

Abstract
Bone remodeling is a highly coordinated process involving bone formation and resorption, and imbalance of this process results in osteoporosis. It has long been recognized that long-term heparin therapy often causes osteoporosis, suggesting that heparan sulfate (HS), the physiological counterpart of heparin, is somehow involved in bone mass regulation. The role of endogenous HS in adult bone, however, remains unclear. To determine the role of HS in bone homeostasis, we conditionally ablated Ext1, which encodes an essential glycosyltransferase for HS biosynthesis, in osteoblasts. Resultant conditional mutant mice developed severe osteopenia. Surprisingly, this phenotype is not due to impairment in bone formation but to enhancement of bone resorption. We show that osteoprotegerin (OPG), which is known as a soluble decoy receptor for RANKL, needs to be associated with the osteoblast surface in order to efficiently inhibit RANKL/RANK signaling and that HS serves as a cell surface binding partner for OPG in this context. We also show that bone mineral density is reduced in patients with multiple hereditary exostoses, a genetic bone disorder caused by heterozygous mutations of Ext1, suggesting that the mechanism revealed in this study may be relevant to low bone mass conditions in humans.
Keywords: Bone Biology
Keywords: Osteoclast/osteoblast biology, Osteoporosis, Proteoglycans
Cell surface heparan sulfate in osteoblasts is a functional component of the RANK-RANKL-mediated bone mass regulation pathway acting as a receptor for osteoprotegerin.
Introduction
Osteoporosis is a common disease characterized by reduced bone mass and increased skeletal fragility. It is a global public health issue affecting more than 200 million people worldwide (1). Fractures deriving from these pathological changes are the most serious aspect of osteoporosis, which causes not only debilitating pains, but also disability and early mortality. The primary pathogenic mechanism of osteoporosis is an imbalance between bone resorption and bone formation, which are mediated by osteoclasts and osteoblasts, respectively.
The key mechanism that regulates bone formation and resorption is transcellular signaling between osteoblasts and osteoclasts (2). This signaling is mediated by RANKL, a cell surface–anchored ligand on activated osteoblasts that stimulates the differentiation of osteoclast precursors, and RANK, the RANKL receptor expressed on osteoclast precursor cells. Additionally, the action of RANKL is negatively regulated by osteoprotegerin (OPG), a decoy receptor expressed by osteoblasts. To maintain bone mass under the physiological range, bone formation and resorption needs to be tightly coupled both in time and space (3). The molecular mechanisms underlying such tight functional coupling, however, have not been completely understood.
Heparan sulfate (HS) is a sulfated linear polysaccharide with a backbone of alternating N-acetylglucosamine and glucuronic acid residues, which is covalently bound to various core proteins to form HS proteoglycans. HS binds various morphogens and growth factors and modulates the activity of these factors. Essentially all key morphogens involved in endochondral ossification, namely BMPs, FGFs, Wnts, and IHH, interact with HS. Functional significance of these interactions has been demonstrated by genetic ablation of the key HS synthetic enzymes in mice. For example, conditional ablation of the Ext1 gene, which encodes a glycosyltransferase essential for the chain elongation step of HS biosynthesis, in the developing limb mesenchyme leads to severe defects in endochondral ossification (4). Moreover, it is clear that HS is relevant to normal skeletal development in humans, as heterozygous mutations of EXT genes cause multiple hereditary exostoses (MHE), one of the most common genetic bone dysplasia syndromes (5).
In contrast to the well-established roles of HS in the developmental aspects of the skeletal system, much less is known about whether HS has physiological functions in the adult skeletal system. The possible role of HS in bone mass regulation has been speculated for some time. For example, the frequent association of osteoporosis with the low-molecular-weight heparin therapy for venous thromboembolism (6–8) has been thought to reflect the physiological function of HS in bone mass regulation. Yet, how endogenous HS acts in bone homeostasis remains elusive because of the lack of appropriate genetic models to address the issue. Against this background, the present study aimed at providing genetic evidence for the physiological significance of endogenous HS in bone mass regulation. We show that removal of HS from osteoblasts leads to severe osteopenia, confirming the physiological significance of HS in the regulation of bone mass. Surprisingly, this phenotype is not due to impairment in bone formation but due to enhancement in bone resorption resulting from dysfunction of OPG. We show that OPG associates with the surface of osteoblasts via interaction with HS and that, in this form, OPG efficiently inhibits RANKL/RANK signaling and osteoclastogenesis. Thus, HS is a functional component of the RANKL/RANK/OPG axis, as it is involved in osteoblast-osteoclast crosstalk. Finally, we show that bone mineral density (BMD) is reduced in patients with MHE, suggesting that the mechanism revealed in this study is relevant to low bone mass conditions in humans.
Results
Osteoblast-targeted Ext1-knockout mice develop osteopenia.
To direct Ext1 ablation to osteoblasts, we employed an osteocalcin-Cre (Oc-Cre) transgene (9). This Cre transgene has been used in a number of studies to interrogate gene function in osteoblasts, and its recombination pattern has been characterized in detail (10–13). Oc-Cre;Ext1flox/flox mice were born and grew without gross morphological abnormalities (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.89624DS1). PCR analysis of DNA samples from Oc-Cre;Ext1flox/flox mice showed that Ext1 ablation occurred in a pattern similar to that of previous reports (Supplemental Figure 1B).
It was noticed that bones of Oc-Cre;Ext1flox/flox mice were brittle. Radiographic examination revealed that bones of Oc-Cre;Ext1flox/flox mice are more translucent than those of WT control mice (Figure 1A). Von Kossa staining of vertebrae showed a marked decrease in bone volume in Oc-Cre;Ext1flox/flox mice at as early as 5 weeks of age (Figure 1B). The decrease was even greater in 3- and 6-month-old animals. Histomorphometric parameters of bone volume (percentage of bone in total measurement area, trabecular number, and trabecular separation) confirmed a significant decrease in bone mass in Oc-Cre;Ext1flox/flox mice compared with WT mice (Figure 1C). The low bone mass phenotype was not limited to vertebrae. Femurs of Oc-Cre;Ext1flox/flox mice also showed a significant decrease in bone volume at as early as 5 weeks of age (Supplemental Figure 2, A and B). While the growth plate of these bones exhibited progressive disorganization in older mutant mice (Supplemental Figure 2A), there were no discernible changes in growth plate morphology at 5 weeks of age, when the low bone mass phenotype was already apparent (Supplemental Figure 2, A and B, 5 weeks), suggesting that the disorganization of the growth plate is a secondary consequence of trabecular bone loss underneath the growth plate. In addition to these phenotypes, Oc-Cre;Ext1flox/flox mice exhibited a few morphological abnormalities, namely a thickening of the metaphysis and a collapse of the secondary ossification center (Supplemental Figure 2A).
