

Abstract
Mesenchymal stem cells (MSCs) can give rise to both adipocytes and osteoblasts, but the molecular mechanisms underlying MSC fate determination remain poorly understood. IκB kinase β (IKKβ), a central coordinator of inflammation and immune responses through activation of NF-κB, has been implicated as a critical molecular link between obesity and metabolic disorders. Here, we show that IKKβ can reciprocally regulate adipocyte and osteoblast differentiation of murine and human MSCs through an NF-κB–independent mechanism. IKKβ is a β-catenin kinase that phosphorylates the conserved degron motif of β-catenin to prime it for β-TrCP–mediated ubiquitination and degradation, thereby increasing adipogenesis and inhibiting osteogenesis in MSCs. Animal studies demonstrated that deficiency of IKKβ in BM mesenchymal stromal cells increased bone mass and decreased BM adipocyte formation in adult mice. In humans, IKKβ expression in adipose tissue was also positively associated with increased adiposity and elevated β-catenin phosphorylation. These findings suggest IKKβ as a key molecular switch that regulates MSC fate, and they provide potentially novel mechanistic insights into the understanding of the cross-regulation between the evolutionarily conserved IKKβ and Wnt/β-catenin signaling pathways. The IKKβ-Wnt axis we uncovered may also have important implications for development, homeostasis, and disease pathogenesis.
Keywords: Cell Biology, Stem cells
Keywords: Adipose tissue, Adult stem cells, NF-kappaB
IKKβ phosphorylates β-catenin to reciprocally regulate adipocyte and osteoblast differentiation of mesenchymal stem cells.
Introduction
Obesity and obesity-associated metabolic disorders such as insulin resistance and type 2 diabetes are rapidly growing public health epidemics, and there is an urgent need to understand the molecular mechanisms underlying these chronic diseases. The expansion of adipose tissue during obesity is through adipocyte hypertrophy and hyperplasia, and adipocyte number can be a major determinant of fat mass in adults (1, 2). Adipose tissue contains abundant adult mesenchymal stem cells (MSCs) that are capable of differentiating into many cell types, including adipocytes, osteoblasts, chondrocytes, and myoblasts, but little is known about the initial signals that trigger the commitment of MSCs to different lineages.
Chronic low-grade inflammation elicited by obesity has been established as a major contributor to the development of type 2 diabetes and atherosclerosis (3, 4). Many inflammatory pathways that contribute to these pathogeneses are regulated by the transcriptional factor NF-κB, a master regulator of the innate and adaptive immune responses (5–7). NF-κB can be rapidly activated in response to various stimuli, including cytokines, infectious agents, ROS, and many free fatty acids (FFAs) (5, 6, 8). In nonstimulated cells, NF-κB dimers are sequestered in the cytoplasm by binding to specific inhibitory proteins — the inhibitors of NF-κB (IκBs). Activation of IκB kinase (IKK), which is composed of 2 catalytic subunits (IKKα and IKKβ) and a regulatory subunit (IKKγ/NEMO), leads to phosphorylation and ubiquitination of IκB, allowing NF-κB to translocate to the nucleus and regulate the expression of its target genes (7, 9). IKKβ is the predominant catalytic subunit of the IKK complex and is required for activation of NF-κB by inflammatory mediators in the canonical or classical activation pathway (5, 7).
It has been demonstrated that high-fat (HF) feeding or overnutrition can lead to activation of IKKβ in many tissues, including liver, adipose tissue, and brain, which contribute to the development of metabolic disorders (10–16). Thus, IKKβ has been implicated as a key molecular link between obesity, inflammation, and metabolic disorders (6, 17). Very recently, we provided potentially novel evidence that IKKβ also functions in adipocyte precursor cells to promote adipocyte differentiation and white adipose tissue expansion in diet-induced obesity (7, 18). However, the role and underlying mechanisms of IKKβ in mediating MSC commitment remain elusive.
In addition to increasing metabolic disorders, obesity can also affect bone homeostasis, leading to abnormal bone density and decreased bone turnover (19–25). For example, studies have found that increased adiposity together with decreased lean mass was associated with lower BM density (BMD) in elderly obese adults (22). Obese premenopausal women with high levels of visceral adipose tissue also had increased BM adipose tissue that was inversely associated with BMD (23). Several other studies have also confirmed the inverse relationship between BM adipose tissue and whole-body BMD (24, 25). However, the interface between fat and bone at the molecular level is poorly understood. MSCs can give rise to both adipocytes and osteoblasts, and strong evidence suggests a reciprocal relationship between adipocyte and osteoblast differentiation (19, 26, 27). However, little is known about the initial signals triggering the commitment of MSCs to different lineages. Here, we show that IKKβ promotes adipogenesis but inhibits osteogenesis in murine and human MSCs through an NF-κB–independent mechanism. IKKβ is a β-catenin kinase that phosphorylates the degron motif of β-catenin to prime it for β-TrCP–mediated ubiquitination and degradation, thereby regulating MSC fate. IKKβ may serve as a key molecular switch that triggers the adipogenic and osteogenic differentiation of MSCs.
Results
IKKβ loss of function impairs adipogenesis but enhances osteogenesis in MSCs.
To investigate the functions of IKKβ in lineage commitment, IKKβ gene was deleted in murine C3H/10T1/2 MSCs (28, 29). CRISPR/CRISPR-associated protein 9–mediated (CRISPR/Cas9-mediated) IKKβ deficiency (Figure 1A) inhibited the differentiation of C3H/10T1/2 MSCs into adipocytes and suppressed the expression of adipogenic genes and adipocyte markers including Zfp423, PPARγ, and adiponectin (Figure 1, B and C). Interestingly, deletion of IKKβ substantially enhanced osteogenesis of C3H/10T1/2 cells and the expression levels of osteogenic markers including runt-related transcription factor 2 (Runx2), alkaline phosphatase (ALP), osteopontin (OPN), and osteocalcin (OC) were induced by IKKβ deficiency (Figure 1, D and E). Consistently, siRNA-mediated temporary IKKβ knockdown also resulted in decreased adipogenesis and enhanced osteogenesis of C3H/10T1/2 cell (Supplemental Figure 1, A–E; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.96660DS1). Further, pharmacological inhibition of IKKβ by BMS-345541 (7, 30) sufficiently inhibited C3H/10T1/2 cells differentiating into adipocytes but increased the osteogenic potential of these cells. (Figure 1, F–I).
Figure 1. Deficiency or inhibition of IKKβ reduces adipogenesis but increases osteogenesis of murine MSCs.

(A) Immunoblotting for IKKβ proteins in control or CRISPR/Cas9-mediated IKKβ-deficient C3H/10T1/2 cells. (B) Oil Red O staining of control and IKKβ-deficient MSCs induced by an adipogenic cocktail. Scale bar: 100 μm. (C) qPCR analysis of mRNA levels of adipogenic genes and adipocyte markers (n = 3). (D) Alkaline phosphatase (ALP) staining of control and IKKβ-deficient C3H/10T1/2 cells induced by an osteogenic cocktail. Scale bar: 100 μm. (E) qPCR analysis of mRNA levels of osteogenic genes and osteoblast markers (n = 3). (F–I) C3H/10T1/2 cells were treated with vehicle control or 5 μM IKKβ inhibitor BMS-345541 and were induced by differentiation media. Oil Red O staining (F) and qPCR analysis (G) of vehicle or BMS-345541–treated C3H/10T1/2 cells induced by an adipogenic cocktail (n = 3). ALP staining (H) and qPCR analysis (I) of vehicle or BMS-345541–treated C3H/10T1/2 cells induced by an osteogenic cocktail (n = 3). Scale bar: 100 μm. Error bars represent ± SEM. Significance was determined by Student’s t test (C, E, G, and I). *P < 0.05; **P < 0.01, ***P < 0.001.
We next sought to test whether IKKβ can regulate adipogenesis and osteogenesis in primary cells. Mouse embryonic fibroblasts (MEFs) have been established as a model to study adipogenesis and osteogenesis in vitro. MEFs were isolated from mice containing loxP-flanked IKKβ alleles (IKKβF/F) mice (7, 18, 31) and were infected with lentivirus expressing Cre, which effectively decreased IKKβ expression (Figure 2A). As anticipated, knockdown of IKKβ inhibited MEFs differentiating into adipocytes and reduced adipogenic gene expression (Figure 2, B and C). By contrast, IKKβ-deficient MEFs had increased osteogenic potential as compared with control MEFs (Figure 2, D and E). In addition to MEFs, Cre-mediated IKKβ deletion also resulted in decreased adipogenesis and enhanced osteogenesis in mouse BM-derived MSCs (BMMSCs) (Supplemental Figure 1, F–J). Collectively, deficiency or pharmacological inhibition of IKKβ inhibits adipogenesis but increases osteogenesis of MSCs.
Figure 2. IKKβ regulates adipocyte and osteoblast differentiation of MEFs.

(A–E) MEFs isolated from IKKβF/F mice were infected with control lentivirus or lentivirus expressing Cre. (A) Immunoblotting for IKKβ proteins in MEFs. (B and C) Oil-red-O staining (B) and qPCR analysis (C) of MEFs induced by an adipogenic cocktail (n = 3). (D and E) ALP staining (D) and qPCR analysis (E) of MEFs induced by an osteogenic cocktail (n = 3). Scale bar: 100 μm. Error bars represent ± SEM. Significance was determined by Student’s t test (C and E). *P < 0.05; **P < 0.01.
IKKβ gain of function increases adipogenesis and inhibits osteogenesis in MSCs.
To test whether ectopic expression of IKKβ can render MSCs competent to undergo adipocyte or osteoblast differentiation, C3H/10T1/2 MSCs were infected by adenovirus expressing WT IKKβ or a kinase-inactive mutant of IKKβ with lysine 44 mutated to methionine (IKKβ KM) (15) (Figure 3A). While IKKβ KM expression inhibited adipogenesis and increased osteogenesis of C3H/10T1/2 cells, overexpression of WT IKKβ enhanced adipocyte differentiation and blocked osteogenic differentiation from C3H/10T1/2 cells (Figure 3, B–E).
Figure 3. Overexpression of IKKβ promotes adipogenesis and decreases osteogenesis of murine MSCs.

