

Abstract
Vertebrates possess 2 proteins with vitamin K oxidoreductase (VKOR) activity: VKORC1, whose vitamin K reduction supports vitamin K–dependent (VKD) protein carboxylation, and VKORC1-like 1 (VKORC1L1), whose function is unknown. VKD proteins include liver-derived coagulation factors, and hemorrhaging and lethality were previously observed in mice lacking either VKORC1 or the γ-glutamyl carboxylase (GGCX) that modifies VKD proteins. Vkorc1–/– mice survived longer (1 week) than Ggcx–/– mice (midembryogenesis or birth), and we assessed whether VKORC1L1 could account for this difference. We found that Vkorc1–/–;Vkorc1l1–/– mice died at birth with severe hemorrhaging, indicating that VKORC1L1 supports carboxylation during the pre- and perinatal periods. Additional studies showed that only VKORC1 sustains hemostasis beyond P7. VKORC1 expression and VKOR activity increased during late embryogenesis and following birth, while VKORC1L1 expression was unchanged. At P0, most (>99%) VKOR activity was due to VKORC1. Prothrombin mRNA, protein, and carboxylation also increased during this period, as did mRNA levels of coagulation factors encoding genes F7, F9, and F10. VKORC1L1 levels in Vkorc1–/– mouse liver may therefore be insufficient for supporting carboxylation beyond day 7. In support of this conclusion, VKORC1L1 overexpression in liver rescued carboxylation and hemostasis in adult Vkorc1–/– mice. These findings establish that VKORC1L1 supports VKD protein carboxylation in vivo.
Keywords: Development, Hematology
Keywords: Coagulation, Mouse models, Thrombin
The VKORC1 paralog, VKORC1L1, supports vitamin K oxidoreduction, γ-carboxylation, and hemostasis in vivo during the pre- and perinatal periods.
Introduction
Vitamin K–dependent (VKD) proteins undergo γ-glutamyl carboxylation characterized by the conversion of specific glutamic acid (Glu) residues to γ-carboxylated Glu (Gla) (reviewed in ref. 1). Carboxylation is essential for the functions of VKD proteins, which include coagulation factors like prothrombin (PT) and factors VII, IX, and X that are produced in the liver. VKD proteins are modified in the ER by the γ-glutamyl carboxylase (GGCX), which requires the vitamin K hydroquinone (KH2) cofactor that is oxygenated to vitamin K 2,3-epoxide (KO) during carboxylation. KO is then recycled by VKORC1, a vitamin K oxidoreductase (VKOR) (2, 3) that is evolutionarily conserved (4). VKORC1 recycles KO to KH2 in 2 steps, wherein KO is first converted to vitamin K quinone (K) and then to KH2 (5). Together, the enzymatic activities of GGCX and VKORC1 form the vitamin K cycle (1). Warfarin, an anticoagulant used by millions of people, suppresses VKD protein carboxylation by inhibiting VKORC1 (3, 6).
Deletion of either Ggcx or Vkorc1 in mice resulted in perinatal or postnatal death caused by bleeding defects (7–9). However, while the Ggcx–/– mice died during midembryogenesis or immediately after birth (P0) (8), most of the Vkorc1–/– mice survived for more than a week following birth (7), suggesting that another gene might compensate for the absence of Vkorc1. All vertebrates possess a single GGCX but have 2 VKORs. VKORC1, and a paralog named VKORC1-like 1 (VKORC1L1) (2), are broadly expressed in tissues (9–11). While VKORC1 is known to support the carboxylation of VKD proteins (3), which are also broadly expressed (1), the role of VKORC1L1 is unknown. VKORC1L1 was shown to reduce KO to K in vitro and to support carboxylation in cells (12, 13). Therefore, we tested whether VKORC1L1 has an in vivo role in VKD protein carboxylation.
Results
Carboxylated VKD proteins are present in Vkorc1–/– mice around birth.
PT carboxylation in WT and Vkorc1–/– mice was monitored to assess the impact of VKORC1 on VKD protein carboxylation. We first established an assay involving immunoprecipitation of carboxylated proteins using rabbit α-Gla antibody, followed by Western blot analysis with appropriate antibodies. The specificity of the α-Gla antibody was validated by testing several VKD proteins that were carboxylated or uncarboxylated (Supplemental Figure 1, A and B; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.96501DS1). When used in the analysis of plasma samples, we found that carboxylated PT was present in the circulation of WT mice at P0 but was reduced and undetectable in Vkorc1–/– mice at P0 and P7, respectively (Figure 1A). Analysis in liver samples showed that PT carboxylation in Vkorc1–/– mice was similar to that of WT mice at embryonic day 18.5 (e18.5) but was reduced at P0 and nearly absent at P7 (Figure 1B). Liver samples were also monitored for the carboxylation of GGCX, which is a VKD protein (14). GGCX carboxylation was reduced as early as e18.5 and was undetectable at P7 using the immunoprecipitation assay. In agreement with this and previous studies in cells (14, 15), we also observed in total liver extracts (i.e., input) that GGCX migrates faster in the absence of Vkorc1 because of its incomplete carboxylation. Therefore, carboxylation is reduced in Vkorc1–/– mice between the pre- and postnatal period but is not completely blunted, suggesting that a second enzyme supports KO reduction during these periods.
Figure 1. Carboxylated vitamin K–dependent (VKD) proteins are present in Vkorc1–/– mice at birth, and hemizygosity at the Vkorc1l1 locus causes premature hemorrhaging and death of Vkorc1–/– mice.