Figure 1. Osteoblast-targeted Ext1 ablation induces osteopenia.

(A) X-ray images of the whole body, femur, and lumbar spine of a 12-week-old Oc-Cre;Ext1flox/flox mouse and a WT littermate. Whole-body images show that there are no gross skeletal abnormalities in Oc-Cre;Ext1flox/flox mice. Images of the femur and lumbar spine show that bones of Oc-Cre;Ext1flox/flox mice are more translucent that those of WT mice. (B) Von Kossa staining of lumbar vertebrae at 5 weeks, 3 months, and 6 months of age. (C) Histomorphometric analysis of bone mass in lumbar vertebrae at 5 weeks, 3 months, and 6 months of age. Data represent the mean ± SD of 5 mice per genotype. CKO, Oc-Cre;Ext1flox/flox; BV/TV, bone volume over tissue volume; Tb.N, trabecular number; Tb.Sp, trabecular separation. ***P < 0.001 by Student’s t test. Scale bar: 5 mm (A, whole body images); 1 mm (A, femur and lumber spine images); 1 mm (B).
Osteoblast development and bone formation are unaffected in Oc-Cre;Ext1flox/flox mice and Ext1-null osteoblasts.
Since Ext1 ablation was targeted to osteoblasts in Oc-Cre;Ext1flox/flox mice, we first examined histomorphometric parameters for osteoblasts. Surprisingly, there were no differences in these parameters; the density of osteoblasts was not different between Oc-Cre;Ext1flox/flox and WT mice (Figure 2A). Furthermore, there was no difference in the dynamic bone formation rate, as measured by the calcein double-labeling assay (Figure 2B). Consistent with these results, cultured Ext1-null osteoblasts exhibited normal levels of mineralization upon induction with β-glycerophosphate and ascorbic acid (Figure 2C). Together, these results indicate that the loss of HS expression does not have adverse cell autonomous effects on the development and osteogenic function of osteoblasts.
Figure 2. Osteoblast development and bone formation are unaltered in Oc-Cre;Ext1flox/flox mice.

(A) Histomorphometric analysis of osteoblasts in 3-month-old Oc-Cre;Ext1flox/flox (CKO) and WT lumber vertebrae (n = 5 WT and 5 CKO). (B) Analysis of bone formation by calcein double labeling in 1-month-old CKO and WT vertebrae. The graph shows bone formation rate (BFR/BS, mm3/mm2/yr) calculated from the calcein double-labeling analysis (n = 3 WT and 4 CKO). (C) In vitro mineralization. Ext1-null (KO) and WT osteoblasts cultured under osteogenic conditions for 3 weeks were stained with alizarin red and extracted with 10% cetylpyridinium chloride; absorbance was measured in the extracts at 570 nm (61) (n = 6 WT and 4 KO). Data represent the mean ± SD. N.Ob., number of osteoblasts per bone surface; n.s., not significant by Student’s t test. Scale bar: 0.5 μm.
Osteoclastogenesis and bone resorption are enhanced in Oc-Cre;Ext1flox/flox mice.
Oc-Cre;Ext1flox/flox mice exhibited marked enhancement in osteoclastogenesis and bone resorption. Tartrate-resistant acid phosphatase (TRAP) staining revealed a striking increase in the number of TRAP-positive osteoclasts in Oc-Cre;Ext1flox/flox mice (Figure 3, A and B). Consistent with enhanced bone resorption, urinary excretion of deoxypyridinoline (DPD), a byproduct of collagen degradation, was significantly increased in Oc-Cre;Ext1flox/flox mice (Figure 3C). Thus, Oc-Cre;Ext1flox/flox mice develop a low bone mass phenotype due to enhancement in osteoclastogenesis and bone resorption.
Figure 3. Osteoclastogenesis and bone resorption are enhanced in Oc-Cre;Ext1flox/flox mice.

(A) Tartrate-resistant acid phosphatase (TRAP) staining of 1-month-old Oc-Cre;Ext1flox/flox (CKO) and WT vertebrae. TRAP-positive osteoclasts (stained purple) are greatly increased in CKO mice. (B) Histomorphometric analysis of osteoclasts in 1-month-old CKO and WT vertebrae. (C) Urinary excretion of DPD (normalized by the level of creatinine in the same urine sample). (D) Cocultures of bone marrow monocytes with either Ext1-null or control primary osteoblasts at 4 days in vitro. Ext1-null and control osteoblasts were prepared by infecting Ext1flox/flox osteoblasts with Cre-expressing adenovirus or control adenovirus, respectively. Note the increase in the number of TRAP-positive giant cells in the coculture with Ext1-null osteoblasts (KO Ob + WT BMM) compared with the coculture with control osteoblasts (WT Ob + WT BMM). (E) Quantification of osteoclasts in cocultures. The number of TRAP-positive cells containing more than 3 nuclei is significantly increased in cocultures with Ext1-null osteoblasts. Data represent the mean ± SD (n = 3 in B, C, and E). T.Ar, osteoclast volume density; B.Pm, osteoclast bone surface density. *P < 0.05, **P < 0.01, ***P < 0.001 by Student’s t test (B, C, and E). Scale bar: 5 μm (A); 0.2 mm (D).
Osteoblasts play a key role in the regulation of osteoclastogenesis by their crosstalk with osteoclasts (14). The above results suggest that reduction in HS expression in osteoblasts disrupts osteoblast-osteoclast crosstalk. To test this possibility, we employed the osteoblast–bone marrow macrophage (osteoblast-BMM) coculture system (15). Ext1-null and control osteoblasts were prepared by infecting Ext1flox/flox osteoblasts with either Cre-expressing adenovirus or GFP-expressing adenovirus and coculturing them with BMMs isolated from WT mice. The number of TRAP-positive cells was increased in cocultures with Ext1-null osteoblasts compared with cocultures with WT osteoblasts (Figure 3D). It was also noted that osteoclasts begin to emerge much earlier in cocultures with Ext1-null osteoblasts than in those with WT osteoblasts (~4 days in Ext1-null cocultures versus ~7 days in WT cocultures), suggesting that loss of HS expression accelerates osteoclastogenesis. Furthermore, the frequency of multinucleated mature osteoclasts was greatly increased in cocultures with Ext1-null osteoblasts (Figure 3E). These results indicate that HS expressed in osteoblasts has cell nonautonomous effects on the osteoclastic differentiation of BMMs.