C3H/10T1/2 cells were infected with control virus or virus expressing WT IKKβ or IKKβ KM. (A) Immunoblotting for IKKβ and phosphorylated IκBα proteins. (B and C) Oil Red O staining (B) and qPCR analysis (C) of C3H/10T1/2 cells induced by an adipogenic cocktail (n = 3). (D and E) ALP staining (D) and qPCR analysis (E) of C3H/10T1/2 cells induced by an osteogenic cocktail (n = 6). Scale bar: 100 μm. Error bars represent ± SEM. Significance was determined by 1-way ANOVA (C and E).*P < 0.05; **P < 0.01, ***P < 0.001.
Many FFAs have been known to activate IKKβ and to induce cellular inflammation (15, 32, 33). Interestingly, treatment with a mixture of FFAs containing myristic, lauric, arachidonic, oleic, and linoleic acids that are known to activate IKKβ (Figure 4A) (15, 32, 34) promoted MSCs differentiating into adipocytes but inhibited osteogenesis of these cells (Figure 4, B–E). However, deficiency of IKKβ abolished the impact of FFAs on adipogenesis and osteogenesis in MSCs (Figure 4, F–I), demonstrating that FFAs regulate MSC lineage commitment through IKKβ signaling. These results suggest that overnutrition-elicited IKKβ activation might be an important trigger to mediate the commitment of MSCs to the adipocyte lineage.
Figure 4. FFAs regulate adipogenesis and osteogenesis of MSCs through IKKβ signaling.

(A) Immunoblotting for phosphorylated and total IKKβ and IκBα proteins of C3H/10T1/2 cells treated with vehicle or 0.5 mM FFAs. (B and C) Oil Red O staining (B) and qPCR analysis (C) of control or FFA-treated C3H/10T1/2 cells induced by an adipogenic cocktail (n = 3). Scale bar: 100 μm. (D and E) ALP staining (D) and qPCR analysis (E) of control or FFA-treated C3H/10T1/2 cells induced by an osteogenic cocktail (n = 3). Scale bar: 100 μm. (F and G) Oil Red O staining (F) and qPCR analysis (G) of vehicle or FFA-treated control or IKKβ-deficient C3H/10T1/2 cells induced by an adipogenic cocktail (n = 3). Scale bar: 100 μm. (H and I) ALP staining (H) and qPCR analysis (I) of vehicle or FFA-treated control or IKKβ-deficient C3H/10T1/2 cells induced by an osteogenic cocktail (n = 3). Scale bar: 100 μm. Error bars represent ± SEM. Significance was determined by Student’s t test (C and E) or 2-way ANOVA (G and I). *P < 0.05; **P < 0.01, ***P < 0.001.
IKKβ regulates β-catenin ubiquitination in MSCs.
We previously demonstrated that canonical Wnt/β-catenin signaling was activated in IKKβ-deficient adipocyte precursor cells (7, 18), likely contributing to decreased adipogenesis in those cells since canonical Wnt/β-catenin signaling inhibits adipogenesis (35, 36). On the other hand, Wnt/β-catenin signaling can stimulate MSC-derived osteogenesis (36–38). Indeed, activation of canonical Wnt signaling by Wnt3a or lithium chloride (LiCl) treatment resulted in decreased adipogenesis but increased osteogenesis of C3H/10T1/2 MSCs (Supplemental Figure 2, A–D). The activity of β-catenin is mostly controlled by its ubiquitination and proteasome-mediated degradation. We then used a proteasome inhibitor, PS-341, to confirm that both Wnt3a and LiCl inhibited β-catenin ubiquitination (Supplemental Figure 2E), leading to accumulation of nuclear β-catenin proteins (Supplemental Figure 2F). By contrast, CRISPR/Cas9-mediated β-catenin deletion enhanced adipogenesis but inhibited osteogenesis in C3H/10T1/2 cells (Supplemental Figure 2, G–K). To determine whether IKKβ regulates MSC differentiation through Wnt/β-catenin signaling, control or β-catenin–deficient C3H/10T1/2 cells were infected by adenovirus expressing IKKβ. Indeed, deficiency of β-catenin diminished the impact of IKKβ expression on adipogenesis and osteogenesis in C3H/10T1/2 cells (Figure 5, A–E), suggesting that Wnt/β-catenin signaling is required for mediating the effects of IKKβ signaling on MSC differentiation.
Figure 5. Deficiency of β-catenin abolishes the impact of IKKβ expression on adipogenesis and osteogenesis of MSCs.

(A) Immunoblotting for β-catenin and IKKβ proteins in control or β-catenin–deficient C3H/10T1/2 cells infected with control or WT IKKβ virus. (B and C) Oil Red O staining (B) and qPCR analysis (C) of control and β-catenin–deficient C3H/10T1/2 cells induced by an adipogenic cocktails (n = 3). Scale bar: 100 μm. (D and E) ALP staining (D) and qPCR analysis (E) of control and β-catenin–deficient C3H/10T1/2 cells induced by an osteogenic cocktails (n = 3). Scale bar: 100 μm. Error bars represent ± SEM. Significance was determined by 2-way ANOVA (C and E). **P < 0.01, ***P < 0.001.
We next determined whether IKKβ can regulate β-catenin ubiquitination in C3H/10T1/2 MSCs. Deficiency of IKKβ in C3H/10T1/2 cells inhibited proteasome-mediated β-catenin ubiquitination (Figure 6A), leading to elevated nuclear β-catenin protein levels (Figure 6B) and increased β-catenin activity (Figure 6C). Further, BMS-345541–mediated inhibition of IKKβ also resulted in decreased β-catenin ubiquitination (Figure 6D) and increased nuclear β-catenin proteins (Figure 6E). While overexpression of WT IKKβ increased β-catenin ubiquitination and diminished nuclear β-catenin protein levels in C3H/10T1/2 cells, overexpression of IKKβ KM decreased β-catenin ubiquitination and increased nuclear β-catenin proteins in those cells (Figure 6, F and G). Interestingly, FFAs substantially increased β-catenin ubiquitination (Figure 6H) and decreased nuclear β-catenin proteins in MSCs (Figure 6I). Moreover, deficiency of IKKβ blocked FFA-induced β-catenin ubiquitination (Figure 6J), which also explains the abolished effects of FFAs on MSC differentiation in IKKβ-deficient C3H/10T1/2 cells (Figure 4, E–H).
Figure 6. IKKβ regulates β-catenin ubiquitination in MSCs.

(A) Control or IKKβ-deficient C3H/10T1/2 cells were treated with vehicle or 100 nM PS-341. β-Catenin proteins were immunoprecipitated with anti–β-catenin antibodies and then probed with anti-ubiquitin antibodies. The whole cell lysates were probed with anti–β-catenin antibodies as an internal control. (B and C) Immunoblotting for nuclear β-catenin proteins (B) and β-catenin reporter activity (C) in control or IKKβ-deficient C3H/10T1/2 cells. (D and E) Immunoblotting for ubiquitinated β-catenin proteins (D) and nuclear β-catenin proteins (E) in control or BMS-345541–treated C3H/10T1/2 cells. (F and G) Immunoblotting for ubiquitinated β-catenin proteins (F) and nuclear β-catenin proteins (G) in C3H/10T1/2 cells infected with control, IKKβ WT, or IKK KM virus. (H and I) Immunoblotting for ubiquitinated β-catenin levels (H) and nuclear β-catenin proteins (I) in control or FFA-treated C3H/10T1/2 cells. (J) Immunoblotting for ubiquitinated β-catenin proteins in control or IKKβ-deficient C3H/10T1/2 cells treated with vehicle or FFAs. Error bars represent ± SEM. Significance was determined by Student’s t test (C). ***P < 0.001.
IKKβ is a β-catenin kinase that phosphorylates the serine-33,-37, and -45 of β-catenin.
In addition to NF-κB–dependent functions, IKKβ also plays key roles in regulating many physiological functions including immunity and cancer through NF-κB–independent pathways by phosphorylating other key proteins (34, 39, 40). Phosphorylation of several critical β-catenin amino acid residues including serine-33 (ser33), ser37, and ser45 by glycogen synthase kinase 3β (GSK3β) and priming kinase casein kinase Iα (CKIα) is essential for ubiquitin ligase–mediated ubiquitination and degradation of β-catenin (41–43). Interestingly, ser33 and ser37 of β-catenin are located within a conserved 6–amino acid sequence that is also found in IKKβ substrates of the IκB family (Figure 7A). Thus, we hypothesized that IKKβ can directly phosphorylate β-catenin to prime it for ubiquitination and proteasome-mediated degradation. We first determined whether IKKβ can directly interact with β-catenin. FLAG-tagged IKKβ and HA-tagged β-catenin proteins were expressed in both C3H/10T1/2 and HEK239T cells, and coimmunoprecipitation assays demonstrated a direct interaction between IKKβ and β-catenin proteins in both cell lines (Figure 7B). We also performed coimmunoprecipitation assays to detect the interaction between endogenous IKKβ and β-catenin proteins in C3H/10T1/2 cells. While there is a weak or undetectable interaction between endogenous IKKβ and β-catenin proteins under the basal condition, IKKβ stimuli such as FFAs and TNFα increased the IKKβ–β-catenin protein interaction in these MSCs (Supplemental Figure 3A).
Figure 7. IKKβ interacts with β-catenin and phosphorylates it at serine-33, -37 and -45.

(A) The sequence of a conserved 6–amino acid motif found in β-catenin and IκB family members. (B) Immunoblotting for FLAG-tagged IKKβ and HA-tagged β-catenin proteins after immunoprecipitation using control IgG or antibodies against FLAG or HA proteins in C3H10T1/2 cells and HEK 293T cells. (C) In vitro phosphorylation of purified GST–β-catenin proteins by IKKβ in the presence of γ-[32P]ATP. (D–F) GST–β-catenin proteins phosphorylated by IKKβ in vitro were tryptic digested and analyzed by mass spectrometry. Figures show the recovered phosphorylated 30-residue fragment of β-catenin (residues 20–49). Tandem mass spectrum of the recovered peptide phosphorylated at ser45 residue (D). The peptides phosphorylated at ser33 and ser37 residues were coeluted. Color codes are used to mark fragment ions that allows distinguishing phosphorylation of ser33 (blue) and ser37 (red) (E). Tandem MS ion traces (10 ppm extraction) for the nonphosphorylated peptide, ser45- and ser33/ser37-phosphorylated peptides, and AUCs shown in italics (F).
