221(1):29–41. doi: 10.1016/j.taap.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gagnon JA, et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One. 2014;9(5):e98186. doi: 10.1371/journal.pone.0098186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.