Loss of HS expression ablates cell surface binding of OPG and impairs its osteoclastogenesis-inhibitory activity.
Osteoclastogenesis is regulated primarily by transcellular signaling mediated by RANK expressed in osteoclast precursors and its membrane-bound ligand RANKL in osteoblasts. RANKL/RANK signaling is negatively regulated by the decoy receptor OPG expressed in osteoblasts (16). We surmise that there are two possible mechanisms by which loss of HS expression in osteoblasts leads to the enhancement of osteoclastogenesis. The first possibility is that loss of HS expression in osteoblasts downregulates OPG expression. Since Wnt proteins bind HS (17) and Wnt signaling is the key pathway that induces OPG expression (15, 18), loss of HS expression may lead to enhanced osteoclastogenesis via downregulation of OPG expression. The second possibility is that HS directly regulates the function of OPG. Since OPG binds heparin, it can associate with cell surface HS (19–21). It is thus conceivable that OPG may physiologically act on RANKL/RANK signaling as a cell surface–associated molecule and that the loss of HS may render OPG nonfunctional. We tested these two possibilities.
As for the first possibility, we analyzed the level of Opg expression in osteoblasts in which Ext1 is ablated by Cre-expressing adenovirus. It has been reported that a 50% reduction in Opg expression in Opg+/– mice results in only modest, statistically insignificant, reduction in bone density (22). Therefore, if the low bone mass phenotype in Oc-Cre;Ext1flox/flox mice is mediated by the downregulation of Opg expression, one would expect a more than 50% reduction in Opg expression in Oc-Cre;Ext1flox/flox mice. However, qRT-PCR analysis showed that Opg mRNA expression was unchanged in Ext1-inactivated osteoblasts both before and after induction with prostaglandin E2 and vitamin D3 (Supplemental Figure 3, A and B). Moreover, the expression of Rankl and Mcsf was not changed in these cells either (Supplemental Figure 3, A and B). Finally, we examined the expression of Opg, Rankl, and Mcsf in vivo by qRT-PCR of bone homogenates and found no significant changes in their expression levels between WT and Oc-Cre;Ext1flox/flox mice (Supplemental Figure 3C). Together, these observations indicate that the first possibility is unlikely.
To test the second possibility, we performed a series of biochemical and cell biological analyses. We first sought to confirm the interaction between OPG and HS and to determine the HS-binding site in OPG. Previous studies have demonstrated that the RANKL-binding site in OPG is located in the N-terminal half of the molecule that is composed of 4 cysteine-rich domains (23), while the heparin-binding site is located in the C-terminal side (20) (Figure 4A). It has also been shown that the RANKL- and heparin-binding activities of OPG are independent of each other — truncated OPG mutants lacking the C-terminal side (and heparin-binding activity) can bind RANKL and inhibit osteoclastogenesis as efficiently as the full-length OPG (OPG-WT) (20). We identified 3 conspicuous basic amino acid clusters, which can potentially serve as HS-binding sites, in the C-terminal side of OPG (namely, 239RIKRR, 314RTRKTCK, and 366RKTMR, referred to as C1, C2, and C3, respectively; Figure 4A). We generated 3 recombinant OPG variants, in which basic amino acids in each of the clusters are mutagenized to neutral amino acids (referred to as OPG-C1, OPG-C2, and OPG-C3; see Methods for amino acid substitutions). These recombinant OPGs were tested for HS binding using heparin-agarose (Figure 4B). OPG-WT was bound by heparin-agarose and eluted with 0.4–0.8 M NaCl. OPG-C1 and OPG-C2 showed similar binding. In contrast, OPG-C3 did not bind heparin-agarose at all, indicating that the third cluster is critical for heparin/HS binding.
Figure 4. Heparan sulfate is required for functional presentation of osteoprotegerin on the surface of osteoblasts.

(A) Structure of osteoprotegerin (OPG) and point mutants generated in this study. The locations of the 3 basic clusters that were mutagenized (C1, C2, C3) are shown. (B) Heparin affinity chromatography of OPG mutants. There is essentially no binding of OPG-C3 to heparin, while OPG-C1 and OPG-C2 bind heparin as efficiently as OPG-WT. (C) Binding of OPG-WT and OPG-C3 to WT (CHO-K1) and HS-deficient (pgsD-677) CHO cells. Cell surface–bound OPG was quantified as described in Methods. (D) Association of endogenous OPG with the surface of WT and Ext1-null (KO) primary osteoblasts. (E) Effect of heparitinase treatment (H’ase) on the association of endogenous OPG with the surface of WT primary osteoblasts. (F) Cell surface localization of endogenous OPG in primary human osteoblasts. Cultures of human osteoblasts were double labeled with anti-OPG monoclonal antibody and SiR actin. Control, staining without primary antibody; αOPG, staining with anti-OPG monoclonal antibody; αOPG + H’ase, staining with anti-OPG monoclonal antibody after heparitinase treatment of cells. Scale bar: 10 μm. (G) HS binding is necessary for OPG to efficiently inhibit osteoclastogenesis. Cocultures of osteoblasts (OB) and bone marrow macrophages (BM) were prepared in the combination of two OPG forms (OPG-WT [WT] and OPG-C3 [C3]) and osteoblasts of two genotypes (WT and KO), as indicated in the marker table on the right, and emergence of TRAP-positive osteoclasts was quantitated. OPGs were added at 0, 1, 10, 100, and 300 ng/ml, as indicated. Results are shown as the percentage of TRAP-positive cells relative to the total number of cells. Note that little osteoclastogenesis-inhibitory activity is detected in the OPG-WT/Ext1-null osteoblast combination (squares) and the OPG-C3/WT osteoblast combination (triangles) at 100 ng/ml, while the OPG-WT/WT osteoblast combination shows a significant inhibitory effects in the range of 1–10 ng/ml and almost complete inhibition at 100 ng/ml (circles). Data represent the mean ± SD (number of cultures tested = 5 in C, 4 in D, and 3 in E and F). *P < 0.05, **P < 0.01, ***P < 0.001 by Student’s t test (D and E); 1-way factorial ANOVA and Bonferroni’s post-hoc multiple comparison test (C); and 2-way factorial ANOVA and Tukey’s post-hoc multiple comparison test (F).