(A) Prothrombin (PT) carboxylation in plasma from WT and Vkorc1–/– (c1–/–) mice at P0 and P7 was assessed by α-Gla antibody immunoprecipitation, followed by Western blot analysis with α-PT antibody. Input represents 4% of serum sample. (B) PT and GGCX carboxylation in e18.5, P0, and P7 livers from WT and Vkorc1–/– mice was tested using α-Gla immunoprecipitation and subsequent Western blot analysis with α-PT and α-GGCX antibodies. β-Actin was used as a loading control. Input represents 2% of total protein extract. (C) Representative picture showing WT, Vkorc1–/–, Vkorc1l1+/– (c1l1+/–), and Vkorc1–/–;Vkorc1l1+/– (c1–/–;c1l1+/–) P0 pups. Arrows indicate abdominal hemorrhages. (D) Shown are Kaplan-Meier survival curves for WT, Vkorc1+/– (c1+/–), Vkorc1+/–, Vkorc1–/–, and Vkorc1–/–;Vkorc1l1+/– mice (n = 7–15; Mantel-Cox test; **P < 0.01; ***P < 0.001). For Western blots, duplicates represent biological replicates and the image is representative of 2 independent experiments.
Vkorc1l1 gene dosage in Vkorc1–/– mice causes hemorrhages and death around birth.
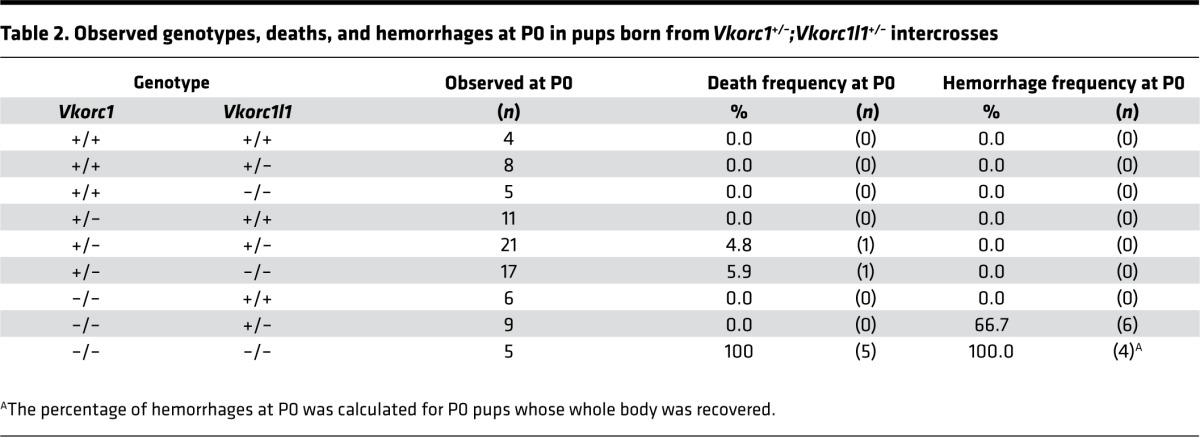
We therefore tested genetically if VKORC1L1 could be compensating for VKORC1 absence around birth. Vkorc1+/–;Vkorc1l1+/– mice were intercrossed to generate pups lacking either 1 or both Vkorc1l1 alleles in the absence of Vkorc1. At the time of weaning (P21), we did not observe mice carrying a homozygous-null allele of Vkorc1, regardless of the Vkorc1l1 genotype (Table 1). At P7, the expected number of Vkorc1–/–;Vkorc1l1+/+ mice (7.41%) was observed; however, no Vkorc1–/–;Vkorc1l1–/– mice were found, and the frequency of Vkorc1–/–;Vkorc1l1+/– pups was 1.23% instead of the expected 12.50%. The genotype frequency at P0 was therefore characterized. All possible genotypes were observed at the expected Mendelian ratio (Table 1), indicating that vitamin K oxidoreduction is dispensable for survival to birth. However, massive intra-abdominal hemorrhaging was observed in Vkorc1–/–;Vkorc1l1+/– mice (Figure 1C), and all Vkorc1–/–;Vkorc1l1–/– pups died soon after birth (Table 2). VKORC1L1 protected against hemorrhaging, which was not observed in Vkorc1–/–;Vkorc1l1+/+ mice and was only found in two-thirds of the Vkorc1–/–;Vkorc1l1+/– newborns (Table 2). Lifespan analysis revealed that all of the Vkorc1–/– mice died before 20 days, as previously reported (7), with a median survival age of 9.5 days (Figure 1D). In contrast, all Vkorc1–/–;Vkorc1l1+/– mice died before 10 days of age, with a median survival age of 1 day. Together, these observations demonstrate that deletion of either 1 or 2 alleles of Vkorc1l1 in a Vkorc1–/– background causes bleeding defects at a much earlier stage compared with the single inactivation of Vkorc1.
Table 1. Expected and observed genotype frequencies following Vkorc1+/–;Vkorc1l1+/– intercrosses.

Table 2. Observed genotypes, deaths, and hemorrhages at P0 in pups born from Vkorc1+/–;Vkorc1l1+/– intercrosses.
VKORC1L1 supports vitamin K oxidoreduction and carboxylation in the perinatal liver.