To determine whether IKKβ can directly phosphorylate β-catenin, we performed in vitro kinase assays using IKKβ and purified GST–β-catenin fusion proteins (Supplemental Figure 3B) together with [32P]-labeled ATP. The results demonstrated that IKKβ can indeed phosphorylate GST–β-catenin proteins (Figure 7C), indicating IKKβ is a β-catenin kinase. In addition to using IKKβ proteins alone, we also used IKKβ antibodies to precipitate IKK protein complex in C3H/10T1/2 cells overexpressing IKKβ. Immunoblotting results confirmed that IKKα proteins were also present in the precipitated protein complex (Supplemental Figure 3C). In vitro kinase assays further demonstrated that the immunoprecipitated IKK protein complex can also phosphorylate β-catenin proteins (Supplemental Figure 3D), which is consistent with kinase assay results using IKKβ proteins alone (Figure 7C).
To map the precise IKKβ phosphorylation sites on β-catenin proteins, GST–β-catenin proteins from the in vitro kinase reaction mixture were purified and analyzed by tandem mass spectrometry (MS/MS). The peptide, containing β-catenin residues 20–49, was identified as phosphorylated, most prominently at serine residues ser33, ser37, and ser45 (Figure 7, D–F). While multiple phosphorylation sites were found to exist simultaneously, the majority of the measured phosphorylation signal (~80%) was associated with phosphorylation of either ser33, ser37, or ser45. Phosphorylation signals of ser33, ser37, and ser45 were also found to be similar (Figure 7F). To confirm the phosphorylation of these key residues of β-catenin proteins by IKKβ, we mutated each serine residue identified above to alanine and used the WT and mutated GST–β-catenin proteins for in vitro kinase assays. Three different antibodies that specifically recognize β-catenin phosphorylated at ser33, ser37, or ser45 were then used to detect the phosphorylated proteins. Immunoblotting analysis confirmed that IKKβ can phosphorylate each of the 3 serines independent of phosphorylation status of the others (Figure 8A). Further, overexpression of WT IKKβ, but not IKKβ KM, increased the phosphorylation of β-catenin in C3H/10T1/2 MSCs (Figure 8B), and treatment with FFAs that activate IKKβ also stimulated β-catenin phosphorylation in MSCs (Figure 8C). In addition to FFAs, other known IKKβ activators such as LPS can also increase β-catenin phosphorylation in control C3H/10T1/2 cells but not in IKKβ-deficient cells (Figure 8D). Further, IKKβ inhibitor BMS-345541 efficiently blocked LPS-induced β-catenin phosphorylation (Supplemental Figure 3E). Taken together, these studies demonstrated that IKKβ is an important β-catenin kinase that can directly phosphorylate 3 key serine residues, ser33, ser37, and ser45, of β-catenin.
Figure 8. IKKβ phosphorylates β-catenin to prime it for β-TrCP-mediated ubiquitination.

(A) GST–β-catenin and indicated mutant proteins were phosphorylated by IKKβ in vitro and analyzed by immunoblotting using anti–phospho-ser33, -ser37 or -ser45 β-catenin antibodies. (B) Immunoblotting for phosphorylated β-catenin proteins in C3H/10T1/2 cells infected with control, WT IKKβ, and IKKβ KM virus. (C) Immunoblotting for phosphorylated β-catenin proteins in C3H/10T1/2 cells treated with vehicle or FFAs. (D) Immunoblotting for phosphorylated β-catenin proteins in control or IKKβ-deficient C3H/10T1/2 cells treated with vehicle or LPS. (E) GST–β-catenin and indicated mutant proteins were phosphorylated by IKKβ in vitro. The reaction substrates were subjected for cell-free ubiquitination assays and analyzed by immunoblotting.
IKKβ-mediated phosphorylation of β-catenin increases its ubiquitination.
Phosphorylated ser33 and ser37 of β-catenin can be recognized by β-TrCP, an E3 ubiquitin ligase component that plays an essential role in the ubiquitination of β-catenin proteins (41, 43). We next examined whether IKKβ-mediated β-catenin phosphorylation is sufficient for β-TrCP–mediated ubiquitination. GST–β-catenin proteins were first subjected to in vitro kinase assays with or without IKKβ, and the reaction substrates were then collected for cell-free ubiquitination assays in the absence or presence of β-TrCP proteins. While there were no ubiquitination products in the absence of IKKβ or β-TrCP, β-catenin phosphorylation by IKKβ was sufficient to induce the β-TrCP–medicated β-catenin ubiquitination (Supplemental Figure 3F). However, mutation of either ser33 or ser37 substantially inhibited β-catenin ubiquitination by β-TrCP (Figure 8E). Mutation of ser45 did not affected β-catenin ubiquitination (Figure 8E), which is consistent with previous studies demonstrating that ser45 phosphorylation is not recognized for β-TrCP binding but rather creates a priming site for GSK-3β phosphorylating ser33 and ser37 (41, 42). Collectively, these results demonstrated that IKKβ phosphorylates the degron motif of β-catenin to prime it for β-TrCP–mediated ubiquitination.
Deficiency of IKKβ decreases adipogenesis and increases osteogenesis in BMMSCs, and increases bone mass in vivo.
To investigate the functions of IKKβ signaling in vivo, we previously generated a mouse model that lacks IKKβ in adipose progenitors, and we demonstrated that deficiency of IKKβ decreased adipogenesis in vivo and protected mice from diet-induced obesity (18). However, it was not clear whether IKKβ also functions in BMMSCs to regulate bone homeostasis in vivo. To determine the impact of IKKβ deficiency on bone homeostasis, we then used a well-defined Prrx1-Cre mouse model that targets BM mesenchymal stromal cells (27, 44) to generate a conditional IKKβ-KO mouse model (Prrx1Cre+IKKβF/F) in which IKKβ is deleted in BMMSCs. Prrx1Cre+IKKβF/F mice were variable and had normal body weight, lean mass, and fat mass at 20 weeks of age under standard laboratory conditions (Supplemental Figure 4, A and B). We then measured the IKKβ expression in BMMSCs of Prrx1Cre+IKKβF/F and IKKβF/F littermates, and we confirmed that IKKβ expression was indeed decreased in BMMSCs of Prrx1Cre+IKKβF/F mice as compared with IKKβF/F mice (Figure 9A). Consistently, BMMSCs derived from Prrx1Cre+IKKβF/F mice had decreased adipogenesis but increased osteogenesis as compared with IKKβF/F mice (Figure 9, B–E). Further, Prrx1Cre-mediated IKKβ deficiency also decreased β-catenin ubiquitination and induced nuclear β-catenin accumulation in BMMSCs (Figure 9, F and G).
Figure 9. Prrx1Cre-mediated IKKβ deficiency decreases adipogenesis and increases osteogenesis in BMMSCs.

(A) Immunoblotting for IKKβ proteins in BMMSCs of IKKβF/F and Prrx1Cre+IKKβF/F mice. (B–G) BMMSCs were isolated from IKKF/F and Prrx1Cre+IKKβF/F mice. Oil Red O staining (B) and qPCR analysis (C) of BMMSCs induced by an adipogenic cocktail (n = 3). ALP staining (D) and qPCR analysis (E) of BMMSCs induced by an osteogenic cocktail (n = 3). Immunoblotting for ubiquitinated β-catenin (F) and nuclear β-catenin proteins (G) of isolated BMMSCs. Scale bar: 100 μm. Error bars represent ± SEM. Significance was determined by Student’s t test (C and E). *P < 0.05; **P < 0.01.
We next performed microCT analyses on the femurs of adult Prrx1Cre+IKKβF/F and IKKβF/F littermates. MicroCT analyses demonstrated that trabecular bone volume was significantly increased in the femur metaphysis of Prrx1Cre+IKKβF/F mice as compared with control IKKβF/F mice (Figure 10, A and B). Prrx1Cre+IKKβF/F mice also had significantly increased trabecular number, BMD, and thickness (Figure 10, C–E), as well as decreased trabecular porosity and bone surface/bone volume (Figure 10, F and G). Trabecular spacing was also decreased but not significantly changed by IKKβ deficiency (Figure 10H). These results suggest increased trabecular bone formation and mass in Prrx1Cre+IKKβF/F mice. Interestingly, deficiency of IKKβ did not affect cortical bone volume, BMD, and porosity, and it only increased cortical thickness as compared with the IKKβF/F mice (Supplemental Figure 4, C–G), indicating that IKKβ acts locally to regulate bone mass. Consistent with the microCT results, histological analysis and immunostaining for the adipocyte marker perilipin also demonstrated that deficiency of IKKβ significantly reduced adipocyte numbers in BM of Prrx1Cre+IKKβF/F mice (Figure 10, I–K). Taken together, Prrx1Cre-mediated IKKβ deficiency increased osteogenesis and inhibited adipogenesis of BMMSCs and increased bone mass in vivo.
Figure 10. Deficiency of IKKβ in BMMSCs increases bone mass in vivo.

(A) MicroCT reconstruction images of trabecular bone from the femur metaphysis of 20-week-old IKKβF/F and Prrx1Cre+IKKβF/F littermate mice. Scale bar: 100 μm. (B–H) MicroCT analyses of trabecular bone volume/total volume (BV/TV) (B), number (C), BM density (BMD) (D), thickness (E), porosity (F), bone surface/bone volume (G), and spacing (H) in the femur metaphysis of IKKβF/F and Prrx1Cre+IKKβF/F mice (n = 5). (I and J) Representative H&E staining (I), IHC for perilipin (J), and quantification of perilipin-positive adipocytes (K) in femurs (n = 5). Scale bar: 500 μm (I) or 100 μm (J). Error bars represent ± SEM. Significance was determined by Student’s t test (B-H, and K). *P < 0.05; **P < 0.01.
IKKβ regulates adipogenesis and osteogenesis in human MSCs.