Using WT CHO cells (CHO-K1) and their HS-deficient variants (pgs-D677), we examined whether OPG associates with the cell surface via interaction with HS. OPG-WT bound to CHO-K1 cells but not to pgs-D677 cells (Figure 4C). OPG-C3 failed to bind both CHO-K1 and pgs-D677 cells (Figure 4C). The requirement of HS for cell surface association of endogenous OPG was examined using WT and Ext1-inactivated osteoblasts. As shown in Figure 4D, the level of cell-associated OPG was significantly lower in Ext1-inactivated osteoblasts compared with in WT osteoblasts. The large majority of cell-associated OPG was released by treatment with heparitinase (Figure 4E). To further confirm cell surface localization of endogenous OPG, primary human osteoblasts were analyzed by immunocytochemistry without cell permeabilization. As shown Figure 4F, OPG immunoreactivity was detected on the surface of osteoblasts. Treatment of cells with heparitinase prior to immunostaining eliminated OPG immunoreactivity, confirming that OPG is associated with cell surface HS. Together, these results demonstrate that OPG associates with the surface of osteoblasts via interaction with HS.
To determine the functional significance of the OPG-HS interaction, we examined whether binding to HS is functionally necessary for OPG to inhibit osteoclastogenesis. For this, we compared the osteoclastogenesis-inhibitory activity of exogenously added OPG-WT and OPG-C3 in osteoblast-BMM cocultures (Figure 4G). OPG-WT inhibited osteoclastogenesis in the concentration range of >1 ng/ml, and inhibition was almost complete at 100 ng/ml (Figure 4G, circles), a result consistent with previous reports using similar assay systems (24–26). In contrast, although nearly as inhibitory as OPG-WT at a very high concentration (300 ng/ml), OPG-C3 showed essentially no inhibitory activity at 10 ng/ml and only a small inhibitory activity at 100 ng/ml (Figure 4G, triangles). Since the RANKL- and heparin-binding activities of OPG are independent of each other (20, 21, 23), this decrease in inhibitory activity is unlikely due to the lack of RANKL-binding activity of OPG-C3. Rather, these results suggest that HS binding to OPG greatly potentiates its osteoclastogenesis-inhibitory activity. Further confirming the importance of the interaction with cell surface HS, OPG-WT did not inhibit osteoclastogenesis in cocultures with Ext1-null osteoblasts at concentrations as high as 100 ng/ml (Figure 4G, squares). Together, these results demonstrate that OPG binds to the osteoblastic surface via HS and that the association greatly potentiates the osteoclastogenesis-inhibitory activity of OPG.
Ext1 heterozygous genotype is associated with low bone mass both in mice and humans.
Although intriguing, the osteoblastic-specific Ext1 homozygous null mutation modeled in Oc-Cre;Ext1flox/flox mice is an artificial condition that does not exist in the general human population. On the other hand, there has been a suggestion that patients with MHE, which is caused by heterozygous mutations of EXT1 or EXT2, exhibit low bone mass (27). Thus, to explore the relevance of the present finding to human pathological conditions, we asked whether heterozygous mutation of Ext1 is associated with low bone mass conditions. First, we examined mice carrying constitutive heterozygous Ext1 mutation (Ext1+/–) and whether they show low bone mass. Von Kossa staining of lumbar vertebrae at 5 weeks and 3 months after birth revealed a substantial reduction in trabecular bone mass in Ext1+/– mice compared with WT mice (Figure 5A). Histomorphometric parameters of bone volume confirmed a significant decrease in bone mass in Oc-Cre;Ext1flox/flox mice compared with that in WT mice (Figure 5B).
Figure 5. The Ext1 heterozygous genotype is associated with low bone mass in mice.

Von Kossa staining (A) and histomorphometry (B) of lumbar vertebrae in heterozygous (Ext1+/–) and WT (Ext1+/+) mice at 5 weeks and 3 months of age. Bone mass of Ext1+/– mice is lower than that of WT (Ext1+/+) mice. Data represent the mean ± SD of 4 mice per genotype. BV/TV, bone volume over tissue volume; Tb.N, trabecular number; Tb.Sp, trabecular separation. **P < 0.01, ***P < 0.001 by Student’s t test. Scale bar: 0.5 mm.
To further examine the significance of HS in bone mass regulation, we asked whether BMD is reduced in individuals with MHE. We determined BMD parameters in 15 Japanese MHE patients by dual-energy x-ray absorptiometry. The BMD values for these MHE patients were significantly lower than the reference values for normal Japanese populations of corresponding age and sex (28) (Table 1), indicating that patients with MHE tend to have low bone mass. To put this observation in clinical perspective, we also analyzed the T-score of BMD. The T-score is the number of SD units above or below the mean BMD value for young sex-matched control subjects, and WHO defines a T-score of between –1 and –2.5 as representing osteopenia and –2.5 or lower as representing osteoporosis (29). According to this criteria, 53% (8 of 15) of MHE patients in this study are considered as having osteopenia (Figure 6). Together with the observation from Ext1 heterozygous null mice, these results indicate that haploinsufficiency of Ext1, which results in an approximately 50% reduction in HS production, is relevant to moderate reduction in bone mass in vivo.
Table 1. Bone mineral density in patients with MHE.
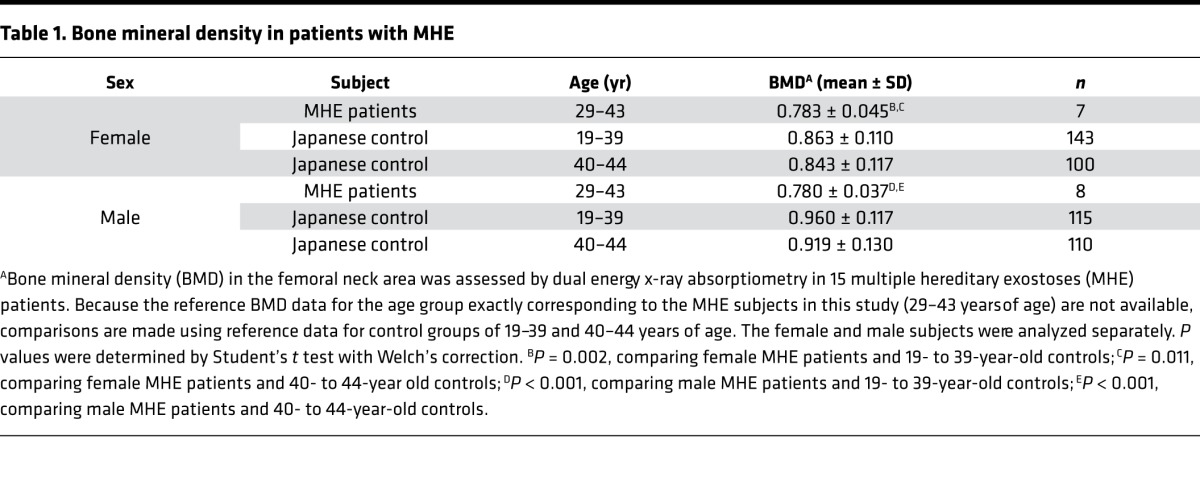
Figure 6. Bone mineral density T-score in patients with multiple hereditary exostoses.