We therefore determined the consequences of deleting Vkorc1 alone or in combination with 1 or 2 alleles of Vkorc1l1 on perinatal liver VKOR activity and VKD protein carboxylation. A VKOR assay, measuring specifically the conversion of vitamin K epoxide to vitamin K (5), revealed VKORC1L1 activity in the liver. The level was low; Vkorc1–/–;Vkorc1l1+/+ P0 livers had 0.4% activity compared with WT livers, and the value was even lower (0.1%) in Vkorc1–/–;Vkorc1l1+/– livers (Figure 2A). P0 livers were also assessed for global VKD protein carboxylation by α-Gla Western blot analysis, which showed that carboxylation was greatly reduced in Vkorc1–/–;Vkorc1l1+/+ versus WT liver and completely blunted in Vkorc1–/–;Vkorc1l1+/– livers (Supplemental Figure 2, A and B). Analysis of specific VKD proteins showed that GGCX and PT carboxylation were abolished or reduced, respectively, in Vkorc1–/–;Vkorc1l1+/– compared with Vkorc1–/–;Vkorc1l1+/+ P0 livers (Figure 2B). P0 Vkorc1–/–;Vkorc1l1–/– neonates could not be measured due to their early lethality, and e18.5 embryo livers were therefore studied. This analysis revealed a gene dosage effect of VKORC1L1 on PT carboxylation, which was full, partial, or absent in Vkorc1–/–;Vkorc1l1+/+, Vkorc1–/–;Vkorc1l1+/–, or Vkorc1–/–;Vkorc1l1–/– livers, respectively (Figure 2C). GGCX carboxylation showed a similar response. These biochemical studies, thus, showed that VKORC1L1 possesses in vivo VKOR activity that supports carboxylation in the liver during the pre- and perinatal periods. Importantly, the VKORC1L1 levels were sufficient for protecting against hemorrhaging at birth and for increasing the survival time of Vkorc1–/– mice (Table 2 and Figure 1D).
Figure 2. Vkorc1 and Vkorc1l1 gene dosage affects vitamin K oxidoreduction and carboxylation in the perinatal mouse liver.

(A) Vitamin K epoxide reductase (VKOR) activity in liver was measured at P0 in controls (WT and Vkorc1l1+/–), Vkorc1–/–, Vkorc1–/–;Vkorc1l1+/–, and Vkorc1l1–/– pups (n = 2–6; mean ± SEM; unpaired, 2-tailed Student’s t test was used to compare Vkorc1–/– and Vkorc1–/–;Vkorc1l1+/– livers; ***P < 0.001). (B) PT and GGCX carboxylation in P0 livers from WT, Vkorc1–/–, Vkorc1–/–;Vkorc1l1+/–, and Vkorc1l1–/– was tested using α-Gla immunoprecipitation and subsequent Western blot analysis with α-PT and α-GGCX antibodies. β-Actin was used as a loading control. Input represents 2% of total protein extract. Duplicates represent biological replicates, and the image is representative of 2 independent experiments. (C) PT and GGCX carboxylation in livers from WT, Vkorc1–/–, Vkorc1–/–;Vkorc1l1+/–, Vkorc1–/–;Vkorc1l1–/– (c1–/–;c1l1–/–), and Vkorc1l1–/– e18.5 embryos were analyzed as in B.
The expression of VKORC1 and VKD coagulation factors increases after birth.
VKORC1L1 only allowed survival for 1 week after birth, which we hypothesized might be due to developmental regulation of some components of the vitamin K cycle. We carried out mRNA analysis to test this hypothesis and showed that Vkorc1 but not Vkorc1l1 expression increased robustly in the liver between P0 and P15 (Figure 3A). Western analysis indicated the strong induction of VKORC1 but not VKORC1L1 protein (Figure 3B). Importantly, VKOR activity significantly increased as liver development progresses (Figure 3C). We also analyzed the expression of several mRNAs encoding VKD coagulation factors (F2, F7, F9, and F10), which all showed increases during the peri- and postnatal periods (Figure 4A), and Ggcx expression followed a similar trend (Figure 4B). Global VKD protein carboxylation also increased, as assessed by α-Gla Western blot analysis (Figure 4C). Finally, both PT protein expression and carboxylation increased in the plasma of WT mice (Figure 4, D and E). Collectively, these results indicate that the level of carboxylated coagulation factors significantly increases around birth and that carboxylation is sustained by upregulation of VKORC1, but not VKORC1L1, in the liver. Developmental upregulation of vitamin K cycle components may be conserved in humans, as well, as F2, F10, VKORC1, and GGCX mRNA levels are higher in adult versus fetal livers (Supplemental Figure 3). The human data are in agreement with the observation that VKD coagulation factor levels are, on average, 75% lower in normal human fetuses compared with adults (16).
Figure 3. VKORC1 expression and VKOR activity increase peri- and postnatally in liver.

(A) Vkorc1 and Vkorc1l1 mRNA expression in C57BL/6J livers at e14.5, e16.5, e18.5, P0, P7, and P15 were measured by quantitative PCR (qPCR) and normalized to Actb (n = 3; mean ± SEM). Copy number was quantified using plasmid DNA containing either Vkorc1 or Vkorc1l1 cDNA as a standard. (B) Protein expression in liver at e16.5, e18.5, P0, P7, and P15 was analyzed by Western blot using antibodies against VKORC1, VKORC1L1, GGCX, or β-actin. Quantification shows arbitrary densitometry units of VKORC1 signal relative to VKORC1L1. (C) VKOR activity in C57BL/6J livers at e16.5, e18.5, P0, P7, and P15 was measured (n = 4; mean ± SEM; 1-way ANOVA tests followed by Bonferroni’s multiple comparisons test; **P < 0.01; ***P < 0.001).
Figure 4. VKD protein expression and carboxylation increase peri- and postnatally in liver and blood.

