

Abstract
NK cell–based immunotherapies have been gaining traction in the clinic for treatment of cancer. IL-15 is currently being used in number of clinical trials to improve NK cell expansion and function. The objective of this study is to evaluate the effect of repetitive IL-15 exposure on NK cells. An in vitro model in which human NK cells are continuously (on on on) or intermittently (on off on) treated with IL-15 was used to explore this question. After treatment, cells were evaluated for proliferation, survival, cell cycle gene expression, function, and metabolic processes. Our data indicate that continuous treatment of NK cells with IL-15 resulted in decreased viability and a cell cycle arrest gene expression pattern. This was associated with diminished signaling, decreased function both in vitro and in vivo, and reduced tumor control. NK cells continuously treated with IL-15 also displayed a reduced mitochondrial respiration profile when compared with NK cells treated intermittently with IL-15. This profile was characterized by a decrease in the spare respiratory capacity that was dependent on fatty acid oxidation (FAO). Limiting the strength of IL-15 signaling via utilization of an mTOR inhibitor rescued NK cell functionality in the group continuously treated with IL-15. The findings presented here show that human NK cells continuously treated with IL-15 undergo a process consistent with exhaustion that is accompanied by a reduction in FAO. These findings should inform IL-15–dosing strategies in NK cell cancer immunotherapeutic settings.
Keywords: Immunology
Keywords: Cytokines, Fatty acid oxidation, NK cells
Continuous exposure to IL-15 leads to diminished function, NK cell exhaustion, and a reduction in fatty acid metabolism.
Introduction
NK cell–based immunotherapy is rapidly evolving due to the ability of NK cells to directly lyse tumor targets, the emergence of antibodies and molecules that mediate NK cell–driven antibody-dependent cell-mediated cytotoxicity (ADCC), and the ability of NK cells to induce inflammatory responses (1). NK cells are being exploited in clinical trials using autologous and allogeneic NK cell infusion strategies alone or with hematopoietic stem cell transplantation. In addition, other modalities of NK cell therapy, such as use of NK cell line products and NK cells transduced with chimeric antigen receptors are on the horizon (2). We have shown that in vivo persistence and expansion of NK cells correlate with antitumor efficacy in patients with advanced AML (3, 4). Among the strategies being evaluated preclinically to address this issue, utilization of cytokines to induce NK cell persistence and expansion seems to dominate current clinical trials. While a number of cytokines have been tested, IL-15 appears to be the most logical choice due to its known physiologic role on NK cell development and homeostasis without stimulating regulatory T cells (4–7). Several clinical trials (NCT02395822, NCT01385423, NCT03388632, NCT01727076, NCT01572493, NCT01021059, NCT01369888, NCT01875601, NCT01885897, NCT02191098, NCT01946789, NCT02989844, NCT03054909, NCT02384954) are underway using IL-15 with dosing strategies, including i.v. bolus (8) and subcutaneous and continuous infusion using monomeric IL-15 (from the NCI) or IL-15 complexes with IL-15Rα transpresentation (https://clinicaltrials.gov/). Although IL-15 therapy is promising, little is known about how to best administer treatment doses of IL-15 to maximize human NK cell function, persistence, and expansion.
In T cell studies, repeated exposure to IL-2 has been shown to induce decreased cytolytic activity (9). The effect of continuous exposure to IL-15 in mouse NK cell studies is a little bit more complicated. In one study, using a transgenic mouse that overexpresses IL-15 by eliminating posttranscriptional checkpoints, NK cells and T cells robustly expanded, resulting in fatal lymphocytic leukemia (10). While mature functional NK cells robustly expanded in this study, NK cells were exposed to large doses of IL-15 from a very early stage, displaying lymphocytosis as early as 3 weeks of age, indicating that this might not be the best model for therapeutic administration of IL-15. In a separate mouse study, in which solid tumors were composed of fibrosarcoma cells engineered to secrete IL-15, NK cells were able to eradicate the large solid tumors (11). Though these findings clearly show the potential of IL-15 in driving solid tumor killing, IL-15 was restricted to the tumor microenvironment (TME), and was not increased systemically, so it is hard to evaluate whether the antitumor function was mediated by NK cells residing in the TME long term or by newly infiltrated NK cells being primed by the IL-15 short term. In a third study, presenting probably the most compelling model for IL-15 treatment in NK immunotherapy, mice received either transient or prolonged treatment with IL-15/IL-15Rα complexes (12). Unlike the previous studies, the prolonged treatment resulted in decreased function of NK cells, suggesting NK cell exhaustion.
Though not as well defined as T cell exhaustion (13, 14), NK cell exhaustion has been previously described. In an experimental setting of prolonged NK cell activation using ionomycin, NK cells exhibited reduced cytolytic function and cytokine production reminiscent of “spent” or exhausted NK cells (15). In a murine model of adoptive transfer, NK cells homed to the tumor environment but failed to eradicate tumor as a result of functional exhaustion characterized by reduced cytolytic and inflammatory activity associated with diminished T-bet and eomesodermin expression (16). However, it is not clear whether or how excessive IL-15 signaling might induce functional NK cell exhaustion in humans.
Immune metabolism has come to the forefront of T cell research in recent years, demonstrating a critical role for metabolic remodeling in T cell development, differentiation, quiescence, activation, and memory formation (17). Central to these studies is a tightly integrated balance between glycolysis and mitochondrial oxidative processes. Recent reports suggest that metabolism is important for NK cells. One group showed a requirement for glucose-driven oxidative phosphorylation in production of IFN-γ via activating NK cell receptors, a requirement bypassed by IL-15 stimulation (18). Another group demonstrated that the mammalian target of rapamycin complex 1 (mTORC1), a key regulator of cellular metabolism, is potently induced after NK cell activation and necessary for NK cell inflammatory and cytolytic function in vitro and in vivo (19). mTORC1 has also been implicated in cytokine-driven NK cell proliferation during development and activation with high concentrations of IL-15 (20). A separate study showed that IL-15 activates mTOR, leading to prolonged antitumor NK cell capacity (21). In both of these studies mTOR inhibition led to decreased NK cell functionality. It should, however, be highlighted that both studies utilized short-term priming through IL-15 and did not explore the effect of continuous dosing. If metabolism is associated with NK cell exhaustion remains unknown. In the current study, we developed a model to evaluate continuous exposure to IL-15 versus a break in IL-15 signaling to evaluate the effect on human NK cell function and metabolism.
Results
Continuous exposure to IL-15 results in proliferation at the expense of survival when compared with intermittent exposure.