Ranges considered osteopenia and osteoporosis according to the WHO criteria are indicated on the graph. The mean value of T-score for the multiple hereditary exostoses group is shown as a horizontal line (n = 15).
Discussion
In this paper, we show that HS plays a physiological role in bone mass regulation, acting as a component of the RANKL/RANK/OPG axis. Our data reveal that HS on the surface of osteoblasts functions as a “receptor” for OPG and that the HS-mediated association with cell surface is important for OPG to efficiently inhibit of RANKL/RANK signaling and osteoclastogenesis.
The RANKL/RANK/OPG system is the key signaling mechanism that regulates osteoclastogenesis and bone resorption (2, 30). Among the 3 components of the system, OPG binds heparin with a physiologically relevant affinity (19). The interaction is mediated by a cluster of basic amino acid residues located in the C-terminal region of the molecule. Both crystallography and truncation analysis indicate that this location is structurally separated from the RANKL-binding site in OPG (23), which is present in the N-terminal half of OPG consisting of cysteine-rich domains. A previous report that truncated OPG mutants lacking the C-terminal domain can inhibit osteoclastogenesis as efficiently as the OPG-WT (20) is also consistent with this notion. Therefore, OPG that is associated with cell surface via HS can simultaneously interact with RANKL and inhibit the RANKL/RANK signaling.
While both RANK and RANKL are transmembrane proteins, OPG is a secretory protein without any membrane anchors. Accordingly, OPG has been thought to act as a soluble inhibitor to RANKL/RANK signaling (31). The results of the present study suggest a different mode of action for OPG — that is, OPG acts primarily as a cell surface–associated protein. Without association with HS, the ability of OPG to inhibit osteoclastogenesis is greatly diminished (Figure 4G). In this context, it is noteworthy that OPG concentrations in human bone marrow fluid are in the range of 0.1–0.2 ng/ml (32), which is far less than the reported concentrations necessary for effective suppression of osteoclastogenesis in vitro (10–100 ng/ml) (24–26). Further supporting the cell surface association of OPG is the localization of OPG in bone. Nakamura et al. (33) showed by immunohistochemistry and immunoelectron microscopy that OPG is associated with bone surfaces, including the surface osteoblasts, where HS proteoglycans are also enriched (34). It is likely that cell surface association raises OPG local concentrations to a level that efficiently counteracts RANKL/RANK signaling in vivo.
Several studies have tried to address the role of HS in bone formation and resorption. Many of these studies examined the effect of exogenously adding heparin in bone cell cultures to define the physiological function of endogenous HS in adult bone. Conclusions from these studies, however, are inconsistent; some studies conclude that the role of HS is to inhibit osteoclastogenesis, while the others suggested the opposite (35–39). Such a discrepancy is most likely due to the use of exogenous soluble heparin to extrapolate to the function of endogenous HS. It has been known that heparin added to cells can have dual effects on the action of HS-binding growth factors. For example, at low concentrations exogenous heparin enhances FGF2 binding to its receptors, while it inhibits binding at high concentrations (40). The results of the present study clearly demonstrate that the physiological role of HS in adult bone is to suppress excessive bone resorption by acting as a functional component of the RANKL/RANK/OPG system. The reported osteoclastogenesis-promoting effect of exogenous heparin (38) is likely caused by the competitive removal of OPG from the cell surface by nonphysiological levels of heparin. In a similar vein, osteoporosis seen in patients under low-molecular-weight heparin treatment is presumably caused by removal of endogenous OPG from bone surfaces.
Since HS binds and modulates the function of various growth factors and morphogens, it is possible that the mechanism revealed by this study is not the sole mechanism by which HS regulates bone mass. For example, conditional knockout in mice of FGF receptor 2 (Fgfr2), in which an impaired longitudinal bone growth is the primary phenotype, also exhibit reduced bone mass (41). However, the reduction in bone mass in these mice is much milder than that in Opg–/– and Oc-Cre;Ext1flox/flox mice, and, moreover, it has been shown that the phenotype is caused by defects in osteoblastogenesis and bone formation, rather than enhanced osteoclastogenesis (41). Inactivation of IHH signaling via Gli2 knockout also causes low bone mass, but this phenotype is also mediated by decreased osteoblastogenesis (42). In the case of BMP signaling, osteoblast-targeted ablation of type I BMP receptors (Bmpr1a, Acvr1) leads to increased bone mass (43–45), a phenotype opposite to that of Oc-Cre;Ext1flox/flox mice. The possibility that loss of HS suppresses Wnt signaling, thereby downregulating Opg expression, has been ruled out by the qRT-PCR results shown in Supplemental Figure 3. Collectively, these observations suggest that the possibility that HS modulates bone mass via these non-OPG pathways seems rather remote. Although this does not rule out the possible contribution of minor, less well-characterized HS-dependent pathways, it is likely that HS is involved in bone mass regulation primarily via modulation of OPG function.
Although not specifically examined in this study, Oc-Cre;Ext1flox/flox mice exhibited a few morphological abnormalities, namely a thickening of the metaphysis and a collapse of the secondary ossification center (Supplemental Figure 2A). The former phenotype was preceded by the development of severe osteopenia in the surrounding area, suggesting that it may be a secondary consequence of bone loss. Meanwhile, it remains to be determined whether the latter phenotype is secondary to osteopenia or a primary developmental defect independent of osteopenia, as abnormalities in secondary ossification centers are not a common feature seen in a number of severely osteopenic mouse lines. Interestingly, a chondrocyte-targeted Ext1 knockout induced by a Col2-based Cre driver causes a delay in secondary ossification center formation (46), suggesting that HS may be functionally involved in the development of a secondary ossification center. This chondrocyte-targeted knockout mouse model also exhibits a reduction in trabecular bone volume (46). Considering that the Col2-Cre transgene exhibits a leaky expression in osteochondroprogenitors (47), this phenotype may be due to Ext1 ablation in the osteoblastic lineage.