(A and B) Relative mRNA levels of F2, F7, F9, F10 (A), and Ggcx (B) were measured by qPCR in C57BL/6J livers at e14.5, e16.5, e18.5, P0, P7, and P15 and normalized to Actb (n = 3; mean ± SEM). (C) Global VKD protein carboxylation in livers from e16.5, e18.5, P0, P7, and P15 C57BL/6J mice was analyzed by Western blot using α-GLA antibody. GADPH was used as a loading control. (D) PT protein and carboxylation in plasma from e16.5, e18.5, P0, P7, and P15 C57BL/6J mice were monitored by immunoprecipitation with α-PT antibody, followed by Western blot analysis with α-Gla or α-PT antibodies. Input represents 4% of serum sample. Duplicates represent biological replicates, and the image is representative of 2 independent experiments. (E) Densitometry values from D were normalized to the e16.5 sample.
Increasing liver VKORC1L1 levels in Vkorc1–/– mice rescues VKD carboxylation.
Altogether, the results presented above suggest that, after 1 week of age, VKORC1L1 expression in the absence of VKORC1 is insufficient in supporting the increased demand for VKD protein carboxylation, causing hemorrhaging and death. This hypothesis was tested by elevating VKORC1L1 levels in the livers of Vkorc1–/– mice and determining the consequence on hemostasis, carboxylation, and survival. We generated APOE-Vkorc1l1 transgenic mice in which the liver-specific regulatory sequences of the human APOE gene were used to express a FLAG-tagged version of VKORC1L1 specifically in hepatocytes (Figure 5A). Two of several independent transgenic lines were selected (APOE-Vkorc1l173 and APOE-Vkorc1l170), which had a 2-fold difference in VKORC1L1-FLAG expression (Figure 5B). Western analysis showed liver-specific expression (Figure 5C), and quantitative PCR (qPCR) revealed ~20-fold higher expression of Vkorc1l1 than Vkorc1 mRNA (Figure 5D). However, quantification analysis by Western blot indicated that the level of VKORC1L1-FLAG protein reached was ~25 times lower (i.e., <4%) than the level of endogenous VKORC1 in WT animals (Figure 5E and Supplemental Figure 4). APOE-Vkorc1l173 transgenic mice were then bred to Vkorc1+/– mice to generate Vkorc1+/–; APOE-Vkorc1l173 animals, which were subsequently backcrossed to Vkorc1+/– mice.
Figure 5. Generation and characterization of APOE-Vkorc1l1 transgenic mice.

(A) Schematic representation of the construct used to generate APOE-Vkorc1l1Tg (APOE-c1l1Tg) mice. Vkorc1l1-3XFLAG was cloned in the pLIV.7 vector containing exon 1 and 2, liver-specific regulatory sequences and 5′ and 3′ flanking sequences of the human APOE gene. (B) VKORC1L1-FLAG expression in livers from WT, APOE-Vkorc1l170 (APOE-c1l170), and APOE-Vkorc1l173 (APOE-c1l173) transgenic mice at 4 weeks of age was assessed by Western blot analysis using α-FLAG antibody. β-Actin was used as a loading control. β-Actin–normalized arbitrary densitometry units of VKORC1L1-FLAG are shown. (C) VKORC1L1-FLAG expression in tissues from WT and APOE-Vkorc1l173 mice was detected by Western blot analysis with α-FLAG antibody. GAPDH was used as a loading control. (D) Gene expression level of Vkorc1 and Vkorc1l1 in livers from WT and APOE-Vkorc1l173 mice at 4 weeks of age was assessed by qPCR and normalized to Actb (n = 3; mean ± SEM). Copy number was quantified using plasmid DNA containing either Vkorc1 or Vkorc1l1 cDNA as a standard. (E) VKORC1 and VKORC1L1-FLAG protein levels in liver from WT and APOE-Vkorc1l173 mice were measured by Western blot using α-VKORC1 and α-FLAG antibodies. VKORC1-FLAG– and VKORC1L1-FLAG–transfected HEK293 cells were used as a standard and β-actin as a loading control (n = 2; mean ± SEM; Supplemental Figure 4).
Progeny from the second cross were then analyzed at P5, a time at which all of the possible genotypes were observed at the expected frequency (Table 3). Increased VKORC1L1 expression was sufficient to support production of active PT; while PT time assessed as the international normalized ratio (INR) was markedly increased in Vkorc1–/– pups, as expected, INR values in Vkorc1–/–; APOE-Vkorc1l173 were comparable with WT mice (Figure 6A). Accordingly, global carboxylation in liver was completely blunted in Vkorc1–/– P5 pups but was rescued to nearly WT levels in Vkorc1–/–; APOE-Vkorc1l173 mice (Figure 6B). In addition, PT and GGCX carboxylation in mouse liver were absent in P5 Vkorc1–/– mice but comparable with WT in Vkorc1–/–; APOE-Vkorc1l173 pups (Figure 6C). VKOR activity was substantially higher in Vkorc1–/–; APOE-Vkorc1l173 than Vkorc1–/– liver (Figure 6D). The activity in Vkorc1–/–; APOE-Vkorc1l173 liver was only 2.6% that of WT values, in agreement with the ~4% VKORC1L1-FLAG expression level found in transgenic livers in comparison with VKORC1 in WT livers (Figure 5E and Supplemental Figure 4). Nonetheless, this level was sufficient for rescuing carboxylation and PT activity in the absence of VKORC1.
Table 3. Expected and observed genotype frequencies following Vkorc1+/–;APOE-Vkorc1l173 and Vkorc1+/– intercrosses.