To investigate the functions of IKKβ in human MSCs, we isolated human adipose stem cells from healthy subjects (34, 45). The cells were then infected with adenovirus expressing IKKβ WT or mutant IKKβ KM (Figure 11A). As expected, overexpression of WT IKKβ or IKKβ KM proteins in human adipose stem cells had similar effects on β-catenin phosphorylation and ubiquitination (Figure 11B), nuclear β-catenin protein levels (Figure 11C), adipogenesis, and osteogenesis (Figure 11, D–G) as murine MSCs. Further, FFA-mediated IKKβ activation also increased β-catenin phosphorylation and ubiquitination (Figure 11, H and I), leading to increased adipogenic potential but reduced osteogenic potential of adipose stem cells (Figure 11, J–M). In addition to human adipose stem cells, overexpression of WT IKKβ or IKKβ KM proteins (Supplemental Figure 5, A–F), pharmacological inhibition of IKKβ (Supplemental Figure 6, A–E), or FFA treatment (Supplemental Figure 6, F–J) resulted in similar effects on β-catenin ubiquitination, adipogenesis, and osteogenesis in human BMMSCs. Collectively, these results demonstrated the important role of IKKβ in regulating adipogenesis and osteogenesis of human MSCs.
Figure 11. IKKβ regulates adipogenesis and osteogenesis of human MSCs.

(A–C) Adipose stem cells were isolated from s.c. adipose tissue of heathy human subjects. Immunoblotting for IKKβ and phosphorylated β-catenin proteins (A), ubiquitinated β-catenin proteins (B), and nuclear β-catenin proteins (C) in human adipose stem cells infected with control, WT IKKβ, and IKKβ KM virus. (D and E) Oil Red O staining (D) and qPCR analysis (E) of human adipose stem cells induced by an adipogenic cocktail (n = 3). Scale bar: 100 μm. (F and G) Alizarin Red S staining (F) and qPCR analysis (G) of human adipose stem cells induced by an osteogenic cocktail (n = 3). Scale bar: 100 μm. (H and I) Immunoblotting for phosphorylated β-catenin proteins (H) and ubiquitinated β-catenin proteins (I) in human adipose stem cells treated with vehicle control or FFAs. (J and K) Oil Red O staining (J) and qPCR analysis (K) of control or FFA-treated human adipose stem cells induced by an adipogenic cocktail (n = 3). Scale bar: 100 μm. (L and M) Alizarin Red S staining (L) and qPCR analysis (M) of control or FFA-treated human adipose stem cells induced by an osteogenic cocktail (n = 3). Scale bar: 100 μm. Error bars represent ± SEM. Significance was determined by Student’s t test (K and M) or 1-way ANOVA (E and G). *P < 0.05; **P < 0.01, ***P < 0.001.
Elevated IKKβ expression and increased β-catenin phosphorylation in adipose tissue of obese humans.
To further determine whether IKKβ signaling is associated with increased adiposity or obesity in humans, a cohort of nondiabetic human subjects with BMI ranged from 24–45 were recruited (Table 1 and Supplemental Table 1), and IKKβ mRNA expression was examined in abdominal s.c. adipose tissue. Interestingly, we observed a significant correlation between adipose IKKβ expression levels and BMI (Figure 12A). IKKβ mRNA levels were significantly higher in obese subjects (BMI > 30) than nonobese subjects (BMI < 30) (Figure 12B). Further, the protein levels of both total and phosphorylated IKKβ were elevated in adipose tissue of obese subjects (Figure 12, C and D) which also had increased phosphorylation of β-catenin on ser33, ser37, and ser45 (Figure 12, C and D). These results indicate the potential impact of IKKβ activation on β-catenin/Wnt signaling in humans.
Table 1. Baseline characteristics of human subjects.
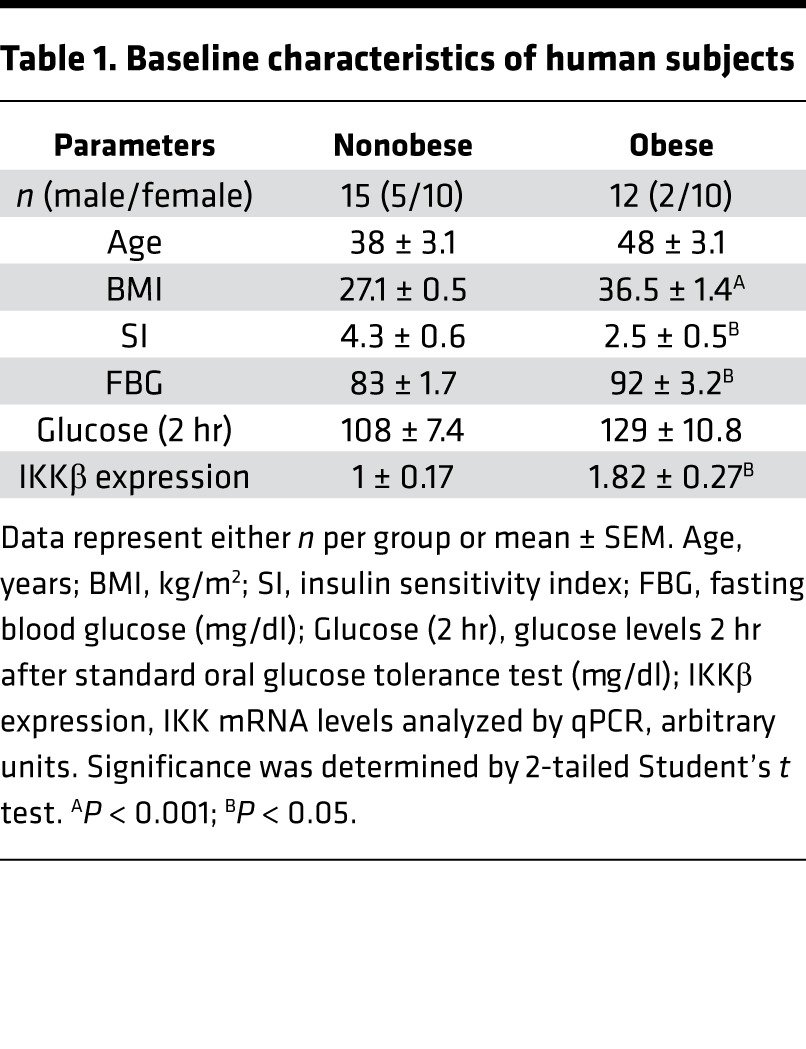
Figure 12. IKKβ expression and β-catenin phosphorylation are elevated in adipose tissue of obese humans.

(A) s.c. adipose tissues were isolated from a cohort of nondiabetic human subjects. Correlation between adipose IKKβ mRNA levels and BMI (n = 27). The correlation was analyzed by Pearson correlation coefficient. (B) IKKβ mRNA levels in adipose tissue of nonobese and obese human subjects (n = 12–15). (C and D) Immunoblotting (C) and densitometric quantification (D) of proteins in adipose tissue of nonobese and obese human subjects (n = 7). Error bars represent ± SEM. Significance was determined by Student’s t test (B and D). *P < 0.05; **P < 0.01, ***P < 0.001. (E) Schematic representation of the mechanism through which IKKβ reciprocally regulates adipogenesis and osteogenesis in MSCs. Activation of IKKβ by stimuli such as FFAs or inflammation cytokines phosphorylates serine-33, -37, and -45 of β-catenin to prime it for β-TrCP–mediated ubiquitination and degradation, leading to increased adipogenic differentiation and reduced osteogenic differentiation of MSCs.
Discussion
Adipocytes and osteoblasts originate from common precursors, MSCs, which can be found in many tissues including fat, BM, muscle, and brain (46, 47). Increasing evidence of the differentiation switching between osteoblasts and adipocytes suggests the plasticity existing between these 2 cell lineages (19, 26, 27, 48). Multiple factors including PPARγ and leptin receptor have been demonstrated to influence the lineage-fate of MSCs (26, 27, 49) but how the different signaling pathways coordinately regulate this process remains poorly understood. We previously reported that IKKβ functions in adipocyte precursor cells to regulate adipocyte differentiation and adipose tissue expansion (7, 18). In the current study, we have uncovered a dual role of IKKβ in the regulation of MSC differentiation (Figure 12E). IKKβ loss of function impaired adipogenesis but enhanced osteogenesis of murine and human MSCs. By contrast, overexpression or activation of IKKβ increased adipogenesis and inhibited osteogenesis of MSCs. Mechanistic studies revealed a network between IKKβ and Wnt/β-catenin signaling, as well as demonstrated that IKKβ is a β-catenin kinase that can directly phosphorylate β-catenin to prime it for ubiquitination. It has been well-recognized that Wnt/β-catenin signaling is essential for osteoblast formation from MSCs but inhibits adipocyte differentiation (35, 36). The effects of IKKβ signaling on adipogenesis and osteogenesis were diminished in β-catenin–deficient MSCs. Therefore, IKKβ-mediated β-catenin phosphorylation and ubiquitination plays an important role in maintaining balance between osteogenic and adipogenic differentiation of MSCs (Figure 12E).
As one of the most studied kinases in regulating inflammation, IKKβ has been well-established as a central mediator of inflammation and immune responses (9, 50). In addition, IKKβ signaling has been shown to be important for stem cell differentiation and hematopoietic cell development (50, 51). For example, the canonical IKKβ/NF-κB pathway is required for initial differentiation of neural stem cells (51). Proinflammatory signaling in adipose tissue has been demonstrated to be important for HF-induced adipogenesis and adipose tissue expansion (52). We recently reported a previously unrecognized role of IKKβ in the regulation of adipocyte differentiation (7, 18). Our current study demonstrates that IKKβ can regulate both adipogenesis and osteogenesis in MSCs from multiple tissues, including adipose tissue, BM, and embryonic tissue. The early signals triggering MSC commitment to adipocyte lineage in response to overnutrition remain elusive. Nutrients are considered naturally inflammatory (3), and many FFAs can activate IKKβ and induce cellular inflammatory responses (15, 32, 34). We demonstrate here, for the first time to our knowledge, that FFA-elicited IKKβ activation enhanced adipogenesis and inhibited osteogenesis of MSCs. Further, deficiency of IKKβ blocked FFAs’ impact on MSC differentiation. In addition to FFAs, IKKβ can also be activated by numerous inflammatory signals, including many cytokines. Therefore, IKKβ may serve as a key molecular switch that triggers the adipogenic and osteogenic differentiation of MSCs upon nutritional challenges or altered inflammation.