To study the effect of continuous (repetitive) supraphysiologic IL-15 dosing on human NK cells, we designed an in vitro model that compares continuous (IL-15cont) and intermittent (IL-15gap) IL-15 signaling (Figure 1A). In this model, magnetically enriched NK cells from healthy donors were put in culture for 3 consecutive 3-day cycles of IL-15 in the IL-15cont group (IL-15 in all cycles), and in the IL-15gap group, the middle 3-day cycle was replaced with media alone (on-off-on schedule). It is important to emphasize that the first and last “on” cycles are identical in both groups, so any differences in phenotype and function are due to differences in the middle cycle condition with or without IL-15. For all experiments, cells were analyzed at the end of culture (day 9). Conditions in which a shorter gap in IL-15 dosing was created, such that IL-15 was administered every other day or every 2 days, were also tested but resulted in fewer differences between the IL-15cont and IL-15gap groups (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.96219DS1) compared with the 3-day cycles (as discussed below). This is important as one considers the translational relevance of this finding and current IL-15 administration dosing schemas (8, 22). Based on the differences with the 3-day cycle schedule, it was studied further.
Figure 1. Continuous IL-15 treatment results in increased proliferation and decreased survival.

(A) Schematic of the experimental system used to study effect of continuous IL-15 treatment (IL-15cont) versus intermittent IL-15 treatment (IL-15gap) on NK cells. Black bars represent IL-15 treatment, white bars represent incubation with media alone. (B) Upon harvest, the number of viable NK cells/ml of culture was assessed (n = 16). (C) NK cells were labeled with CellTrace Dye (Molecular Probes), and proliferation was assessed at day 9. Representative histogram showing CellTrace dilution in CD56+CD3– NK cells in the IL-15cont group (light gray) and the IL-15gap group (dark gray). (D) The proliferation index was calculated for NK cells based on the CellTrace dilution data utilizing Flowjo software (n = 4). (E) NK cells were assessed for cell death by gating to assess the percentage of cells incorporating Live/Dead Fixable dye (n = 12). (F) Fas and (G) FasL MFI was assessed on NK cells (n = 12). Paired t test was used to compare samples. *P ≤ 0.05; ***P < 0.001.
The IL-15cont group contained increased numbers of NK cells when compared with the IL-15gap group, indicating that, in this culture, continuous treatment resulted in enhanced expansion of NK cells (Figure 1B). These findings were confirmed with CellTrace dye dilution data showing greater proliferation in the IL-15cont group (Figure 1, C and D). In contrast, when viability was evaluated more cell death was noted in the IL-15cont group (Figure 1E). When exploring possible contributors to the increased cell death, a potent increase in Fas expression was seen in the IL-15cont group, but no differences were noted in FasL expression (Figure 1, F and G). These results indicate that continuous IL-15 signaling results in increased proliferation but decreased viability, consistent with a Fas-mediated mechanism indicative of activation-induced cell death.
Since IL-15 signaling is generally thought to be mediated by IL-15 transpresented with IL-15Rα to the IL-2/IL-15Rβ (CD122) and the common γ chain (CD132), it is important to evaluate if the differences in proliferation and survival were the result of altered CD122 and CD132 expression. To answer this question, CD122 and CD132 were evaluated on IL-15cont– and IL-15gap–treated NK cells. Neither CD122 expression (Figure 2A) nor CD132 expression (Figure 2B) differed after IL-15cont versus IL-15gap treatment, indicating that the changes in proliferation and viability are not mediated by IL-15 receptor complex components.
Figure 2. NK cells continuously treated with IL-15 do not differ in complex expression but display different cell cycle profiles than those that receive a gap in treatment.

(A) CD122 and (B) CD132 MFI were assessed on NK cells treated with IL-15cont and IL-15gap conditions for 9 days (n = 5). (C) Differences in cell cycle–related genes were explored on day 9 using a PCR array. Heat map displaying expression of significantly changed genes on individual donors treated with IL-15cont or IL-15gap methodology. Paired t tests were used to compare samples.
Continuous exposure to IL-15 changes the cell cycle profile when compared with intermittent exposure.
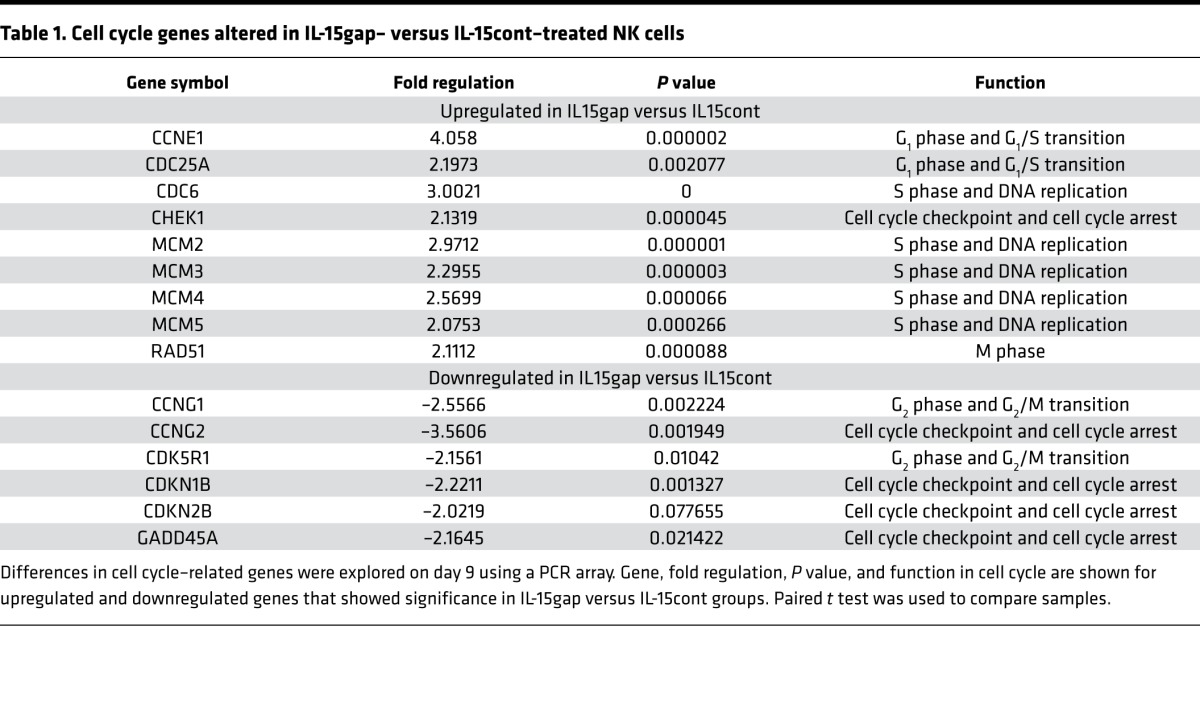
Given the differences in proliferation between the IL-15cont group and the IL-15gap group, we hypothesized potent cell cycle differences between the two groups. To test this question we performed a cell cycle gene array with IL-15cont and IL-15gap cells harvested at day 9 of culture. Contrary to expectations, the IL-15gap group was enriched with genes in the G1/S transition as well as genes involved in DNA replication, while the IL-15cont group was enriched for genes in cell cycle checkpoint and cell cycle arrest (Figure 2C and Table 1), indicating that at day 9 of culture the IL-15cont NK cells are transitioning to a more arrested state. This is consistent with the reduced NK cell viability (Figure 1E) and the concept that continuous IL-15 exposure results in more cellular damage and stress, which the cell attempts to control by upregulating checkpoints and entering cell cycle arrest (23). Failure to repair the damage properly likely results in activation of programmed cell death pathways.