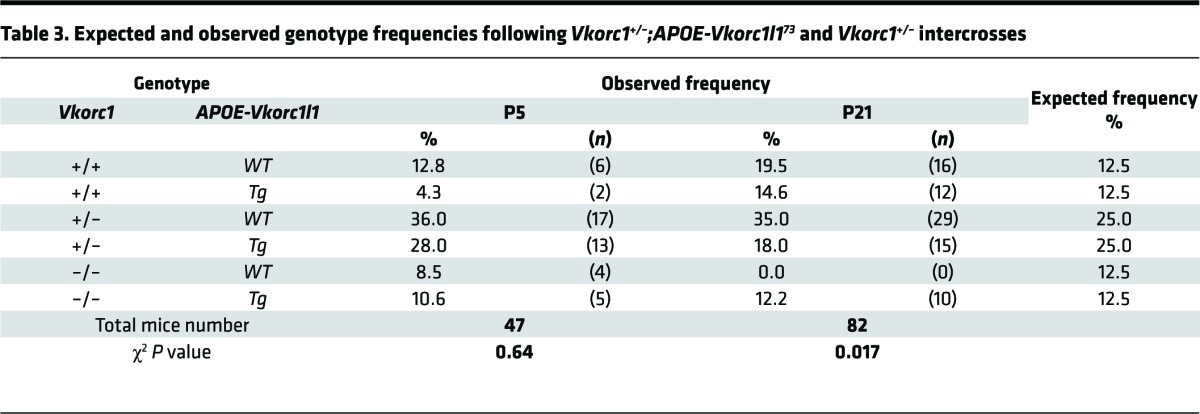
Figure 6. Increased VKORC1L1 levels in the absence of Vkorc1 rescues thrombin activity, carboxylation, and survival.

(A) Thrombin activities in WT, Vkorc1–/–, Vkorc1–/–;APOE-Vkorc1l173, and APOE-Vkorc1l173 P5 pups were measured using a Coagucheck and are shown as international normalized ratio (INR) (n = 3-6; mean ± SEM). The limit of detection of the Coagucheck ranges from 0.8 to 8 and is depicted by dashed lines. (B) The global carboxylation profile in livers from WT, Vkorc1–/–, Vkorc1–/–;APOE-Vkorc1l173, and APOE-Vkorc1l173 P5 pups was analyzed by Western blot using α-Gla antibody. β-Actin was used as a loading control. Arrows show VKD proteins, while asterisks indicate nonspecific bands. (C) PT and GGCX carboxylation in WT, Vkorc1–/–, Vkorc1–/–;APOE-Vkorc1l173, and APOE-Vkorc1l173 P5 animals were measured by α-Gla immunoprecipitation followed by Western blot analysis using α-PT and α-GGCX antibodies. β-Actin was used as a loading control. Input represents 2% of total protein extract. (D) VKOR activity was measured in livers from WT, Vkorc1–/–, Vkorc1–/–;APOE-Vkorc1l173, and APOE-Vkorc1l173 P5 pups (n = 4; mean ± SEM; unpaired, 2-tailed Student’s t test was used to compare Vkorc1–/– and Vkorc1–/–; APOE-Vkorc1l173 livers; ***P < 0.001). (E) Kaplan-Meier survival curves were determined from birth to 5 months of age for WT, Vkorc1–/–, Vkorc1–/–;APOE-Vkorc1l173, and APOE-Vkorc1l173 mice (n = 6–14; Mantel-Cox test; ***P < 0.001). (F) The INR was determined for WT and Vkorc1–/–;APOE-Vkorc1l173 mice at 16 weeks of age (n = 4–7, mean ± SEM; unpaired, 2-tailed Student’s t test; **P < 0.01). For Western blot, duplicates represent biological replicates and the image is representative of 2 independent experiments.
Increasing liver VKORC1L1 levels supports survival of Vkorc1–/– mice to adulthood.
We next investigated whether elevated VKORC1L1 levels in liver rescue the hemostatic defects and lethality associated with VKORC1 deficiency beyond 1 week of age. In contrast to Vkorc1–/– mice that all died before 20 days, the Vkorc1–/–; APOE-Vkorc1l173 mice reached weaning with the expected Mendelian frequency (12.5%), and most survived past 6 months of age without any noticeable phenotype (Table 3 and Figure 6E). Both APOE-Vkorc1l1 transgenic lines tested rescued the postnatal lethality of VKORC1 deficiency (Table 3 and Supplemental Table 1). Finally, INRs were within the normal range in 4-month-old adult Vkorc1–/–; APOE-Vkorc1l173 mice (Figure 6F). Therefore, VKORC1L1 expressed at sufficient levels prevents bleeding and lethality in adult mice, even in the absence of VKORC1.
Discussion
In summary, our results establish for the first time to our knowledge that VKORC1L1 supports VKD protein carboxylation in vivo. The study focused on VKORC1L1 function in liver because of the difference in timing of hemorrhaging and lethality in Vkorc1–/– and Ggcx–/– mice (7, 8). While VKORC1L1 expression and activity are much lower than that of VKORC1 in liver, it may have a more dominant role in some extrahepatic tissues. VKORC1L1 and VKORC1 are both broadly expressed, as are VKD proteins that have multiple functions besides hemostasis, and Vkorc1l1 mRNA levels are higher than that of Vkorc1 in some tissues (e.g., brain and testes) (10, 11). It should be noted that the results do not rule out alternative roles for VKORC1L1 — for example, in oxidative stress, as previously suggested (12).