Previous studies have implicated canonical NF-κB signaling in bone homeostasis. Activation of NF-κB signaling inhibits osteoblastic bone formation through regulating Fos-related antigen-1 expression (53). NF-κB can transcriptionally regulate the expression of SMAD ubiquitination regulatory factors, which also target β-catenin to inhibit osteogenesis (38). More recently, Wang et al. also demonstrated that NF-κB can upregulate miR150-3p that target β-catenin to inhibit osteogenesis in MSCs (54). NF-κB p65-p50 heterodimer has been shown to directly bind to the promoters of OC and bone sialoprotein (Bsp) and to inhibit β-catenin–induced transcription of OC and Bsp in osteoblasts (55). Interestingly, IKKβ — but not IKKα — is an important survival factor for osteoclasts and is required for inflammation-induced bone loss (56). Therefore, the role of IKKβ in bone homeostasis is complex, and IKKβ may regulate both osteogenesis and osteoclastogenesis in vivo through NF-κB–dependent and NF-κB–independent mechanisms. Future studies are required to dissect the detailed mechanisms of the crosstalk between IKKβ and Wnt/β-catenin signaling and its implication in bone homeostasis and other diseases.
Although much attention has been focused on the NF-κB–dependent functions of IKKβ, IKKβ can regulate inflammation, apoptosis, cell proliferation, and metabolic homeostasis through NF-κB–independent mechanisms by phosphorylating other key molecules such as SNAP-23, IRF7, p53, BAD, and XBP1 (39, 40, 57). For example, IKKβ can inhibit cell death in an NF-κB–independent manner by phosphorylating BAD, a BH3-only proapoptotic protein, to prime it for inactivation (40). We also found that IKKβ phosphorylation of BAD is important for adipocyte survival in obesity (34). A recent study demonstrated that IKKβ can phosphorylate XBP1 to regulate glucose homeostasis in the liver (57). We and others previously demonstrated that IKKβ affects β-catenin ubiquitination and degradation in different cell types (7, 18, 38), but the precise mechanisms have not been elucidated. β-Catenin degradation is regulated by the ubiquitin-proteasome pathway, and phosphorylation of the key serine residues ser33 and ser37 of β-catenin degron motif by GSK3β is required for β-TrCP–mediated ubiquitination. Intriguingly, the degron motif of β-catenin is highly conserved in both β-catenin and IKKβ substrates of the IκB family members. Our results convincingly demonstrate that IKKβ can phosphorylate ser33 and ser37 of β-catenin and mutation of either serine can decrease β-TrCP-mediated β-catenin ubiquitination. In addition to these 2 serines, IKKβ can also phosphorylate ser45 of β-catenin. Although ser45 does not serve as a recognition site for the β-TrCP–ubiquitin ligase complex, phosphorylation of ser45 primes β-catenin for GSK-3β phosphorylating ser33 and ser37, leading to the initiation of β-catenin phosphorylation–ubiquitination cascade (42). However, ser45 phosphorylation is not required for IKKβ phosphorylating ser33 or ser37 in vitro, as IKKβ can still phosphorylate ser33 and ser37 of GST–β-catenin (S45A) mutant proteins. It would be interesting to study whether ser45 phosphorylation affects IKKβ phosphorylating ser33 and ser37 in vivo in the future. The functions of a complex of GSK3β, APC, axin, and CKIα in the regulation of β-catenin phosphorylation and degradation have been extensively studied by many groups (41–43). Our findings demonstrate that IKKβ is an important β-catenin kinase that regulates canonical Wnt/β-catenin signaling. Since IKKβ can be activated by numerous signals, including many inflammatory cytokines, these findings may provide a connection between inflammation and Wnt signaling, which may lead to new insights into the pathogenesis of multiple human diseases, including but not limited to obesity, osteoporosis, and cancer.
In conclusion, our studies have uncovered a critical role of IKKβ in the regulation of MSC fate and have provided potentially novel mechanistic insights on the crosstalk between IKKβ and Wnt/β-catenin signaling. We suggest that IKKβ serves as a molecular switch for the Wnt pathway in response to inflammatory stimuli and excessive nutrients. Both IKK and Wnt/β-catenin signaling are evolutionarily conserved pathways, and this IKKβ-Wnt axis we uncovered will have important implications for numerous biological phenomena across many species. Findings from our studies will hopefully stimulate further investigations of IKKβ and Wnt/β-catenin crosstalk, as well as the perspective of the IKKβ-Wnt axis in development, homeostasis, and disease pathogenesis.
Methods
Mice.
To delete IKKβ in BM mesenchymal stromal cells, mice containing loxP-flanked IKKβ alleles (IKKβF/F) (7, 18, 31) were crossed with Prrx1-Cre transgenic mice (27, 44) (The Jackson Laboratory; catalog 005584) to generate Prrx1Cre+IKKβF/F mice. All skeleton analyses were performed on the femurs of 20-week-old mice unless otherwise specified. All animals were housed in a specific pathogen-free environment with a light-dark cycle, under a protocol approved by the University of Kentucky IACUC.
Human subjects.
The human samples used in this manuscript were described previously (58), and the protocols were approved by the IRB at the University of Kentucky. Briefly, abdominal s.c. adipose tissue was obtained from 27 human subjects (BMI ranges from 24–44.7) (Table 1 and Supplemental Table 1). Participants were assigned in 2 groups, nonobese group (BMI ≤ 30; n = 15, male 5, female 10) and obese group (BMI > 30; n = 12, male 2, female 10). Insulin sensitivity index (SI) was measured by frequently sampled i.v. glucose tolerance test. All participants were generally healthy, with no evidence of chronic inflammatory disease; they participated in normal activities of daily living (ADL) without significant exercise and were weight stable (±3%) for at least 3 months. No subjects were employed in jobs that involved prolonged outdoor activity, none were taking any medication likely to change adipocyte metabolism, and there were no abnormalities in liver enzymes, creatinine, or thyroid-stimulating hormone (TSH). All subjects were initially assessed with a standard oral glucose tolerance test to rule out diabetes. White adipose tissue surgical biopsies (s.c.) were operated from the anterior abdomen under local anesthesia within 1 hr of coming inside.
Cell lines.
Mouse C3H10T1/2 cells (ATCC CRL-3268), human HEK293T cells (ATCC CRL-3216), and human BMMSCs (ATCC PCS-500-012) were purchased from ATCC and cultured following the manufacturer’s instruction. MEFs, mouse BMMSCs, and human adipose stem cells were isolated as described below. All of the above cells were cultured at 37°C in a humidified atmosphere with 5% CO2.
Primary cell isolation.
MEFs were isolated as previously described (59). Briefly, the embryos between E13 and E15 were minced and typsinized for 20 minutes, after the heads, tails, limbs, and most of the internal organs were removed. The MEFs were then cultured in DMEM containing 10% FBS (MilliporeSigma, F4135). Mouse BMMSCs were isolated from the femur and tibia of mice as previously described (60) and cultured in DMEM containing 15% FBS. Human HEK293T cells were purchased from ATCC and maintained in DMEM with 10% FBS. Human adipose stem cells were isolated from the s.c. adipose tissue from human subjects as previously described (34). The adipose tissue was from a collagenase (MilliporeSigma, C6885) digestion of the lipoaspirate of patients undergoing liposuction of s.c. fat. These subjects were generally healthy and were young women undergoing cosmetic procedures, and this method of collection was approved by the University of Kentucky IRB. All of above cells were cultured at 37°C in a humidified atmosphere with 5% CO2.
Total protein extraction from cells.
Cells were lysed in lysis buffer (Cell Signaling Technology, 9803) containing protease inhibitor cocktails (Roche Diagnostics, 11836153001) and phosphatase inhibitor cocktails (MilliporeSigma, P5726 and P0044). After incubation on ice for 30 minutes with frequent vortex, cell lysates were centrifuged at 16,000 g for 15 minutes at 4°C and the supernatants were collected. Protein concentrations were measured by using BCA protein assay kit (Thermo Fisher Scientific, 23225). Proteins were denatured by boiling at 100°C for 5 minutes in 1× Laemmli buffer (Bio-Rad, 161-0737). The lysates were cooled to room temperature before loading to Western blot gel.
Total protein extraction from tissues.
Human adipose tissues (100 mg) were homogenized with Bullet Blender (Next Advance, BBX24) in 0.5 ml of ice-cold lysis buffer (Cell Signaling Technology, 9803) containing protease inhibitor cocktails (Roche Diagnostics, 11836153001) and phosphatase inhibitor cocktails (MilliporeSigma, P5726 and P0044). After homogenization, lysates were centrifuged at 16,000 g for 15 minutes at 4°C. The top lipid layer was carefully removed, and the remaining supernatant was centrifuged twice to completely remove the lipid. Protein concentrations were measured by using BCA protein assay kit (Thermo Fisher Scientific, 23225). Proteins were denatured in 1× Laemmli buffer (Bio-Rad, 161-0737) by boiling at 100°C for 5 minutes. The lysates were cooled to room temperature before loading for Western blot analysis.
Nuclear protein extraction.
Nuclear proteins were isolated by utilizing Nuclear Extract kit (Thermo Fisher Scientific, 78833) following the manufacturer’s instruction. Briefly, the cells were collected into buffer CER I, and were vortexed vigorously for 15 seconds, followed by a 10-minute incubation on ice. The ice-cold buffer CER II was added to the mixture, and the tubes were vortexed for 5 seconds. The samples were incubated on ice for 1 minute, vortexed for another 5 seconds, and centrifuged for 5 minutes at the speed of 16,000 g at 4°C. The supernatant containing cytoplasmic extract was immediately transferred into a new prechilled tube and further lysed in 1× Laemmli buffer. The pellet (nuclear fraction) was incubated in ice-cold NER buffer for total 40 minutes and vortexed for 15 seconds every 10 minutes. The lysate was centrifuged at maximum speed for 10 minutes at 4°C in order to collect the supernatant (nuclear extract).
Western blotting.