We found that BMD was reduced in patients with MHE (see Table 1 and Figure 6). Previously, Lemos et al. (27) reported an association between MHE and osteoporosis in an Australian kindred carrying an EXT1 splice site mutation. Our present data suggest that the low bone mass condition is not specific for this splice site mutation but more widespread among patients with MHE. In addition, these observations suggest that the mode of action of HS revealed in this study is relevant to bone mass regulation in humans. A question of a more general significance is whether impairment in HS expression underlies the pathogenesis of primary (nonsyndromic) osteoporosis. Besides overt mutations in EXT genes, factors that can potentially cause impairment in HS expression include weaker genetic variants of genes involved in biosynthesis of HS, such as those encoding HS sulfotransferases; epigenetic suppression of these genes; nutritional changes affecting the production of properly sulfated HS chains; and combinations of these factors. For instance, miR-24 is one of a few microRNAs whose expression is elevated in the bone tissue of osteoporotic patients (48), and one of the targets of miR-24 is NDST1, which catalyzes the key sulfation step of HS biosynthesis (49). miR-24 actually reduces HS sulfation and HS affinity to its ligands (50). At any rate, the level of osteoblastic HS expression decreases in an age-dependent manner (51), although its mechanistic basis is unknown. Thus, it is possible that impaired HS expression plays a role in the age-dependent decrease in bone mass and osteoporosis as a susceptibility factor.
In conclusion, this study demonstrated that HS plays a physiological role in the regulation of bone mass by acting as a functional component of OPG-mediated inhibition of osteoclastogenesis. This study also revealed that OPG, which has been thought to act as a soluble decoy receptor, actually functions as a cell surface–associated molecule. Our findings may open an avenue of study toward the identification of a drug target for osteoporosis.
Methods
Conditional knockout mice.
Conditional Ext1-knockout mice were generated by crossing the Ext1flox allele (52) and Oc-Cre transgene (9); they were a gift from Thomas Clemens (Johns Hopkins University, Baltimore, Maryland, USA). Littermates that inherited the incomplete combination of the above alleles were used as WT controls. A constitutive Ext1-null allele (Ext1–) was created by crossing Ext1flox/flox mice with the germline deleter Meox2-Cre mice (53). Rosa26-lacZ mice (54) were obtained from The Jackson Laboratory. All experiments were performed with mice with a complete C57BL/6J background. Genotyping of the mice was performed by PCR as previously described (55). Radiographic examination was performed on a Faxitron MX-20 DC4 (Faxitron X-Ray LLC) using an energy of 26 kV and an exposure time of 10 seconds.
Histological analysis.
All histological analyses were performed on undecalcified sections. Lumbar vertebrae were harvested, immediately frozen in dry ice, and embedded in SCEM compound (Section-Lab Co.). The cut surface was covered with an adhesive film (Cryofilm type IIC9, Section-Lab Co.), and frozen sections (5 μm) were prepared on a cryostat (CM3050, Leica Microsystems) according to a method described previously (56, 57). Femurs from 3-month-old mice were fixed in 70% ethanol and embedded in glycol methacrylate without decalcification. Histomorphometric analysis was performed by the Ito Bone Science Institute (Niigata, Japan) on serial sections as described previously (58). Von Kossa, TRAP, and Safranin O staining were performed as described previously (15, 59). For assessment of dynamic bone formation, calcein double labeling was performed as described previously (60).
Cell culture.
Cultures of primary mouse osteoblasts were prepared as described previously (61). Briefly, calvarial bones from P3 Ext1flox/flox mice were incubated 3 times for 10 minutes in PBS containing 4 mM EDTA. Specimens were subjected to seven 10-minute digestions with PBS containing 180 μg/ml collagenase type 2. The last 5 digests were pooled and seeded in αMEM containing 10% FCS. For the induction of mineralization, primary osteoblasts were cultured in αMEM supplemented with β-glycerophosphate (10 mM) and 50 μg/ml ascorbic acid for 3 weeks (62). To analyze calcium deposition in these cultures, standard alizarin red staining (63) was used. For culturing osteoclasts, femurs and tibiae of 5- to 8-week-old C57BL/6J mice were cut, and cells were flushed into a petri dish with αMEM containing 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine. Filtered cells (100-μm sieve size) were centrifuged after a red blood cell lysis step (150 mM NH4Cl, 0.1 mM EDTA, 10 mM KHCO3, pH 7.2) and plated in cell culture dishes. After incubation for 48 hours, nonadherent cells (BMMs) were collected and induced to differentiate into osteoclasts in αMEM containing 20 ng/ml M-CSF, 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine for 48 hours. Osteoblast/osteoclast cocultures were prepared as described previously (64). Briefly, primary osteoblasts were seeded in 48-well plates at a concentration of 1 × 105 cells/ml. After a 24-hour incubation, BMMs were seeded on primary osteoblasts at a concentration of 1 × 105 cells/ml and treated with 10–8 M 1α,25(OH)2D3 and 10–6 M prostaglandin E2 for 5 days. Osteoclasts in cocultures were identified by TRAP staining. Inactivation of Ext1 in osteoblasts was achieved by infecting cells with Cre-expressing adenovirus. Adenovirus containing a Cre expression cassette (Ad-Cre) and adenovirus containing a control insert (Ad-GFP) were generated by using the pAd/CMV/V5-DEST Gateway Vector Kit (ThermoFisher Scientific), and viral particles were produced and concentrated in the Viral Core Facility at Sanford Burnham Prebys Medical Discovery Institute. For transduction, 3 × 105 primary osteoblasts in a 6-well plate were infected with Ad-Cre or Ad-GFP particles at a multiplicity of infection of 10. After 24 hours, cells were fed with fresh medium and cultured for another 24 hours before being used for experiments. The HS-deficient CHO subline pgs-D677 (65) and the parental CHO-K1 cells were cultured as described previously (66). Primary human osteoblasts isolated from the distal femur (CC-2538, lot 419229) were purchased from Lonza. Digestion of cell surface HS was performed by incubating live cells with 5 mIU/ml heparitinase (heparinase III) (MilliporeSigma, H8891) for 2 hours at 37°C.
Quantitative PCR.