Importantly, our data also revealed that blood coagulation is a developmentally regulated process. We found that VKD clotting factors and the machinery responsible for their activation both increased peri- and postnatally. These results are of interest regarding newborn hemorrhaging, which has been attributed solely to vitamin K deficiency ameliorated by supplementation at birth. Indeed, our data suggest that, in some cases, the hemorrhagic response could also be due to low VKORC1 activity in the liver. These findings also raise questions about the role of carboxylated proteins during embryogenesis. Of interest, for example, is whether upregulation of VKD clotting factors is important because the neonate/adult is more exposed to injury than the fetus, and whether other VKD proteins known to be expressed in adult liver (e.g., GAS6) are also developmentally regulated.
Interestingly, all of the Vkorc1–/–;Vkorc1l1–/– mice survived until birth, while previous studies showed that about half of Ggcx–/– mice died mid-embryogenesis (8). GGCX function requires KH2, and the results therefore suggest that an additional reductase supports carboxylation during embryogenesis. A quinone reductase that reduces K to KH2 is present in adult mouse liver (7). The carboxylase converts KH2 to KO that the quinone reductase cannot recycle, and vitamin K levels from maternal transfer are low (17). Consequently, a quinone reductase could only support a limited amount of VKD protein carboxylation. In this regard, it is worth noting that many VKD clotting factors also function in signaling, activating protease-activated receptors (PARs) thought to be important to embryonic development (18). PT, factor X, and PAR1 deficiency in mice all result in approximately half of the embryos dying midgestation (18–20), which may reflect the role of these VKD proteases in signaling. The amounts of carboxylated protein required for signaling would be substantially less then that needed for clotting, and so a quinone reductase could be functionally significant. We note that quinone reductase activity in adult liver only supports blood clotting in the context of administration of high doses of vitamin K (7, 21), which were not used here; therefore, the presence of the quinone reductase in embryonic liver should not affect blood coagulation and the conclusion of the current study.
Methods
Animal models.
Vkorc1+/– and Vkorc1l1+/– mice, generated in our laboratory, were maintained on a C57BL/6J background (backcrossed for more than 10 generations with C57BL/6J mice from the Jackson Laboratory) and described previously (9) were crossed to generate Vkorc1+/–;Vkorc1l1+/– mice, which were further intercrossed to generate e16.5, e18.5, P0, P7, and P15 mice of the various studied genotypes. Mouse Vkorc1l1 cDNA was cloned in the p3XFLAG-CMV-14 vector, and the SpeI site was then removed by mutagenesis. APOE-Vkorc1l1 transgene was generated by cloning the 3XFLAG-tagged version of mouse Vkorc1l1 cDNA into the pLIV.7 plasmid, which contains the liver-specific human APOE promoter and regulatory sequences (22) (see Supplemental Table 2 for a list of the oligos and restriction sites used in the cloning). The APOE-Vkorc1l1 transgenic construct was excised from the plasmid using SpeI and SacII unique restriction sites, purified, and microinjected into mouse fertilized oocytes from F2 (C3H/HeNHs × C57BL/6J). Transgenic and mutant mice were identified by specific PCR (Supplemental Table 2). Each transgenic founder was successively crossed with heterozygous Vkorc1+/– mice to generate Vkorc1+/–;APOE-Vkorc1l1 transgenic progenies. These progenies were then backcrossed with Vkorc1+/– mice to produce controls, Vkorc1–/– and Vkorc1–/–;APOE-Vkorc1l1 mice (genetic background: ~87.5% C57BL/6J, ~12.5% C3H/HeNHs). Both males and females were used in this study.
Plasmid constructs.
GGCX-FLAG and PT-FLAG plasmids were generated by PCR amplification and cloning in the HindIII and BamHI sites of p3XFLAG-CMV-14 vector. PRRG2-FLAG plasmid was generated by PCR amplification and cloning in the EcoRI and KpnI sites of p3XFLAG-CMV-14 vector. GAS6-Myc and MGP-Myc were generated by PCR amplification and cloning in the EcoRI and XbaI sites of pcDNA3.1/myc-His B. Sequences of the primers used for cloning are included in Supplemental Table 2.
Generation of rabbit polyclonalα-Gla antibodies. The α-Gla antibody was developed in collaboration with Covance Inc. Rabbits were immunized with the H2N-G(Gla)G(dPEG4)G(Gla)G(dPEG4)C-OH synthetic peptide, and rabbit immunoglobulins were affinity purified against H2N-G(Gla)G and depleted against GEGGE peptides. The specificity of the antibodies toward carboxylated residues was tested by immunoprecipitation and Western blot (Supplemental Figure 1, A and B).
Immunoprecipitation and Western blot assays.
Livers were homogenized in lysis buffer containing: 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA (pH 8.0), 1 mM EGTA, 2.5 mM NaPyrophosphate, 1 mM β-glycerophosphate, 10 mM NaF, 1% Triton, 1 mM PMSF, and protease inhibitors (EDTA-free protease inhibitor cocktail tablets, Roche Diagnostics). For α-Gla immunoprecipitation, 200 μg of liver extracts from each condition were incubated overnight with rotation at 4°C with 12 μg of rabbit α-Gla antibodies followed by 3 hours incubation with 40 μl of Protein A-Agarose beads (Roche Diagnostics). Immunoprecipitated proteins were washed 4 times with lysis buffer, resuspended in loading buffer, and heated at 70°C for 10 minutes before being resolved by SDS-PAGE on 7.5% polyacrylamide Tris-Glycine gels. Proteins were detected in the immunoprecipitated and input (4%) fractions by Western blot following standard procedures using sheep α-PT (Thermo Fisher Scientific, PA1-43062), rabbit α-GGCX (Proteintech, 16209-1-AP), and mouse α–β-actin (MilliporeSigma, A5441) antibodies.