Table 1. Cell cycle genes altered in IL-15gap– versus IL-15cont–treated NK cells.
A gap in IL-15 treatment results in enhanced function when compared with continuous treatment.
The next goal was to determine the effect of repetitive IL-15 treatment on human NK cell function. IL-15cont– and IL-15gap–treated NK cells were exposed to K562 cell targets for 4 hours and evaluated for CD107a degranulation and IFN-γ production. IL-15gap treatment significantly increased NK cell degranulation compared with IL-15cont treatment (Figure 3A). Similarly, IL-15gap NK cells displayed over a 3-fold increase in IFN-γ–producing cells (Figure 3B). Degranulation and cytokine production on a per cell basis was also modestly increased (data not shown). This difference in functionality was not limited to natural cytotoxicity, as activation via cytokine receptors also induced higher degranulation and inflammatory cytokine production (Figure 3, C and D). Of note, when activated via cytokines (IL-12/18), the IL-15gap–treated NK cells markedly increased IFN-γ production on a per cell basis compared with IL-15cont–treated NK cells, nearly doubling the MFI (Figure 3E). One possible explanation for the difference in function noted in Figure 3 is that less mature NK cells predominantly expanded in the IL-15cont group. To test this hypothesis, NK cells were labeled with a proliferation dye at the beginning of culture, and function was evaluated based on proliferation (high vs. low). As shown in Supplemental Figure 1, B and C, proliferation within the IL-15cont or IL-15gap groups did not correlate with decreased function. This indicated that the differential function between conditions was not explained by proliferation of less mature NK cells.
Figure 3. Continuous IL-15 treatment results in functional impairment of NK cells.

(A and B) IL-15cont– and IL-15gap–treated NK cells were incubated with K562 targets (E/T = 2:1) for 4 hours or (C–E) IL-12/IL-18 cytokines (10/100 ng/ml, respectively) overnight (n = 16). (A and C) Percentages of surface CD107a and (B and D) intracellular IFN-γ were measured. (E) IFN-γ MFI was also measured on the IFN-γ+ NK cells for the IL-12/18 stimulation. Cytotoxicity was measured on chromium-51–loaded (F) K562, (G) HL-60, and (H) Raji targets incubated with IL-15cont and IL- 15gap NK cells for 4 hours at the noted effector-to-target ratios (n = 6). Rituximab (10 μg/ml) was added to mediate ADCC in the Raji assay. Paired t test was used to do all comparisons. *P ≤ 0.05; **P < 0.01; ***P < 0.001.
Continuous IL-15 exposure leads to a marked decrease in NK cell cytolytic activity in vitro when compared with intermittent exposure.
Influence of IL-15 treatment on NK cell cytolytic capacity was evaluated next. IL-15cont– and IL-15gap–treated NK cells were incubated with K562 (chronic myelogenous leukemia) or HL-60 (promyelocytic leukemia) chromium-loaded targets, and specific lysis was measured after 4 hours (Figure 3, F and G). At all effector-to-target (E/T) ratios tested the IL-15cont treatment potently diminished NK cell natural cytotoxicity compared with the IL-15gap treatment. To measure the effect on ADCC, NK cells were incubated with chromium-loaded Raji (Burkitt’s lymphoma) targets and rituximab (Figure 3H). IL-15gap induced significantly greater ADCC than IL-15cont. These data indicate that continuous IL-15 exposure is detrimental to direct NK cell cytolytic activity.
Continuous IL-15 exposure reduces the ability of NK cells to control tumor in a xenogeneic mouse model when compared with intermittent exposure.
Given the reduced ability of IL-15cont NK cells to kill tumors in vitro, we next evaluated function in an in vivo xenogeneic model. Briefly, NSG mice were injected with HL-60luc tumor cells 3 days prior to injection of NK cells (1 million i.v.) previously exposed to the IL-15cont or IL-15gap strategy. Bioluminescent imaging was performed 14 days after injection of NK cells, while bleeds were carried out at days 6, 13, and 20. Though mice injected with IL-15cont NK cells exhibited some tumor control compared with the HL-60luc (tumor alone) group, only mice treated with IL-15gap NK cells induced potent antitumor activity (Figure 4A and Supplemental Figure 1D). To evaluate if other factors were operant in the difference of tumor control, NK cell numbers were analyzed from a constant volume blood draw at the noted time points. At all times tested, the IL-15gap group contained significantly more NK cells than the IL-15cont group, with maximal expansion noted at day 13 (Figure 4B). While these data are at odds with the expansion data shown in Figure 1B, it is worth highlighting that these cells were injected into the mice after the 9-day culture (IL-15cont or IL-15gap). By this time point, the IL-15cont NK cells were enriched for cell cycle checkpoint and arrest genes (Figure 2 and Table 1), likely limiting further expansion. In concordance with this, the viability of NK cells circulating in vivo from the IL-15gap group was greater at all time points measured compared with the IL-15cont condition (Figure 4C). Finally, to evaluate if continuous exposure to IL-15 could effect immunotherapeutic antibody use/efficacy, CD16 levels were analyzed on NK cells circulating in vivo. CD16 is the NK cell activating receptor needed to mediate ADCC via therapeutic antibodies. The IL-15cont group showed less CD16 expression at all time points (Figure 4D). These data show that continuous exposure to IL-15 can have potent negative effects on tumor control in vivo.
Figure 4. Continuous IL-15 treatment reduces NK cell in vivo efficacy in a xenogeneic model of AML.

NSG mice were injected (i.v.) with HL-60luc cells and either IL-15cont or IL-15gap NK cells 3 days later. (A) Day 14 luminescence (p/s/cm2/sr) for mice engrafted with tumor alone, tumor plus IL-15cont NK cells, or tumor plus IL-15gap NK cells. (B) Quantification of human NK cells (CD45+CD56+CD3–) within 100 μl blood of mice at noted time points. Events were collected in the flow cytometer at a constant rate for 60 seconds (n = 4–5). (C) Human NK cell viability in the blood of the mice was measured by exclusion of Live/Dead dye (Molecular Probes) at noted days. (D) CD16 expression, measured by MFI, was assessed at noted time points in CD45+LiveDead– NK cells in the blood from the mice. One-way ANOVA (B) and unpaired t tests (C and D) were used to do comparisons. *P ≤ 0.05; **P < 0.01; ***P < 0.001.