Total RNA was extracted from cultured cells using the RNeasy kit with DNAse (Qiagen). To isolate total RNA from bone homogenates, femurs were harvested from 5-week-old mice. After flushing the bone marrow with PBS and scraping off the periosteum with a scalpel, bones were cut into small pieces, frozen in liquid nitrogen, and pulverized using a tissue homogenizer in Trizol reagent (67). Total RNA was isolated from bone homogenates with the RNeasy kit with DNAse (Qiagen). cDNA was synthesized using SuperScript VILO Master Mix (Invitrogen), and qRT-PCR was performed by using the TaqMan Gene Expression Master Mix (Applied Biosciences). Gene-specific primer pairs and TaqMan probes were purchased from Applied Biosystems. All values were normalized to Gapdh levels, and qRT-PCR data were expressed as fold increases compared with controls.
Biochemical methods.
Concentrations of OPG protein in culture supernatants were measured using an ELISA kit from R&D Systems. DPD in urine was measured using the MicroVue DPD enzyme immunoassay kit (Quidel), and results were normalized by the level of creatinine in the same urine sample measured with a colorimetric assay kit (Cayman Chemical).
Recombinant proteins.
Full-length mouse Opg cDNA was amplified by RT-PCR and ligated to pcDNA3 (ThermoFisher) containing a FLAG peptide sequence downstream of multiple cloning sites for C-terminal fusion. Three mutants of OPG, in which the basic amino acid residues in the putative HS-binding sites were mutagenized (OPG-C1, OPG-C2, OPG-C3; see Figure 4A), were created by using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies). Amino acid sequences of these OPG mutants are (before and after mutagenesis): OPG-C1, 239RIKRR→SINQS; OPG-C2, 314RTRKTCK→STTNTCN; OPG-C3, 366RKTMR→SNTMS. FLAG-tagged OPG proteins were produced by transfecting these expression vectors into 293T cells. After transfection, cells were cultured for 3 days, and then the culture supernatants were harvested. The concentrations of recombinant OPGs in the culture supernatants were determined by ELISA.
Analysis of OPG binding to heparin-agarose and cell surface HS.
To analyze binding of OPGs to heparin, the 293T culture supernatants containing FLAG-tagged OPGs were applied to heparin-agarose (Amersham) and fractionated as described previously (66). To analyze the association of endogenous OPG with osteoblasts, primary osteoblasts were seeded in 48-well plates at a concentration of 1 × 105 cells/well. After 72 hours of incubation in αMEM, cell surface–associated OPG was released with 200 μl cold 1 M NaCl for 5 minutes (68). After salt extraction, the cell layer was extracted with 1% Triton X-100 in PBS to determine the level of total protein in cells. To analyze binding of recombinant OPGs to WT and HS-deficient CHO cells, CHO-K1 and pgs-D677 cells were cultured in a 48-well plate. When cultures become confluent, culture supernatants containing 5 ng/ml OPG-WT or OPG-μC3 were added to the cells. After a 30-minute incubation at 4°C, OPGs in the cell-associated fraction was collected and quantified as described above. For immunocytochemical detection of endogenous OPG on the surface of human osteoblasts, cells were cultured on collagen-coated coverslips in αMEM containing 10% FBS. Prior to fixation, cells were incubated with 1 μM SiR-actin (Spirochrome, catalog no. CY-SC001) for 2 hours to label F-actin and then treated with or without heparitinase as described above. Cells were fixed with 4% paraformaldehyde/4% sucrose in PBS. Localization of endogenous OPG was examined by staining with a mouse monoclonal anti-OPG (clone 98A1071, Novus Biologicals, NB100-56505) and an Alexa Fluor 488–conjugated anti-mouse IgG (Jackson Immuno Research, catalog no. 715-545-151) without permeabilization. Stained cells were examined on a Zeiss LSM710 laser point scanning confocal microscope.
BMD measurement.
All the MHE patients included in this study were Japanese and from unrelated families. They were diagnosed as having MHE and followed up at the Department of Orthopedic Surgery in the Gifu University Hospital. Information, such as age, ethnicity, menstrual status, medication history, and disease history, was obtained via questionnaire. The mean age was 38.50 ± 1.79 years (range, 29–43 years; n = 8) for male and 34.29 ± 1.94 years (range: 29–43 years; n = 7) for female subjects. None of the female subjects were postmenopausal at the time of BMD measurement. The femoral neck was examined using a dual-energy x-ray absorptiometry unit (Lunar DPX-NT, GE Healthcare). BMD and T-score were obtained from the measurements using the manufacturer’s software (enCORE, GE Healthcare). BMD data were compared with the reference values for normal Japanese populations of corresponding age and sex published by the Japanese Society for Bone and Mineral Research (28). The enCORE reference database was used to determine the T-score for the femoral neck. Osteopenia and osteoporosis were defined according to WHO criteria (29).
Statistics.
Statistical significance was determined by following tests. Unpaired 2-tailed Student’s t test was performed for histomorphometric analysis in lumbar vertebrae (Figure 1), in femurs (Supplemental Figure 2B), and of osteoblasts (Figure 2A); analysis of bone formation by calcein double labeling (Figure 2B); in vitro mineralization (Figure 2C); histomorphometric analysis of osteoclasts (Figure 3B); urinary excretion of DPD (Figure 3C); analysis of osteoclastogenesis in cocultures (Figure 3E); association of endogenous OPG with primary osteoblasts (Figure 4D); analysis of the effect of heparitinase treatment on association of endogenous OPG with primary osteoblasts (Figure 4E); histomorphometric analysis of lumber vertebrae in WT and Ext1 heterozygous mice (Figure 5B); and analysis of body weight of WT and Ext1 CKO mice (Supplemental Figure 1A). Unpaired 2-tailed Student’s t test with Welch’s correction was used for BMD of MHE patients (Table 1). For data regarding OPG binding to CHO cell lines (Figure 4C), 1-way factorial ANOVA and Bonferroni’s post-hoc multiple comparison test were used. For data regarding osteoclastogenesis-inhibitory activity of OPGs in osteoblast-BMM cocultures (Figure 4G), 2-way factorial ANOVA and Tukey’s post-hoc multiple comparison test were used. A P value less than 0.05 was considered significant.
Study approval.
All protocols for animal experiments were approved by the IACUC of the Sanford Burnham Prebys Medical Discovery Institute in accordance with NIH guidelines. All work with human clinical specimens was approved by the Medical Ethics Committees of Gifu University. Written informed consent was obtained from all patients prior to their inclusion in the study.
Author contributions
YY conceived the research; SN, TI, FI, and IT performed the research; SN, TI, FI, KM, and YY analyzed data; KM, KS, and HA designed and coordinated the BMD measurement study; and SN and YY wrote the paper.