Mouse blood was collected using heparinized capillaries and EDTA-treated tubes, and cells were removed from plasma by centrifuging at 800 g for 5 minutes at room temperature. For α-PT immunoprecipitation, 2 μl of plasma was diluted in 500 μl of lysis buffer for each condition. Samples were precleared using 40 μl of protein G-Agarose beads (Roche Diagnostics) for 2 hours at 4°C before adding 14 μg of α-PT antibodies and incubating at 4°C overnight with rotation. For α-Gla immunoprecipitation, 2 μl of plasma was diluted in 500 μl of lysis buffer, and samples were precleared using 40 μl of protein A-Agarose beads for 2 hours at 4°C before adding 14 μg of α-Gla antibodies. Immunoprecipitated proteins were washed 4 times with lysis buffer, resuspended in loading buffer, and heated at 70°C for 10 minutes before being resolved on SDS-PAGE and detected by Western blot using rabbit α-Gla and sheep α-PT antibodies.
HEK293 cells (ATCC), cultured in EMEM supplemented with 10% heat-inactivated FBS, were transfected using Lipofectamine 2000 per manufacturer’s instructions (Invitrogen) with plasmids containing either GGCX-FLAG, PT-FLAG, PRRG2-FLAG, GAS6-Myc, or MGP-Myc in presence of warfarin (Santa Cruz Biotechnology Inc., 50 μM) or vitamin K1 (MilliporeSigma, 22 μM). Two days after transfection, cells were harvested in lysis buffer, and 200 μg of each condition was used for α-Gla immunoprecipitation as described above. GGCX-FLAG, PT-FLAG, and PRRG2-FLAG were detected using mouse α-FLAG M2 antibodies (MilliporeSigma, F1804). GAS6-Myc and MGP-Myc were detected using mouse α-Myc antibodies (Cell Signaling Technologies, 2276).
Other antibodies used in this study for Western blot include: rabbit α-VKORC1 generated in our laboratory (9), rabbit α-VKORC1L1 (Abcam, ab116508), and rabbit α-GAPDH (Cell Signaling Technologies, 5174). To detect PRRG2, MGP, VKORC1, and VKORC1L1, proteins were resolved on 10% polyacrylamide Tris-Tricine gels.
VKOR activity assay.
Mouse liver samples were homogenized in 25 mM Tris (pH 7.4), 250 mM sucrose, and 1 mM phenylmethylsulfonyl fluoride using a tenbroeck tissue grinder to lyse cells, followed by centrifugation at 2,000 g for 15 minutes, all at 4°C. Supernatant protein concentrations were determined using a BCA assay (Thermo Fisher Scientific), and protein samples were then diluted to 2 mg/ml with homogenization buffer. Vitamin KO reductase activity was determined as before (5), except for the use of larger volumes (400 μl) and times of incubation (4 hours) in the assay.
PT time measurement.
Mouse blood was collected following tail cutting with a scalpel, and PT time was measured as INR using a Coagucheck (Roche Diagnostics).
Gene expression.
Livers were harvested from mice and homogenized in guanidium thiocyanate lysis solution, and total RNA was isolated according to Chomczynski and Sacchi (23). Total mRNA was then DNaseI treated (Invitrogen) and reverse transcribed with random and oligo dT primers using M-MLV reverse transcriptase (Invitrogen). Gene expression was measured using SYBR Green qPCR Master Mix (BiMake) with gene-specific primers (Supplemental Table 2) using ViiA 7 Real-Time PCR System (Applied Biosystems). Expression levels were normalized to Actb expression levels.
Statistics.
Statistical analyses were performed using GraphPad prism software. Results are given as means ± SEM. For single measurement, unpaired, 2-tailed Student’s t test was used. Grouped analysis was performed using 1-way ANOVA, followed by Bonferroni’s multiple comparisons test. For survival analysis, Mantel-Cox test was used. In all figures, **P < 0.01; ***P < 0.001. For all Western blots, each lane represents a biological replicate and the image is representative of at least 2 independent experiments.
Study approval.
All animal use complied with the guidelines of the Canadian Committee for Animal Protection and was approved by IRCM institutional animal care committee.
Author contributions
JL performed most experiments with crucial help from MAR (VKOR assays) and MF (mice generation). JL and MF conceived the research. MF, KLB, and JL wrote the manuscript with suggestions from MAR. All authors discussed the results and the manuscript.
Supplementary Material
Acknowledgments
We are grateful to M. Trudel and K. Runge for their critical reading of the manuscript. We thank J. Bonneau and B.A. Reddy for mouse genotyping and the generation of plasmid constructs. This work was supported by funding from the CRC program (MF), the CIHR (MF, MOP-133652) and by NIH (KLB, R01 HL81093 and HL55666).
Version 1. 01/11/2018
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2017;2(24):e96501. https://doi.org/10.1172/jci.insight.96501.
Contributor Information
Julie Lacombe, Email: julie.lacombe@ircm.qc.ca.
Mark A. Rishavy, Email: rishavm@ccf.org.
Mathieu Ferron, Email: mathieu.ferron@ircm.qc.ca.