NK signaling is negatively affected by continuous IL-15 exposure when compared with intermittent exposure.
To better understand the mechanism underlying the differential in function between IL-15cont and IL-15gap strategies, an intracellular signaling panel was evaluated by Phosflow. Several signaling proteins showed decreased phosphorylation in the IL-15cont group compared with the IL-15gap group upon PMA and ionomycin treatment (Figure 5A and Supplemental Figure 2A). Phosphorylated STAT5, a measure of IL-15 signaling, displayed a trend for decreased expression in the IL-15cont groups after a 15-minute stimulation with IL-15 (Figure 5B and Supplemental Figure 2B). In agreement with this finding, pS6, used to evaluate mTOR signaling that has been demonstrated to be essential for IL-15 signaling on NK cells (20), also showed decreased phosphorylation in the IL-15cont group. STAT5 signaling was evaluated earlier, at day 6 of culture, but no substantial differences were seen at this early time point, suggesting that IL-15 exposure intervals are critical to this finding. To evaluate negative regulators of IL-15 signaling, SOCS1– SOCS3 gene expression was assessed (Supplemental Figure 2D). SOCS2, whose knockdown has been shown decrease NK cell function via reduced regulation of Pyk2 (24), showed mild upregulation in the IL-15gap group. In contrast, SOCS1, which has been shown to be induced upon IL-15 signaling on T cells (25), was robustly increased in the IL-15gap group, highlighting the increased sensitivity to IL-15 signaling in this group versus the IL-15cont group. However, the dominant role of SOCS1 and SOCS3 signaling downstream of IL-15 has recently been questioned on NK cells (26). This study showed that CIS, encoded by the CISH gene, was the primary negative regulator of IL-15 signaling for NK cells, which was also addressed in our system. CISH was variably upregulated in all IL-15gap NK cells tested in comparison to IL-15cont (Figure 5C). While this might seem counterintuitive, CISH was also induced downstream of common γ chain cytokines (27–29), and the increased expression likely reflects increased signaling strength in the IL-15gap group (highlighted in Figure 5, A and B). These data would indicate that the negative regulator CIS is not the dominant determinant of functional differences between the IL-15cont– and IL-15gap–treated NK cells.
Figure 5. Continuous IL-15 treatment results in reduced NK cell signaling.

The indicated phospho-protein expression was evaluated by flow cytometry on IL-15cont or IL- 15gap NK cells activated with (A) PMA/ionomycin or (B) recombinant IL-15 (n = 6). (C) Fold CISH mRNA induction, normalized to GAPDH (n = 4). (D) Intracellular T-bet (n = 6) or (E) eomesodermin protein expression was evaluated by flow cytometry (n = 4). (F) Fold CPT1A mRNA induction, normalized to GAPDH (n = 4). (G) Intracellular CPT1a MFI on NK cells (n = 7). Paired t tests were used to do all comparisons. *P ≤ 0.05; **P < 0.01; ***P < 0.001.
T-bet protein expression, which is upregulated upon functional maturation of NK cells and has been shown to be downregulated in tumor-induced NK cell exhaustion (16), was decreased in the IL-15cont group in agreement with the decrease in function seen in this group (Figure 5D). However, eomesodermin expression, which was also shown to be downregulated in ref. 16, was not altered by IL-15cont treatment (Figure 5E). Since the mTOR pathway is known to effect metabolism, carnitine palmitoyltransferase 1a (CPT1a) expression (Figure 5, F and G) was evaluated to discern if IL-15cont could also effect the fatty acid oxidation (FAO) metabolic component (30). CPT1a is a mitochondrial protein that poses the rate-limiting step in β-oxidation of fatty acids, thus low expression decreases FAO while high expression enhances it. The IL-15cont–treated NK cells consistently expressed less CPT1a, indicating a possible defect in FAO.
Continuous IL-15 signaling disrupts FAO when compared with intermittent exposure.
Real-time NK cell metabolism was explored next. NK cells were exposed to IL-15cont and IL-15gap conditions and evaluated for both glycolytic and mitochondrial function in a previously described Seahorse assay (20). Although no statistically significant differences were found in glycolytic metabolism (Supplemental Figure 3, A–D), IL-15gap–treated cells showed a clearly enhanced oxidative pattern when compared with IL-15cont–treated NK cells (Figure 6, A and B). This was highlighted by strong increases in basal respiration measured after addition of glucose. ATP production, elucidated from subtraction of basal respiration and respiration after addition of oligomycin, was also higher in the IL-15gap group. Maximal mitochondrial respiration, measured after addition of carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) and sodium pyruvate, was shown to be defective in the IL-15cont group as well. Of particular note, mitochondrial spare respiratory capacity (SRC), which can be used as a measure of the cell’s fitness and has been shown to be important in memory T cell formation (31), was much higher in the IL-15gap group than in the IL-15cont NK cells. Since SRC can be fueled by mitochondrial FAO, these data correlate well with the CPT1a expression findings. Following up on this concept, IL-15cont and IL-15gap NK cells were treated with etomoxir, which inhibits CPT1 function, to evaluate the input of FAO on the SRC of these cells (Figure 6C). As expected, basal respiration, measured prior to addition of etomoxir, and ATP production were not affected (Figure 6D), but maximal respiration and SRC were drastically decreased in the IL-15gap group and minimally altered in the IL-15cont group (Figure 6D). This indicated that the increased SRC noted in the IL-15gap group is mediated by FAO, while FAO is all but absent in the IL-15cont group.
Figure 6. Continuous IL-15 treatment alters NK cell metabolism by mitigating fatty acid oxidation required for enhanced NK cell function.

(A) IL-15cont and IL-15gap NK cells were immobilized on plates, and the oxygen consumption rate (OCR) was measured in real time in an XFe24 analyzer after injection of glucose, oligomycin, FCCP plus sodium pyruvate, and rotenone/antimycin A. Spare respiratory capacity (SRC) represents change from basal oxygen consumption to maximal oxygen consumption (n = 6). (B) Graphical analysis of basal respiration, ATP production, maximal respiration, and SRC derived from A (n = 6). (C) Loss of FAO in this system was measured by addition of the CPT-1 inhibitor etomoxir (n = 6). (D) Graphical analysis of basal respiration, ATP production, maximal respiration, and SRC derived from C (n = 6). (E) Rapamycin was added throughout IL-15cont treatment, and CD107a (left) and IFN-γ (right) expression were evaluated (n = 12). Paired t test was used to do all comparisons. *P ≤ 0.05; **P < 0.01; ***P < 0.001.
Limiting IL-15 signal strength restores functionality in the continuously treated NK cells.