Western blotting was performed as previously described (7, 18). Briefly, cell or tissue lysate samples, prepared as described above, were resolved on SDS-PAGE. Proteins were then transferred to nitrocellulose (NC) membrane. The membrane was blocked in phosphate-buffered saline solution with 0.05% Tween 20 (PBST, pH 7.4) containing 5% BSA (MilliporeSigma, A9647) for 1–3 hours and then incubated with primary antibody in PBST containing 5% BSA at 4°C overnight. After the incubation, the membrane was washed 4 times with PBST and incubated with secondary antibody in PBST with 5% nonfat dry milk (Bio-Rad, 170-6404) for 1 hour at room temperature. After subsequent 3-time washing in PBST, the membrane was washed once in PBS and developed using Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, 32209) and exposed to CL-XPosure films (Thermo Fisher Scientific, 34099). Western blots were probed with the primary antibodies at a dilution of 1:1,000 except for the following antibodies: anti–β-catenin (MilliporeSigma, C2206, 1:8,000), anti–β-actin (MilliporeSigma, A2066, 1:5,000), and anti-ubiquitin (Santa Cruz Biotechnology Inc., sc-8017 HRP, 1:300). Anti-IKKβ (catalog 2678), anti–pIKKβ (Ser176/180, catalog 2697), anti–pβ-catenin (Ser45, catalog 9564), anti-FLAG (catalog 8146), anti-HA (catalog 2367), anti-TFIIB (catalog 4169), anti–NF-κB p65 (catalog 3034), and anti–pNF-κB p65 (Ser536, catalog 3033) antibodies were purchased from Cell Signaling Technology. Anti–pβ-catenin (Ser33, catalog ab73153), and anti–pβ-catenin (Ser37, catalog ab47335) antibodies were purchased from Abcam.
Virus production and transduction.
Adenovirus particles expressing GFP control, WT IKKβ, and IKKβ KM were provided by Haiyan Xu (Brown University, Providence, RI, USA) (15). Viral particles were used for the infection of HEK293T cells (ATCC) for amplification. After 48-hour infection, HEK293T cells were collected and lysed by frequent freezing and thawing. Supernatant that contains viral particles was further used for purification by using Virabind adenovirus purification kit (Cell Biolabs, VPK-100). For lentivirus production and amplification, pPACKH1 HIV Lentivector Packaging Kit (System Biosciences, LV500A-1) was used as per the manufacturer’s instruction. The plasmids for lentiviral-Cre (LV-Cre) and lentiviral-LacZ (LV-lac) were purchased from Addgene. For the virus-mediated overexpression, cells in 6-well plates were washed twice with PBS and treated with 2 ml of serum-free media containing 5 mg/ml of polybrene and virus. The plates were sealed with parafilm, placed in microplate adaptors, and centrifuged for 1 hour at the speed of 700 g at room temperature. Parafilm was removed after centrifugation, and the plates were placed in the 37°C tissue culture incubator for 4 hours, followed by adding 2 ml of complete media to each well. After an overnight incubation, the virus-containing media was removed and replaced with fresh complete media. The cells were used for further experiments after 2 days.
Adipogenic differentiation.
Adipogenesis protocol of mouse cells was developed as previously described (7, 18). Briefly, 2 days after the cells were confluent, DMEM supplemented with 10% FBS, 1 μM dexamethasone, 0.5 mM isobutylmethylxanthine, and 10 μg/ml insulin (5 μg/ml for C3H10T1/2 cell) were utilized as the induction media to induce adipogenesis. For C3H10T1/2, MEF, and mouse BMMSC differentiation, 1 μM of rosiglitazone was added into the above induction media. Dexamethasone (D4902), isobutylmethylxanthine (I5879), insulin (I2643), and rosiglitazone (R2408) were purchased from MilliporeSigma. Three days later, the media was changed into maintenance media (induction media without dexamethasone and isobutylmethylxanthine). Dexamethasone (1 μM) was added into the maintenance media for BMMSC differentiation. For the FFA treatment experiments, insulin was removed from the induction media and the maintenance media in the presence of 0.5 mM FFA mixture containing myristic, lauric, arachidonic, oleic, and linoleic acids (15, 32, 34). IKKβ inhibitor BMS-345541 (5 μM, MilliporeSigma, B9935) was used to inhibit the activity of IKKβ during adipogenesis. Wnt inducers Wnt3a (100 ng/ml, R&D Systems, 5036-WN-010) and LiCl (20 mM) were included in the induction and maintenance media to activate Wnt/β-catenin signaling. Every 2 days, the maintenance media was replaced until the cells are ready for Oil-Red O staining. For the differentiation of human adipose stem cells and BMMSCs, the protocol was followed as previously described (34). The 2-day postconfluent cells were induced with DMEM/F10 supplemented with 3% FBS, 15 mM HEPES (MilliporeSigma), 33 μM Biotin (MilliporeSigma), 17 μM panthothenate (MilliporeSigma), 1 μM of dexamethasone, 100 nM human insulin, 0.25 mM isobutylmethylxanthine, and 1 μM rosiglitazone. Three days later, the media was changed to maintenance media containing DMEM/F10 supplemented with 3% FBS, 15 mM HEPES, 33 μM Biotin, 17 μM panthothenate, 1 μM dexamethasone, and 100 nM human insulin. The maintenance media was replaced every 3 days until the cells were ready for analysis.
Osteogenic differentiation.
Osteogenic media (MEM containing 10% FBS, 100 nM dexamethasone, 50 μM L-ascorbic acid, and 10 mM β-glycerophosphate) was used to incubate the cells after confluency. L-ascorbic acid (A4403) and β-blycerophosphate (G9422) were purchased from MilliporeSigma. The media was replaced every 3 days for 2 weeks. BMS-345541 (5 μM), 0.5 mM FFA mixture, 100 ng/ml Wnt3a, or 20 mM LiCl was added into the osteogenic media to study their impact on osteogenesis. The ALP staining was used to determine the osteogenesis of C3H10T1/2, mouse or human BMMSC, and mouse SV cells (26). Briefly, cultured plates were rinsed with PBS, fixed in 10% formalin buffered with PBS, and stained with 0.1 M Tris-HCl (pH 9.5) solution containing 0.1 M of NaCl, 0.05 M of MgCl2, and NBT/BCIP (MilliporeSigma) as a substrate. Mineralization of the extracellular matrix in human adipose stem cells was visualized by Alizarin Red S staining. Briefly, the cultured plates were rinsed with PBS, fixed in 10% buffered formalin, and stained with 2% Alizarin red S (pH 4.0) (MilliporeSigma).
CRISPR/Cas9-KO in cells.
The CRISPR/Cas9 method was used to delete IKKβ or β-catenin in C3H/10T1/2 cells. Briefly, The lentiCRISPR plasmid (Addgene plasmid 52961), containing hCas9 and single-guide RNA (sgRNA), was digested with BbsI, and a pair of annealed oligonucleotides for IKKβ (5′-CACCGGCAGCCGTTGGGGCCATACT-3′) or β-catenin (5′-CACCGATGAGCAGCGTCAAACTGCG-3′) for targeting site was cloned into the guide RNA according to the previous protocol (61). The lentivirus was prepared in HEK293T cells, and the viral supernatant was then used to infect C3H10T1/2 cells. The cells were selected for 7 days with 1 μg/ml puromycin, and single clones were expanded and screened by Western blot analysis. Further, the Transgenomic Surveyor Mutation Detection Kit (Thermo Fisher Scientific, 706020) and genomic DNA sequencing were used to verify genomic mutations within the locus of IKKβ or β-catenin. For LPS treatment, the β-catenin–deficient-C3H/10T1/2 cells were treated with LPS (500 ng/ml, MilliporeSigma, L4391) together with PS-341 (100 nM, Cell Signaling Technology, VCA-1003) for 4 hours in order to detect the phosphorylation of β-catenin by immunoblotting.
Cell transfection.
For siRNA transfection assays, 0.2 μM of IKKβ siRNAs (MilliporeSigma, SASI_Mm01_00132411 and 00132412) were transfected into 2 × 106 cells via Amaxa Cell Line Nucleofector kit V (Lonza VCA-1003) following the manufacturer’s protocol. The cells were then cultured with complete media for 2–3 days until they were ready for further analysis. For β-catenin reporter activity assays, cells were transfected with the TOP-flash reporter plasmids to evaluate the activity of β-catenin, as well as β-gal plasmids as a transfection control, by using Amaxa Cell Line Nucleofector kit V (Lonza) (18). Two days after transfection, the cells were lysed and the activities of luciferase and β-gal were measured as previously described (18).
Recombinant β-catenin proteins.
GST-tagged full-length human β-catenin plasmid (pGEX-Bcatfl) was obtained Addgene (plasmid 24193, a gift from David Rimm at Yale University, New Haven, CT). The mutant plasmids were generated by using QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies). To express the recombinant proteins, the pGEX-Bcatfl plasmid was transformed into E. coli Express BL21 (DE3) Chemically Competent Cells (Lucigen, 60401), and a single colony was inoculated into 5 ml of Luria-Bertani (LB) medium containing ampicillin. After overnight culture, 0.5 ml of bacterial culture was inoculated into 50 ml of LB medium containing ampicillin to incubate with shaking at 37°C until the OD600 reached 0.6–0.8. Isopropyl β-D-1-thiogalactopyranoside (IPTG, 0.5 mM) was used to induce the recombinant protein expression at 37°C for 4 hours. The cell pellet was collected by centrifugation for 15 minutes at the speed of 5,000 g at 4°C and lysed by sonication on ice. The bacterial lysates were used for protein purification via Glutathione Sepharose 4B (GE Healthcare, 17075601) following the manufacturer’s instruction.
IKKβ kinase assays.
For cell-free kinase assay, 100 ng of recombinant human IKKβ proteins (SignalChem, I03-18G) were incubated with purified GST-tagged full-length human β-catenin protein and 150 nCi/nmol of γ-[32P] ATP (PerkinElmer, BLU002A250UC) with a total 100 nM of ATP in kinase buffer (50 mM Tris-HCl, pH 7.7, 10 mM MgCl2, 1 mM DTT, and protease inhibitor cocktail). Kinase reactions were incubated for 60 minutes at 30°C and denatured in Laemmli sample buffer (Bio-Rad), followed by autoradiography or Western blot analysis.
LC-MS/MS analysis of β-catenin phosphorylation.