References
- 1.Berkner KL. Vitamin K-dependent carboxylation. Vitam Horm. 2008;78:131–156. doi: 10.1016/S0083-6729(07)00007-6. [DOI] [PubMed] [Google Scholar]
- 2.Rost S, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004;427(6974):537–541. doi: 10.1038/nature02214. [DOI] [PubMed] [Google Scholar]
- 3.Li T, Chang CY, Jin DY, Lin PJ, Khvorova A, Stafford DW. Identification of the gene for vitamin K epoxide reductase. Nature. 2004;427(6974):541–544. doi: 10.1038/nature02254. [DOI] [PubMed] [Google Scholar]
- 4.Li W, Schulman S, Dutton RJ, Boyd D, Beckwith J, Rapoport TA. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature. 2010;463(7280):507–512. doi: 10.1038/nature08720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rishavy MA, Hallgren KW, Wilson LA, Usubalieva A, Runge KW, Berkner KL. The vitamin K oxidoreductase is a multimer that efficiently reduces vitamin K epoxide to hydroquinone to allow vitamin K-dependent protein carboxylation. J Biol Chem. 2013;288(44):31556–31566. doi: 10.1074/jbc.M113.497297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen G, et al. Warfarin traps human vitamin K epoxide reductase in an intermediate state during electron transfer. Nat Struct Mol Biol. 2017;24(1):69–76. doi: 10.1038/nsmb.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spohn G, et al. VKORC1 deficiency in mice causes early postnatal lethality due to severe bleeding. Thromb Haemost. 2009;101(6):1044–1050. [PubMed] [Google Scholar]
- 8.Zhu A, et al. Fatal hemorrhage in mice lacking gamma-glutamyl carboxylase. Blood. 2007;109(12):5270–5275. doi: 10.1182/blood-2006-12-064188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferron M, Lacombe J, Germain A, Oury F, Karsenty G. GGCX and VKORC1 inhibit osteocalcin endocrine functions. J Cell Biol. 2015;208(6):761–776. doi: 10.1083/jcb.201409111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hammed A, Matagrin B, Spohn G, Prouillac C, Benoit E, Lattard V. VKORC1L1, an enzyme rescuing the vitamin K 2,3-epoxide reductase activity in some extrahepatic tissues during anticoagulation therapy. J Biol Chem. 2013;288(40):28733–28742. doi: 10.1074/jbc.M113.457119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caspers M, et al. Two enzymes catalyze vitamin K 2,3-epoxide reductase activity in mouse: VKORC1 is highly expressed in exocrine tissues while VKORC1L1 is highly expressed in brain. Thromb Res. 2015;135(5):977–983. doi: 10.1016/j.thromres.2015.01.025. [DOI] [PubMed] [Google Scholar]
- 12.Westhofen P, et al. Human vitamin K 2,3-epoxide reductase complex subunit 1-like 1 (VKORC1L1) mediates vitamin K-dependent intracellular antioxidant function. J Biol Chem. 2011;286(17):15085–15094. doi: 10.1074/jbc.M110.210971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tie JK, Jin DY, Stafford DW. Conserved loop cysteines of vitamin K epoxide reductase complex subunit 1-like 1 (VKORC1L1) are involved in its active site regeneration. J Biol Chem. 2014;289(13):9396–9407. doi: 10.1074/jbc.M113.534446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berkner KL, Pudota BN. Vitamin K-dependent carboxylation of the carboxylase. Proc Natl Acad Sci USA. 1998;95(2):466–471. doi: 10.1073/pnas.95.2.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hallgren KW, Zhang D, Kinter M, Willard B, Berkner KL. Methylation of γ-carboxylated Glu (Gla) allows detection by liquid chromatography-mass spectrometry and the identification of Gla residues in the γ-glutamyl carboxylase. J Proteome Res. 2013;12(6):2365–2374. doi: 10.1021/pr3003722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forestier F, Daffos F, Rainaut M, Solé Y, Amiral J. Vitamin K dependent proteins in fetal hemostasis at mid trimester of pregnancy. Thromb Haemost. 1985;53(3):401–403. [PubMed] [Google Scholar]
- 17.Shearer MJ, Rahim S, Barkhan P, Stimmler L. Plasma vitamin K1 in mothers and their newborn babies. Lancet. 1982;2(8296):460–463. doi: 10.1016/s0140-6736(82)90493-7. [DOI] [PubMed] [Google Scholar]
- 18.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3(8):1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- 19.Xue J, et al. Incomplete embryonic lethality and fatal neonatal hemorrhage caused by prothrombin deficiency in mice. Proc Natl Acad Sci USA. 1998;95(13):7603–7607. doi: 10.1073/pnas.95.13.7603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dewerchin M, et al. Blood coagulation factor X deficiency causes partial embryonic lethality and fatal neonatal bleeding in mice. Thromb Haemost. 2000;83(2):185–190. [PubMed] [Google Scholar]
- 21.Ingram BO, Turbyfill JL, Bledsoe PJ, Jaiswal AK, Stafford DW. Assessment of the contribution of NAD(P)H-dependent quinone oxidoreductase 1 (NQO1) to the reduction of vitamin K in wild-type and NQO1-deficient mice. Biochem J. 2013;456(1):47–54. doi: 10.1042/BJ20130639. [DOI] [PubMed] [Google Scholar]
- 22.Allan CM, Taylor S, Taylor JM. Two hepatic enhancers, HCR.1 and HCR.2, coordinate the liver expression of the entire human apolipoprotein E/C-I/C-IV/C-II gene cluster. J Biol Chem. 1997;272(46):29113–29119. doi: 10.1074/jbc.272.46.29113. [DOI] [PubMed] [Google Scholar]
- 23.Chomczynski P, Sacchi N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat Protoc. 2006;1(2):581–585. doi: 10.1038/nprot.2006.83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.