Finally, since our data indicate that persistent supraphysiologic IL-15 signaling, in the IL-15cont–treated NK cells, induces functional exhaustion and a change in metabolism through downstream signals, it stands to reason that limiting those signals would decrease exhaustion. To test this hypothesis we treated NK cells with the IL-15cont culture with or without rapamycin, which blocks mTOR function downstream of IL-15. Unlike stronger mTOR inhibitors like Torin 1, which would essentially completely shut down mTOR signaling, rapamycin is more permissive, allowing for diminished signaling downstream of IL-15 (32, 33). Additionally, mTOR signaling limits FAO through the inhibition of CPT1a (34), thus rapamycin treatment might also amplify FAO in the IL-15cont group. NK cell function was evaluated after culture for 9 days with IL-15cont treatment with or without rapamycin (Figure 6E). Treatment with rapamycin resulted in a rescue of IL-15cont NK function that remarkably resembled the functional profile of IL-15gap–treated NK cells (Figure 3A). To evaluate if rapamycin limited mTOR signals downstream of IL-15, leading to glycolysis, the glycolytic reserve of IL-15cont– and IL-15cont plus rapamycin–treated NK cells was evaluated by Seahorse. The glycolytic reserve was significantly decreased by rapamycin treatment (Supplemental Figure 3E), as expected, and there was a slight increase in SRC in the rapamycin-treated IL-15cont NK cells (Supplemental Figure 3F). This supports the premise that a strong, persistent, mTOR-based signal downstream of IL-15 is a critical component of cytokine-induced functional exhaustion. Likewise, these data might allude to the role of IL-15–induced mTOR in limiting FAO, loss of which could be the culprit of the functional exhaustion on NK cells exposed to supraphysiologic IL-15.
Discussion
IL-15 is currently being tested clinically in a number of formats to enhance immunotherapy for several indications (2, 8, 35), so it is critical to understand what repetitive, continuous, supraphysiologic dosing does to immune effectors. In the current study, we describe the effect of continuous IL-15 treatment on human NK cells. Our findings indicate that continuous treatment with IL-15 results in a functional NK cell change consistent with exhaustion. This is supported by a number of biological defects. First, though the IL-15cont–treated NK cells seemed to initially display better proliferation and expansion during the 9 days of treatment, they were more susceptible to cell death. This is consistent with cell cycle gene expression data showing that the IL-15cont NK cells were enriched for expression of cell cycle checkpoint and arrest genes, indicating that at day 9 of culture the IL-15cont NK cells were transitioning to an arrested state due to cellular stress. Induction of cell cycle checkpoints has been shown to allow cells to cope with cellular stress, often culminating with apoptosis (23). In agreement with this cell cycle finding, our data show a defect in IL-15cont NK cell expansion/survival when the cells were placed in a xenogeneic mouse model. The IL-15cont NK cells also displayed markedly diminished cytolytic and inflammatory function, which results in reduced tumor killing in vitro and in vivo. The diminished function with continuous IL-15 exposure was explained by decreased phosphorylation of a number of key signaling components in the IL-15cont group, indicating that continuous IL-15 exhausts human NK cells. The T-box transcription factor T-bet was also reduced with continuous IL-15, in agreement with a previous study displaying NK cell exhaustion after transfer into tumor-bearing mice (16). Eomesodermin expression, however, was not altered, perhaps indicating a difference in the signaling constraints of cytokine-induced exhaustion versus those of tumor-induced exhaustion. It should be made clear that repetitive dosing, without the benefit of a break in dosing, is responsible for the induction of exhaustion. Treating NK cells with a single dose of IL-15 for a week (data not shown) or with IL-15gap for 9 days, where the middle 3 days have no IL-15 present, does not result in exhaustion. Though some studies have evaluated overexposure to IL-15 on mouse NK cells, the resulting data were contradictory likely due to intricacies within the systems (10–12). Our system better mimics the outcomes of repetitive therapeutic IL-15 dosing on NK cells, and to our knowledge this is the first report of IL-15–mediated exhaustion on human NK cells.
Our data indicate an association between metabolism and function. CPT1a, a key mediator of FAO, was reduced in IL-15cont–treated cells. Further evaluation identified a potent defect in mitochondrial oxidative respiration in the IL-15cont NK cells when compared with the IL-15gap NK cells, which were better able to cope with mitochondrial stress. When the IL-15gap NK cells were treated with an inhibitor of CPT1, thus inhibiting FAO, the exhausted metabolic phenotype mimicked IL-15cont conditions. There was no further effect of the CPT1 inhibitor on IL-15cont–treated NK cells, supporting the specificity of the FAO pathway in maintaining potent NK cell SRC.
The effect of IL-15–mediated signal strength on cytokine-driven exhaustion was evaluated using a permissible mTOR inhibitor, rapamycin, to decrease downstream signals in the IL-15cont culture. The basic idea behind this experiment is that if repetitive IL-15 dosing in the IL-15cont cultures is generating an excessive signal that induces functional exhaustion then limiting that signal should rescue NK cell function. Given the role of mTOR signaling in tuning downstream IL-15 signals on NK cells and its effect in immune metabolism, it seemed like a perfect target for modulation in our system (20). Though stronger mTOR inhibitors, like Torin 1, are readily available, we utilized a weaker inhibitor (rapamycin), as complete inhibition of the mTOR pathway would just result in NK cell death within our 9 day assay. Function in rapamycin-treated IL-15cont NK cells was rescued to levels similar to those seen in the IL-15gap group, indicating that signal strength via mTOR is likely the culprit in the functional exhaustion. Though other studies have highlighted a positive role for mTOR signaling in NK cell function (19, 21), NK cells were treated with IL-15 for a short duration (12–48 hours), thus inducing only a brief priming stimulus. These studies did not explore continuous dosing with multiple doses of IL-15, and thus, the resulting functional exhaustion and negative input of excessive mTOR signals was not seen. The goal of the present study was precisely to explore that concept. It should also be pointed out that mTOR signaling can decrease CPT1a expression and, thus, FAO (34). Therefore, by treating with rapamycin, we might be indirectly inducing FAO. Supporting this notion, we saw a slight increase in SRC in the rapamycin-treated IL-15cont NK cells. It is not entirely clear if the effect of mTOR on CPT1a expression is mediated by IL-15 signal strength, but the concept that rapamycin might be decreasing exhaustion and rescuing function through an induction of CPT1a and FAO fits well with the Seahorse data, showing better FAO in the functional IL-15gap culture versus the exhausted IL-15cont culture. Taken together, our data demonstrate a mechanistic role for mTOR-mediated signal strength and FAO in IL-15–induced human NK cell exhaustion. It is possible that STAT5 phosphorylation and translocation could be an important component of this mechanism, but a previous short-term study, in which NK cells were triggered with high and low doses of IL-15, showed that, while both high and low doses induced STAT5 phosphorylation, only high doses boosted bioenergetic metabolism (20). This suggests that STAT5 phosphorylation alone is not the main driver of changes in metabolism. However, that study did not focus specifically on FAO. STAT5 has been shown to be involved in regulation of acetyl-CoA carboxylase (ACC), a rate-limiting enzyme for FAO that limits CPT1A via induction of malonyl-CoA (36). Both CPT1A and ACC are regulated by AMPK, which has clear interactions with the mTOR pathway (37). Future mechanistic studies are planned to explore the interplay among IL-15 signaling, STAT5 translocation, mTOR and AMPK activation, modulation of CPT1A and ACC, and the downstream result on FAO.