After in vitro phosphorylation by IKKβ protein (SignalChem) in kinase buffer containing 100 nM of ATP, the human β-catenin proteins were separated by SDS-PAGE and excised followed by trypsinization as described previously (62). Extracted peptides were analyzed by LC-MS/MS (Ultimate 3000 nano-HPLC system coupled to a Q-Exactive Plus mass spectrometer, Thermo Fisher Scientific). Peptides were separated on a C18 column (12 cm/75 μm, 3 μm beads, Nikkyo Tecnologies) at 200 nl/min with a gradient increasing from 1% buffer B/95% buffer A to 45% buffer B/55% buffer A in 137 minutes (buffer A, 0.1% formic acid; buffer B, 0.1% formic acid in acetonitrile) and analyzed in a data dependent (DDA) manner. The peptide AAVSHWQQQSYLDSGIHSGATTTAPSLSGK was found to be phosphorylated and targeted in a follow-up parallel reaction monitoring (PRM) experiment (63). The peptide in its unphosphorylated form as well as with 1, 2, 3 and 4 phosphorylations in charge states 3+ and 4+ were targeted in the PRM experiment. Tandem MS spectra were recorded at 17,500 resolution with m/z 100 as lowest mass. Normalized collision energy was set at 27, with AGC target and maximum injection time being 2 × 105, and 60 ms, respectively. DDA data were extracted and queried against UniProt’s Human database (http://www.uniprot.org/) concatenated (December 2014) with common contaminants using Proteome Discoverer 1.4 (Thermo Fisher Scientific) and MASCOT 2.5.1 (Matrix Science). Acetyl (Protein N-term), Oxidation (Methionine), and phosphorylation of serine, threonine, and tyrosine were allowed as variable modifications. All cysteines were treated as being carbamidomethylated. Both 10 ppm and 20 mDa were used as mass accuracy for precursors and fragment ions, respectively. Matched peptides were filtered using 1% FDR calculated by Percolator and, in addition, requiring that a peptide was matched as rank 1 and that precursor mass accuracy was better than 5 ppm. PhosphoRS v. 3.0 was used to in the assignment of phosphorylated residues. PRM data were analyzed using Skyline v. 3.5. Second stage of mass spectrometry (MS2) data were used to validate and to estimate phosphorylation stoichiometry. First stage of mass spectrometry (MS1) data were used to estimate total phosphorylation. Multiple charge states for each peptide were used for quantitation.
Immunoprecipitation.
For immunoprecipitation experiments, cells were incubated with 100 nM of PS-341 (Cell Signaling Technology, VCA-1003) for 4 hours to block the proteasome-mediated ubiquitination. The whole-cell lysates were then isolated using lysis buffer (Cell Signaling Technology) containing protease inhibitor cocktails (Roche Diagnostics), incubated with anti–β-catenin antibodies (MilliporeSigma, C2206) overnight at 4°C, and then incubated with Protein A Agarose beads (Roche Diagnostics, 11134515001) for another 5 hours at 4°C. The samples were washed with lysis buffer, followed by 4-time wash with ice-cold PBS, and eluted into 2× Laemmli buffer (Bio-Rad) by boiling at 100°C for 5 minutes. The supernatant was analyzed by Western blot using anti-ubiquitin monoclonal antibodies (Santa Cruz Biotechnology Inc., sc-8017 HRP) (7). For pull-down experiment, HA-tagged β-catenin plasmid (a gift from Brett Spear (University of Kentucky, Lexington, KY); ref. 64) and FLAG-tagged IKKβ plasmid (pcDNA-Ikkβ-FLAG WT [Addgene plasmid 23298] was a gift from Warner Greene, University of California, San Francisco, CA ) were cotransfected into C3H10T1/2 or HEK293T cells (ATCC). FLAG-IKKβ–containing proteins were immunoprecipitated with an anti-FLAG antibody M2 agarose (MilliporeSigma, F2555) and then immunoblotted using an anti-FLAG (1:2,000 dilution, Cell Signaling Technology, 8146) or anti-HA antibody (1:2,000 dilution, Cell Signaling Technology, 2367). In a reciprocal fashion, HA–β-catenin–containing proteins were immunoprecipitated with the anti-HA antibody (Cell Signaling Technology, clone 2367) and then immunoblotted using the anti-FLAG or anti-HA antibody. To detect the endogenous IKKβ and β-catenin interactions, C3H/10T1/2 cells were treated with vehicle control, FFA or TNFα (20 ng/ml, MilliporeSigma, T7539) for 4 hours. The cell lysates were immunoprecipitated with anti–IKKβ antibodies (Cell Signaling Technology, 2678) or anti–β-catenin antibodies (MilliporeSigma, C2206), and then immunoblotted with anti–β-catenin antibodies (1:8,000 dilution) and anti-IKKβ antibodies (1:1,000 dilution) (Cell Signaling Technology, clone 2678). For the endogenous IKK protein complex pull-down experiment, HEK293T cells were treated with WT IKKβ adenovirus for 48 hours. The endogenous IKK protein complex in the cell lysate were immunoprecipitated with anti-IKKβ antibodies (Cell Signaling Technology, 2678) and then immunoblotted with anti-IKKβ antibodies (1:2,000 dilution) and anti-IKKα antibody (1:1,000 dilution, Cell Signaling Technology, 2682) for confirmation. The immunoprecipitated IKK protein complex was used for kinase assay by incubating with purified GST–β-catenin proteins as described above in the IKKβ kinase assays section.
Ubiquitination assays.
For the cell-free ubiquitination assay, the purified β-catenin proteins or its mutants were first incubated with the IKKβ protein (SignalChem) in kinase buffer (50 mM Tris-HCl, pH 7.7, 10 mM MgCl2, 1 mM DTT, and protease inhibitor cocktail) containing 100 nM of ATP for 60 minutes at 30°C. The phosphorylated β-catenin proteins or its mutants were then mixed with E1 ubiquitin-activating enzyme, E2 (UbcH5), Mg-ATP solution, ubiquitin, and ubiquitination buffer (Enzo Life Science) in the presence of purified β-TrCP (p4489 FLAG-β-TrCP plasmid [Addgene plasmid 10865], a gift from Peter Howley, Harvard Medical School, Boston, MA) by FLAG immunoprecipitation. The reaction was incubated at 37oC for 1 hour, followed by SDS-PAGE and Western blotting analysis to detect the ubiquitinated β-catenin utilizing anti-ubiquitin antibodies (Santa Cruz Biotechnology Inc., sc-8017 HRP). For cell-based ubiquitination assay, the cells were treated with 100 nM of proteasome inhibitor PS-341 for 4 hours. Total cell lysates were isolated and immunoprecipitated with anti–β-catenin antibodies (MilliporeSigma) in order to estimate ubiquitinated β-catenin by Western blot analysis using anti-ubiquitin antibody (Santa Cruz Biotechnology Inc.).
MicroCT analyses.
High-resolution microCT was performed at the MicroCT laboratory of the Texas A&M University Baylor College of Dentistry using a ScanCo MicroCT 35 Scanner as described previously (27). Briefly, femurs were dissected, fixed in 10% formalin, and stored in 70% ethanol at 4°C. Trabecular bone parameters were measured in the distal metaphysis of the femurs. The bottom of the distal growth plate was used as the initiation of the slices analysis, and 100 slices (3.5 μm/slice) were analyzed toward the proximal end of the femur. Cortical bone parameters were measured by analyzing 100 slices (3.5 μm/slice) in middiaphysis femurs.
IHC.
Dissected femurs were fixed in 10% neutral buffered formalin for 2 days and decalcified in 10% EDTA for 5 days. Samples were further processed for paraffin-embedded section, and H&E staining was performed following standard protocol. For IHC, deparaffinized and rehydrated sections were treated with sodium citrate buffer (Vector Laboratories, H-3300) for antigen retrieval and incubated with DAKO Dual Endogenous Enzyme blocking reagent (Agilent Technologies, S2003) for quenching endogenous peroxidase. Sections were blocked in 0.1% Triton X-100 in PBS with 10% normal goat serum (Vector Laboratories, S-1000) and stained overnight at 4°C with anti-perilipin antibody (1:500 dilution, Cell Signaling Technology, 3470). Sections were then incubated with biotinylated goat anti–rabbit IgG antibodies (Vector Laboratories, BA-1000) for 1 hour and VECTASTAIN ABC reagent (Vector Laboratories, PK-6100) for 30 minutes at room temperature, and were visualized by the avidin-biotin–complex method using the DAB Peroxidase Substrate Kit (Vector Laboratories, SK-4105). Counterstaining was performed using nuclear fast red (MilliporeSigma, N3020) following the manufacturer’s instruction. The number of adipocytes was determined via counting perilipin-positive cells based on the IHC images.
RNA isolation and quantitative PCR analysis.
Total RNA was isolated from mouse tissues or cells using TRIzol Reagent (Thermo Fisher Scientific, 15596026) and quantitative PCR (qPCR) was performed using gene-specific primers and the SYBR Green PCR kit (Bio-Rad, 170-8886) as previously described (7). The sequences of primer sets used in this study are listed in Supplemental Table 2.
Statistics.
All data are presented as the mean ± SEM. Individual pairwise comparisons were analyzed by 2-sample, 2-tailed Student’s t test unless otherwise noted, with P < 0.05 regarded as significant. The statistical significance of differences among more than 2 groups was assessed using 1-way ANOVA with Dunnett’s test. Two-way ANOVA was used when multiple comparisons were made, followed by a Bonferroni multiple comparisons test. The correlation of human BMI and IKKβ was analyzed by Pearson correlation coefficient. Two-way ANOVA was done using SigmaPlot 13.0. The other statistics were analyzed using GraphPad Prism.
Study approval.
All animal studies were performed in compliance with the IACUC protocol approved by the University of Kentucky. All human studies were approved by IRB at the University of Kentucky, and written informed consent was obtained from each participant.
Author contributions
CZ conceptualized and designed the research. YS performed most of the experiments and analyzed the data with the help from ZL, SHP, SET, and BZ. JPF and HM performed the proteomics experiments. PAK provided human tissue samples and adipose stem cells. CZ, YS, and HM wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Michael Karin at UCSD for IKKβF/F mice; Haiyan Xu at Brown University for IKKβ vectors; Wendy Katz for tissue sectioning and staining; Taesik Gwag for technique support; and Hartmut Malluche for valuable input. We also acknowledge the support of the MicroCT Laboratory at Texas A&M University Baylor College of Dentistry. This work was supported in part by NIH grants (R01HL123358, R01ES023470, R01HL131925, R01DK107646, R21ES022745, P20GM103527, and UL1TR001998). The Proteomics Resource Center at The Rockefeller University also received funding from the Leona M. and Harry B. Helmsley Charitable Trust for mass spectrometer instrumentation.