Rapamycin-treated IL-15cont NK cells expanded less, as evident by decreased cell numbers, without a change in viability (Supplemental Figure 3, G and H), similarly to the IL-15gap–treated NK cells, supporting a role for IL-15 signal strength and FAO to determine NK cell function that appears to inversely correlate with proliferation. FAO has been shown to play an important role in generation of the memory T cell pool, where memory T cells limit their division until challenge (30). In a recent study evaluating murine NK cell memory pool contraction during viral responses, rapamycin increased autophagic processes, which helped formation of the memory pool (38). Given the prominent role that IL-2, whose signaling outcomes mimic IL-15’s, has during NK cell–based CMV responses, it is tempting to speculate that the memory pool is formed by cells that have not undergone IL-2–mediated exhaustion and that FAO, present in the remaining healthy mitochondria after mitophagy, has a prominent role in formation of the memory pool. While there is no evidence that the NK cells generated with the IL-15gap treatment are indeed memory NK cells, the differential in FAO with the IL-15cont group might reveal an important requirement for establishment of longer-lived, more functional NK cells.
In physiologic settings, IL-15–mediated functional exhaustion of NK cells can be seen during endometriosis, where endometrial stromal cells secrete IL-15, which promotes immune escape via inactivation of NK cell cytolytic function (39). One could also foresee inflammatory settings, in which IL-15–mediated exhaustion of NK cells can be beneficial by capping their response. However, in the therapeutic setting where IL-15 is being used to enhance patient immune responses to tumor, inducing NK cell exhaustion is clearly detrimental (4, 5, 40). There are currently a number of clinical trials (ref. 35 and https://clinicaltrials.gov/, see above) testing utilization of monomeric IL-15 (from the NCI) or an IL-15N72D/IL-15Rα-Fc super-agonist complex, termed ALT-803 (from Altor Bioscience). While the settings vary from treatment of hematological malignancies to solid tumors, the goal of IL-15 treatment in all of these trials is the same — to drive expansion and priming of immune cells (NK cells and CD8+ T cells). Depending on the drug, administration can be achieved through repetitive single i.v. doses, continuous infusion, or subcutaneous dosing (5, 6, 8). One of these clinical studies, in which rhIL-15 (0.3–3 μg/kg by i.v. bolus) was injected for 12 consecutive days, showed a >10-fold in vivo expansion of NK cells (8). Pharmacokinetics from this study showed a half-life of NCI IL-15 of 2.4–2.7 hours, which is important to interpret how much exposure NK cells have to the cytokine. In addition, recently published studies using this same IL-15 product administered at 2 μg/kg subcutaneously show an extended Tmax, lower Cmax, and longer half-life (22). However, while both studies showed short on-off serum concentrations within a 24-hour period with daily dosing, neither study looked at the effect of longer intervals of rest, which we plan to study in future clinical trials to fully understand the effect of IL-15 dosing, exhaustion, and NK cell function. Our findings indicate that the methodology of dosing will likely have great effect in the NK cell responses. While weekly subcutaneous dosing might elicit strong NK cell expansion and function, repetitive i.v. dosing or continuous infusion might have an opposite effect by driving NK cell exhaustion; albeit drug half-life and availability will have an important role in this process. Altering IL-15–dosing regimens to limit exhaustion or coupling them with known approved drugs that can induce FAO has the potential to greatly enhance IL-15–based NK cell immunotherapies.
Methods
Cell isolation.
Buffy coats collected from healthy donors were obtained from Memorial Blood Bank (Minneapolis, Minnesota, USA). Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Ficoll-Paque (GE Healthcare). NK cells were obtained from PBMCs using an NK cell enrichment magnetic bead kit (StemCell Technologies). Isolation with this method yielded NK cell purity of 91% ± 1.5%.
Cell lines.
K562, Raji, HL-60, and HL-60luc cell lines were all cultured in RPMI-1640 with 10% fetal bovine serum (Gibco) (termed RPMI-10 henceforth). Cell lines were purchased from ATCC in 2013 (HL-60) and 2015 (Raji and K562) and authenticated by the University of Arizona Genetics Core in March 2017.
In vitro culture conditions.
300,000 NK cells, resuspended in RPMI-10, were plated per well in a 96-well round-bottom plate. Plates were spun down, supernatant was removed, and cells were resuspended in RPMI-10 with 10 ng/ml IL-15 (R&D Systems) for both the IL-15cont and the IL-15gap groups. After 3 days (day 3), wells were spun down, washed twice, and then resuspended in RPMI-10 with 10 ng/ml IL-15 for the IL-15cont or RPMI-10 alone for the IL-15gap. Three days later (day 6), cells were spun down, washed twice, and both groups were resuspended in RPMI-10 with 10 ng/ml IL-15 for the final 3 days. Cells were harvested at day 9, counted, and used for assays. In experiments looking at rescue of FAO in the IL-15cont treatment group, rapamycin (1 nM) was added to the cultures throughout the 9 days.
Proliferation assays.
NK cells from healthy donors were labeled with CellTrace Violet Cell Proliferation Dye (ThermoFisher Scientific), per the manufacturer’s protocol, and placed in the IL-15cont or IL-15gap conditions for 9 days. Viable NK cells (CD56+CD3–) were then analyzed for dilution of dye.
Flow cytometry analysis.
Fluorochrome-conjugated antibodies were purchased from BioLegend (anti-CD56 [catalog no. 318318], CD16 [catalog no. 302018], Fas [catalog no. 305622], FasL [catalog no. 306407], CD107a [catalog no. 328606], IFN-γ [catalog no. 502532], and CD45 [catalog no. 304042]), BD Biosciences (anti-CD3 [catalog no. 562280], pSLP76 [catalog no. 558439], p38 [catalog no. 563569], pNF-κB/p65 [catalog no. 560335], pAKT [catalog no. 560858], pS6 [catalog no. 561457], pSTAT5 [catalog no. 612598], pLCK [catalog no. 558552], and T-bet [catalog no. 564142]), eBioscience (anti-Eomesodermin [catalog no. 50-4877-42]), and Abcam (anti-CPT1a [ab171449]). For intracellular Phosflow, 500,000 cells were treated with PMA (25 ng/ml) and ionomycin (1 μM) or IL-15 (10 ng/ml) for 15 minutes and fixed immediately followed by permeabilization with BD Phosflow Perm Buffer III prior to intracellular staining. Cell cycle analysis was carried out with the BD Cell Cycle/DNA Kit (BD Biosciences) per the manufacturer’s recommendations. Cells were run on a LSRII flow cytometer (BD Biosciences), and data were analyzed with FlowJo software (Tree Star Inc.).
Quantitative RT-PCR.
RNA was processed from IL-15cont and IL-15gap (day 9) NK cells as described previously (41). For cell cycle gene analysis, transcripts were evaluated using the Human Cell Cycle RT2 Profiler PCR Array (Qiagen). CPT1a, SOCS1, SOCS2, and SOCS3 transcripts were analyzed using a TaqMan probe (Applied Biosystems) and normalized to GAPDH.
CD107a and IFN-γ/TNF-αassays. IL-15cont or IL-15gap NK cells (day 9) were incubated overnight with IL-12 (10 ng/ml, R&D Systems) and IL-18 (100 ng/ml, R&D Systems), or cocultured for 4 hours with K562 tumor targets at 2:1 E/T ratio. Within the 4-hour incubation, antiCD107a-FITC antibody was added during the first hour, followed by GolgiStop and GolgiPlug (BD Biosciences) incubation for the following 3 hours. Cells were then washed, stained with LiveDead viability stain (ThermoFisher Scientific), stained with surface antibodies, fixed, permeabilized, and stained intracellularly with anti-IFN-γ-BV421.
Chromium-51 release assays.
Chromium-51 was incubated with tumor targets (K562, HL-60, and Raji) for 1 hour at 37°C, washed 3 times, and cocultured with NK cells (IL-15cont or IL-15gap) at 3 E/T ratios (10:1, 5:1, and 2.5:1). In the case of the Raji assay, rituximab (10 μg/ml) was also added to induce ADCC. 5% Triton-X 100 was used to achieve total lysis. After 4 hours, supernatant was harvested and analyzed in a PerkinElmer 1470 Automatic Gamma Counter. Specific lysis was calculated using the following equation: % cytolytic NK activity (chromium-51 release) = 100 × (test release − spontaneous release)/(maximal release − spontaneous release).
In vivo mouse study and imaging.
NOD/SCID/γc–/– (NSG) mice (Jackson Labs) were sublethally irradiated (275 cGy) and xenografted i.v. with 750,000 firefly luciferase-expressing HL-60 human acute promyelocytic leukemia cells (day –3). At day 0, mice were given i.v. 1 × 106 IL-15cont or IL-15gap NK cells; they were harvested at day 9 of culture. 2 μg IL-15 (NCI) was injected i.p. per mouse on that day and every 7 days following to induce basal maintenance of the NK cells. Retro orbital bleeds, 150 μl, were carried out at day 6, 13, and 20 to assess human cell content. Mice were injected with 100 μl of 30 mg/ml luciferin substrate 10 minutes prior to imaging and then anesthetized via inhalation of isoflurane gas. Assessment of the presence of tumor cells by bioluminescent imaging (BLI) was carried out at day 14 using the Xenogen IVIS imaging system and analyzed with Living Image 2.5 software (Caliper Life Science).
Metabolic studies.
IL-15cont and IL-15gap NK cells were harvested at day 9 of culture and resuspended in Seahorse XF Assay Medium (Agilent Technologies). One million cells/well were immobilized with Poly-L-Lysine (MilliporeSigma). The extracellular acidification rate and the oxygen consumption rate were measured (pmoles/min) in real time in an XFe24 analyzer after injection of glucose (10 mM), oligomycin (1 μM), FCCP (1 μM) plus sodium pyruvate (1 mM), and rotenone/antimycin A (0.5 μM). SRC was calculated from the change from basal oxygen consumption, after addition of glucose, to maximal oxygen consumption, after addition of FCCP. In experiments measuring the input of FAO, glucose was added to the media prior to beginning measurements. This was the followed by injection of the CPT-1 inhibitor etomoxir, injection of oligomycin, injection of FCCP plus sodium pyruvate, and final injection of rotenone/antimycin A.
Statistics.
All statistical tests (noted in figure legends) were carried out and analyzed with Graphpad Prism software. All t tests were 2-tailed. Error bars represent the mean ± SEM. P values of less than 0.05 were considered significant.
Study approval.
Blood from healthy donors was obtained after receipt of written informed consent at Memorial Blood Bank. Use of PBMCs from donors was approved by the Committee on the Use of Human Subjects in Research at the University of Minnesota (IRB 9709M00134) in accordance with the Declaration of Helsinki. Animals studies were conducted in accordance with a protocol reviewed and approved by the University of Minnesota institutional animal care and use committee (IACUC 1506-A32639).
Author contributions
Scientific concepts and design of research studies were conceived by MF, MAG, JT, BRB, and JSM. Experiments and data acquisition were conducted by AJL, RM, SC, PH, and LB. The manuscript was written and edited by MF, AJL, BRB, and JSM.
Supplementary Material
Acknowledgments
We wish to acknowledge the flow cytometry core and the University Imaging Centers at the University of Minnesota for excellent service and technical support. This work was supported by the US Department of Defense (CA150085 to MF) as well as the following NIH grants: P01 CA111412 (to MF and JSM), P01 CA65493 (to BRB, JT, MF, and JSM), R35 CA197292 (to MF and JSM), R01 HL122216 (to MF, JT, and JSM), R01 HL56067 (to BRB), and R01 HL11879 (to BRB).
Version 1. 02/08/2018
Electronic publication
Footnotes
Conflict of interest: JSM serves on the scientific advisory boards of Celgene, GT BioPharma, and Fate Therapeutics and has received research funds and/or clinical trial support from these relationships. BRB declares a financial conflict with Tmunity and Kadmon Corp.
Reference information: JCI Insight. 2018;3(3):e96219. https://doi.org/10.1172/jci.insight.96219.
Contributor Information
Martin Felices, Email: mfelices@umn.edu.
Alexander J. Lenvik, Email: lenvi009@umn.edu.
Ron McElmurry, Email: mcelm001@umn.edu.
Sami Chu, Email: chus@umn.edu.
Peter Hinderlie, Email: phoward@umn.edu.
Laura Bendzick, Email: green711@umn.edu.
Melissa A. Geller, Email: gelle005@umn.edu.
Jakub Tolar, Email: tolar003@umn.edu.
Bruce R. Blazar, Email: blaza001@umn.edu.