Version 1. 01/25/2018
Electronic publication
Footnotes
Zun Liu’s present address is: Department of Human Evolutionary Biology, Harvard University, Cambridge, Massachusetts, USA.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2018;3(2):e96660. https://doi.org/10.1172/jci.insight.96660.
Contributor Information
Yipeng Sui, Email: yipeng.sui@uky.edu.
Zun Liu, Email: zun.liu@uky.edu.
Se-Hyung Park, Email: sh.park@uky.edu.
Beibei Zhu, Email: bzhu2@email.uky.edu.
Henrik Molina, Email: hmolina@rockefeller.edu.
Changcheng Zhou, Email: c.zhou@uky.edu.
References
- 1.Rodeheffer MS, Birsoy K, Friedman JM. Identification of white adipocyte progenitor cells in vivo. Cell. 2008;135(2):240–249. doi: 10.1016/j.cell.2008.09.036. [DOI] [PubMed] [Google Scholar]
- 2.Spalding KL, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453(7196):783–787. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- 3.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 4.Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. 2008;8(12):923–934. doi: 10.1038/nri2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 6.Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011;13(1):11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sui Y, et al. IKKβ links vascular inflammation to obesity and atherosclerosis. J Exp Med. 2014;211(5):869–886. doi: 10.1084/jem.20131281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou C, et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest. 2006;116(8):2280–2289. doi: 10.1172/JCI26283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell. 2017;168(1-2):37–57. doi: 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293(5535):1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 11.Kim JK, et al. Prevention of fat-induced insulin resistance by salicylate. J Clin Invest. 2001;108(3):437–446. doi: 10.1172/JCI11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arkan MC, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11(2):191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 13.Cai D, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11(2):183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135(1):61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiao P, et al. FFA-induced adipocyte inflammation and insulin resistance: involvement of ER stress and IKKβ pathways. Obesity (Silver Spring) 2011;19(3):483–491. doi: 10.1038/oby.2010.200. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Tang Y, Cai D. IKKβ/NF-κB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat Cell Biol. 2012;14(10):999–1012. doi: 10.1038/ncb2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Solinas G, Karin M. JNK1 and IKKbeta: molecular links between obesity and metabolic dysfunction. FASEB J. 2010;24(8):2596–2611. doi: 10.1096/fj.09-151340. [DOI] [PubMed] [Google Scholar]
- 18.Helsley RN, et al. Targeting IκB kinase β in Adipocyte Lineage Cells for Treatment of Obesity and Metabolic Dysfunctions. Stem Cells. 2016;34(7):1883–1895. doi: 10.1002/stem.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosen CJ, Bouxsein ML. Mechanisms of disease: is osteoporosis the obesity of bone? Nat Clin Pract Rheumatol. 2006;2(1):35–43. doi: 10.1038/ncprheum0070. [DOI] [PubMed] [Google Scholar]
- 20.Fazeli PK, et al. Marrow fat and bone--new perspectives. J Clin Endocrinol Metab. 2013;98(3):935–945. doi: 10.1210/jc.2012-3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lecka-Czernik B, Stechschulte LA. Bone and fat: a relationship of different shades. Arch Biochem Biophys. 2014;561:124–129. doi: 10.1016/j.abb.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 22.Aguirre L, Napoli N, Waters D, Qualls C, Villareal DT, Armamento-Villareal R. Increasing adiposity is associated with higher adipokine levels and lower bone mineral density in obese older adults. J Clin Endocrinol Metab. 2014;99(9):3290–3297. doi: 10.1210/jc.2013-3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bredella MA, et al. Vertebral bone marrow fat is positively associated with visceral fat and inversely associated with IGF-1 in obese women. Obesity (Silver Spring) 2011;19(1):49–53. doi: 10.1038/oby.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen W, et al. Relationship between MRI-measured bone marrow adipose tissue and hip and spine bone mineral density in African-American and Caucasian participants: the CARDIA study. J Clin Endocrinol Metab. 2012;97(4):1337–1346. doi: 10.1210/jc.2011-2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen W, Chen J, Punyanitya M, Shapses S, Heshka S, Heymsfield SB. MRI-measured bone marrow adipose tissue is inversely related to DXA-measured bone mineral in Caucasian women. Osteoporos Int. 2007;18(5):641–647. doi: 10.1007/s00198-006-0285-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akune T, et al. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113(6):846–855. doi: 10.1172/JCI19900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yue R, Zhou BO, Shimada IS, Zhao Z, Morrison SJ. Leptin Receptor Promotes Adipogenesis and Reduces Osteogenesis by Regulating Mesenchymal Stromal Cells in Adult Bone Marrow. Cell Stem Cell. 2016;18(6):782–796. doi: 10.1016/j.stem.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 28.Tang QQ, Lane MD. Adipogenesis: from stem cell to adipocyte. Annu Rev Biochem. 2012;81:715–736. doi: 10.1146/annurev-biochem-052110-115718. [DOI] [PubMed] [Google Scholar]
- 29.McDonald ME, Li C, Bian H, Smith BD, Layne MD, Farmer SR. Myocardin-related transcription factor A regulates conversion of progenitors to beige adipocytes. Cell. 2015;160(1-2):105–118. doi: 10.1016/j.cell.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burke JR, et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J Biol Chem. 2003;278(3):1450–1456. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]
- 31.Li ZW, Omori SA, Labuda T, Karin M, Rickert RC. IKK beta is required for peripheral B cell survival and proliferation. J Immunol. 2003;170(9):4630–4637. doi: 10.4049/jimmunol.170.9.4630. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen MT, et al. JNK and tumor necrosis factor-alpha mediate free fatty acid-induced insulin resistance in 3T3-L1 adipocytes. J Biol Chem. 2005;280(42):35361–35371. doi: 10.1074/jbc.M504611200. [DOI] [PubMed] [Google Scholar]
- 33.Van Beek M, et al. Bcl10 links saturated fat overnutrition with hepatocellular NF-kB activation and insulin resistance. Cell Rep. 2012;1(5):444–452. doi: 10.1016/j.celrep.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park SH, et al. IKKβ Is Essential for Adipocyte Survival and Adaptive Adipose Remodeling in Obesity. Diabetes. 2016;65(6):1616–1629. doi: 10.2337/db15-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ross SE, et al. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289(5481):950–953. doi: 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- 36.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7(12):885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 37.Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116(5):1202–1209. doi: 10.1172/JCI28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang J, et al. NF-κB inhibits osteogenic differentiation of mesenchymal stem cells by promoting β-catenin degradation. Proc Natl Acad Sci USA. 2013;110(23):9469–9474. doi: 10.1073/pnas.1300532110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12(8):695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 40.Yan J, et al. Inactivation of BAD by IKK inhibits TNFα-induced apoptosis independently of NF-κB activation. Cell. 2013;152(1-2):304–315. doi: 10.1016/j.cell.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu C, et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108(6):837–847. doi: 10.1016/S0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 42.Amit S, et al. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 2002;16(9):1066–1076. doi: 10.1101/gad.230302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 44.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33(2):77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 45.Spencer M, et al. Adipose tissue extracellular matrix and vascular abnormalities in obesity and insulin resistance. J Clin Endocrinol Metab. 2011;96(12):E1990–E1998. doi: 10.1210/jc.2011-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pittenger MF, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 47.Crisan M, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3(3):301–313. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 48.Beresford JN, Bennett JH, Devlin C, Leboy PS, Owen ME. Evidence for an inverse relationship between the differentiation of adipocytic and osteogenic cells in rat marrow stromal cell cultures. J Cell Sci. 1992;102(Pt 2):341–351. doi: 10.1242/jcs.102.2.341. [DOI] [PubMed] [Google Scholar]
- 49.Cao Y, et al. S-nitrosoglutathione reductase-dependent PPARγ denitrosylation participates in MSC-derived adipogenesis and osteogenesis. J Clin Invest. 2015;125(4):1679–1691. doi: 10.1172/JCI73780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y, et al. Nuclear factor kappa B signaling initiates early differentiation of neural stem cells. Stem Cells. 2012;30(3):510–524. doi: 10.1002/stem.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wernstedt Asterholm I, et al. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014;20(1):103–118. doi: 10.1016/j.cmet.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chang J, et al. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med. 2009;15(6):682–689. doi: 10.1038/nm.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang N, et al. TNF-α-induced NF-κB activation upregulates microRNA-150-3p and inhibits osteogenesis of mesenchymal stem cells by targeting β-catenin. Open Biol. 2016;6(3):150258. doi: 10.1098/rsob.150258. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Tarapore RS, et al. NF-κB Has a Direct Role in Inhibiting Bmp- and Wnt-Induced Matrix Protein Expression. J Bone Miner Res. 2016;31(1):52–64. doi: 10.1002/jbmr.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ruocco MG, et al. I{kappa}B kinase (IKK){beta}, but not IKK{alpha}, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J Exp Med. 2005;201(10):1677–1687. doi: 10.1084/jem.20042081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu J, et al. Inflammation Improves Glucose Homeostasis through IKKβ-XBP1s Interaction. Cell. 2016;167(4):1052–1066.e18. doi: 10.1016/j.cell.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kern PA, et al. The effects of temperature and seasons on subcutaneous white adipose tissue in humans: evidence for thermogenic gene induction. J Clin Endocrinol Metab. 2014;99(12):E2772–E2779. doi: 10.1210/jc.2014-2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Singhal PK, et al. Mouse embryonic fibroblasts exhibit extensive developmental and phenotypic diversity. Proc Natl Acad Sci USA. 2016;113(1):122–127. doi: 10.1073/pnas.1522401112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soleimani M, Nadri S. A protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. Nat Protoc. 2009;4(1):102–106. doi: 10.1038/nprot.2008.221. [DOI] [PubMed] [Google Scholar]
- 61.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11(8):783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68(5):850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 63.Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics. 2012;11(11):1475–1488. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gedaly R, et al. Targeting the Wnt/β-catenin signaling pathway in liver cancer stem cells and hepatocellular carcinoma cell lines with FH535. PLoS One. 2014;9(6):e99272. doi: 10.1371/journal.pone.0099272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.