References
- 1.Murphy WJ, Parham P, Miller JS. NK cells--from bench to clinic. Biol Blood Marrow Transplant. 2012;18(1 Suppl):S2–S7. doi: 10.1016/j.bbmt.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davis ZB, Felices M, Verneris MR, Miller JS. Natural killer cell adoptive transfer therapy: exploiting the first line of defense against cancer. Cancer J. 2015;21(6):486–491. doi: 10.1097/PPO.0000000000000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller JS, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):3051–3057. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- 4.Bachanova V, Miller JS. NK cells in therapy of cancer. Crit Rev Oncog. 2014;19(1-2):133–141. doi: 10.1615/CritRevOncog.2014011091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patidar M, Yadav N, Dalai SK. Interleukin 15: A key cytokine for immunotherapy. Cytokine Growth Factor Rev. 2016;31:49–59. doi: 10.1016/j.cytogfr.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Pilipow K, Roberto A, Roederer M, Waldmann TA, Mavilio D, Lugli E. IL15 and T-cell stemness in T-cell-based cancer immunotherapy. Cancer Res. 2015;75(24):5187–5193. doi: 10.1158/0008-5472.CAN-15-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Romee R, Leong JW, Fehniger TA. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica (Cairo) 2014;2014:205796. doi: 10.1155/2014/205796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conlon KC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. 2015;33(1):74–82. doi: 10.1200/JCO.2014.57.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shih CC, Truitt RL. Downregulation of L3T4+ cytotoxic T lymphocytes by interleukin-2. Science. 1987;238(4825):344–347. doi: 10.1126/science.2443976. [DOI] [PubMed] [Google Scholar]
- 10.Fehniger TA, et al. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J Exp Med. 2001;193(2):219–231. doi: 10.1084/jem.193.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu RB, et al. Densely granulated murine NK cells eradicate large solid tumors. Cancer Res. 2012;72(8):1964–1974. doi: 10.1158/0008-5472.CAN-11-3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elpek KG, Rubinstein MP, Bellemare-Pelletier A, Goldrath AW, Turley SJ. Mature natural killer cells with phenotypic and functional alterations accumulate upon sustained stimulation with IL-15/IL-15Ralpha complexes. Proc Natl Acad Sci USA. 2010;107(50):21647–21652. doi: 10.1073/pnas.1012128107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 14.Wherry EJ, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 15.Romera-Cárdenas G, Thomas LM, Lopez-Cobo S, García-Cuesta EM, Long EO, Reyburn HT. Ionomycin treatment renders NK cells hyporesponsive. PLoS One. 2016;11(3):e0150998. doi: 10.1371/journal.pone.0150998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gill S, et al. Rapid development of exhaustion and down-regulation of eomesodermin limit the antitumor activity of adoptively transferred murine natural killer cells. Blood. 2012;119(24):5758–5768. doi: 10.1182/blood-2012-03-415364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212(9):1345–1360. doi: 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keppel MP, Saucier N, Mah AY, Vogel TP, Cooper MA. Activation-specific metabolic requirements for NK Cell IFN-γ production. J Immunol. 2015;194(4):1954–1962. doi: 10.4049/jimmunol.1402099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donnelly RP, et al. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J Immunol. 2014;193(9):4477–4484. doi: 10.4049/jimmunol.1401558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marçais A, et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. 2014;15(8):749–757. doi: 10.1038/ni.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mao Y, et al. IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood. 2016;128(11):1475–1489. doi: 10.1182/blood-2016-02-698027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller JS, et al. A first-in-human phase 1 study of subcutaneous outpatient recombinant human IL-15 (rhIL-15) in adults with advanced solid tumors. Clin Cancer Res. doi: 10.1158/1078-0432. [published online ahead of print December 4, 2017]. https://doi.org/10.1158/1078-0432.CCR-17-2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pietenpol JA, Stewart ZA. Cell cycle checkpoint signaling: cell cycle arrest versus apoptosis. Toxicology. 2002;181-182:475–481. doi: 10.1016/S0300-483X(02)00460-2. [DOI] [PubMed] [Google Scholar]
- 24.Lee SH, et al. Suppressor of cytokine signaling 2 regulates IL-15-primed human NK cell function via control of phosphorylated Pyk2. J Immunol. 2010;185(2):917–928. doi: 10.4049/jimmunol.1000784. [DOI] [PubMed] [Google Scholar]
- 25.Ilangumaran S, et al. Suppressor of cytokine signaling 1 attenuates IL-15 receptor signaling in CD8+ thymocytes. Blood. 2003;102(12):4115–4122. doi: 10.1182/blood-2003-01-0175. [DOI] [PubMed] [Google Scholar]
- 26.Delconte RB, et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol. 2016;17(7):816–824. doi: 10.1038/ni.3470. [DOI] [PubMed] [Google Scholar]
- 27.Yoshimura A, et al. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. 1995;14(12):2816–2826. doi: 10.1002/j.1460-2075.1995.tb07281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsumoto A, et al. CIS, a cytokine inducible SH2 protein, is a target of the JAK-STAT5 pathway and modulates STAT5 activation. Blood. 1997;89(9):3148–3154. [PubMed] [Google Scholar]
- 29.Aman MJ, et al. CIS associates with the interleukin-2 receptor beta chain and inhibits interleukin-2-dependent signaling. J Biol Chem. 1999;274(42):30266–30272. doi: 10.1074/jbc.274.42.30266. [DOI] [PubMed] [Google Scholar]
- 30.van der Windt GJ, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36(1):68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Q, et al. Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benzo[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J Med Chem. 2010;53(19):7146–7155. doi: 10.1021/jm101144f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thoreen CC, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284(12):8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem. 2006;281(49):37372–37380. doi: 10.1074/jbc.M608372200. [DOI] [PubMed] [Google Scholar]
- 35.Pulliam SR, Uzhachenko RV, Adunyah SE, Shanker A. Common gamma chain cytokines in combinatorial immune strategies against cancer. Immunol Lett. 2016;169:61–72. doi: 10.1016/j.imlet.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao J, Molenaar AJ, Wheeler TT, Seyfert HM. STAT5 binding contributes to lactational stimulation of promoter III expressing the bovine acetyl-CoA carboxylase alpha-encoding gene in the mammary gland. J Mol Endocrinol. 2002;29(1):73–88. doi: 10.1677/jme.0.0290073. [DOI] [PubMed] [Google Scholar]
- 37.Xu J, Ji J, Yan XH. Cross-talk between AMPK and mTOR in regulating energy balance. Crit Rev Food Sci Nutr. 2012;52(5):373–381. doi: 10.1080/10408398.2010.500245. [DOI] [PubMed] [Google Scholar]
- 38.O’Sullivan TE, Johnson LR, Kang HH, Sun JC. BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory. Immunity. 2015;43(2):331–342. doi: 10.1016/j.immuni.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu JJ, et al. IL15 promotes growth and invasion of endometrial stromal cells and inhibits killing activity of NK cells in endometriosis. Reproduction. 2016;152(2):151–160. doi: 10.1530/REP-16-0089. [DOI] [PubMed] [Google Scholar]
- 40.Rhode PR, et al. Comparison of the superagonist complex, ALT-803, to IL15 as cancer immunotherapeutics in animal models. Cancer Immunol Res. 2016;4(1):49–60. doi: 10.1158/2326-6066.CIR-15-0093-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foley B, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood. 2012;119(11):2665–2674. doi: 10.1182/blood-2011-10-386995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.