Supplementary Material
Acknowledgments
This work was supported by NIH grant R01 AR055670 (to YY). SN was the recipient of the Uehara Memorial Foundation Fellowship. TI was the recipient of the Postdoctoral Fellowship for Research Abroad from Japan Society for the Promotion of Science. We thank Thomas Clemens for providing Oc-Cre mice and the MHE Research Foundation for support and encouragement.
Version 1. 02/08/2018
Electronic publication
Funding Statement
S.N. was the recipient of Uehara Memorial Foundation Fellowship.
T.I. was the recipient of Postdoctoral Fellowship for Research Abroad from Japan Society for the Promotion of Science.
Footnotes
Conflict of interest: YY is a recipient of a research grant from Clementia Pharmaceuticals.
Reference information: JCI Insight. 2018;3(3):e89624. https://doi.org/10.1172/jci.insight.89624.
Contributor Information
Satoshi Nozawa, Email: noza618@gmail.com.
Toshihiro Inubushi, Email: tinubushi@sbpdiscovery.org.
Fumitoshi Irie, Email: firie@sbpdiscovery.org.
Iori Takigami, Email: ioritigers@yahoo.co.jp.
Kazu Matsumoto, Email: mkazuu@gifu-u.ac.jp.
Katsuji Shimizu, Email: katsuji.spine@gmail.com.
Haruhiko Akiyama, Email: hakiyama@gifu-u.ac.jp.
Yu Yamaguchi, Email: yyamaguchi@sbpdiscovery.org.
References
- 1.Reginster JY, Burlet N. Osteoporosis: a still increasing prevalence. Bone. 2006;38(2 Suppl 1):S4–S9. doi: 10.1016/j.bone.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 2.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 3.Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13(7):791–801. doi: 10.1038/nm1593. [DOI] [PubMed] [Google Scholar]
- 4.Matsumoto Y, Matsumoto K, Irie F, Fukushi J, Stallcup WB, Yamaguchi Y. Conditional ablation of the heparan sulfate-synthesizing enzyme Ext1 leads to dysregulation of bone morphogenic protein signaling and severe skeletal defects. J Biol Chem. 2010;285(25):19227–19234. doi: 10.1074/jbc.M110.105338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stieber JR, Dormans JP. Manifestations of hereditary multiple exostoses. J Am Acad Orthop Surg. 2005;13(2):110–120. doi: 10.5435/00124635-200503000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Avioli LV. Heparin-induced osteopenia: an appraisal. Adv Exp Med Biol. 1975;52:375–387. doi: 10.1007/978-1-4684-0946-8_33. [DOI] [PubMed] [Google Scholar]
- 7.Murphy MS, John PR, Mayer AD, Buckels JA, Kelly DA. Heparin therapy and bone fractures. Lancet. 1992;340(8827):1098. doi: 10.1016/0140-6736(92)93118-7. [DOI] [PubMed] [Google Scholar]
- 8.Jaffe MD, Willis PW. MULTIPLE FRACTURES ASSOCIATED WITH LONG-TERM SODIUM HEPARIN THERAPY. JAMA. 1965;193:158–160. doi: 10.1001/jama.1965.03090020072024. [DOI] [PubMed] [Google Scholar]
- 9.Zhang M, et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277(46):44005–44012. doi: 10.1074/jbc.M208265200. [DOI] [PubMed] [Google Scholar]
- 10.Holmen SL, et al. Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem. 2005;280(22):21162–21168. doi: 10.1074/jbc.M501900200. [DOI] [PubMed] [Google Scholar]
- 11.Yeo H, et al. Conditional disruption of calcineurin B1 in osteoblasts increases bone formation and reduces bone resorption. J Biol Chem. 2007;282(48):35318–35327. doi: 10.1074/jbc.M702435200. [DOI] [PubMed] [Google Scholar]
- 12.Riddle RC, et al. Lrp5 and Lrp6 exert overlapping functions in osteoblasts during postnatal bone acquisition. PLoS ONE. 2013;8(5):e63323. doi: 10.1371/journal.pone.0063323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan B, et al. Aberrant Phex function in osteoblasts and osteocytes alone underlies murine X-linked hypophosphatemia. J Clin Invest. 2008;118(2):722–734. doi: 10.1172/JCI32702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2(4):389–406. doi: 10.1016/S1534-5807(02)00157-0. [DOI] [PubMed] [Google Scholar]
- 15.Glass DA, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8(5):751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 16.Wada T, Nakashima T, Hiroshi N, Penninger JM. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med. 2006;12(1):17–25. doi: 10.1016/j.molmed.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 17.Lin X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development. 2004;131(24):6009–6021. doi: 10.1242/dev.01522. [DOI] [PubMed] [Google Scholar]
- 18.O’Brien CA. Control of RANKL gene expression. Bone. 2010;46(4):911–919. doi: 10.1016/j.bone.2009.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Théoleyre S, et al. Characterization of osteoprotegerin binding to glycosaminoglycans by surface plasmon resonance: role in the interactions with receptor activator of nuclear factor kappaB ligand (RANKL) and RANK. Biochem Biophys Res Commun. 2006;347(2):460–467. doi: 10.1016/j.bbrc.2006.06.120. [DOI] [PubMed] [Google Scholar]
- 20.Yamaguchi K, et al. Characterization of structural domains of human osteoclastogenesis inhibitory factor. J Biol Chem. 1998;273(9):5117–5123. doi: 10.1074/jbc.273.9.5117. [DOI] [PubMed] [Google Scholar]
- 21.Li M, Yang S, Xu D. Heparan sulfate regulates the structure and function of osteoprotegerin in osteoclastogenesis. J Biol Chem. 2016;291(46):24160–24171. doi: 10.1074/jbc.M116.751974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizuno A, et al. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun. 1998;247(3):610–615. doi: 10.1006/bbrc.1998.8697. [DOI] [PubMed] [Google Scholar]
- 23.Nelson CA, Warren JT, Wang MW, Teitelbaum SL, Fremont DH. RANKL employs distinct binding modes to engage RANK and the osteoprotegerin decoy receptor. Structure. 2012;20(11):1971–1982. doi: 10.1016/j.str.2012.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haynes DR, Crotti TN, Loric M, Bain GI, Atkins GJ, Findlay DM. Osteoprotegerin and receptor activator of nuclear factor kappaB ligand (RANKL) regulate osteoclast formation by cells in the human rheumatoid arthritic joint. Rheumatology (Oxford) 2001;40(6):623–630. doi: 10.1093/rheumatology/40.6.623. [DOI] [PubMed] [Google Scholar]
- 25.Simonet WS, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89(2):309–319. doi: 10.1016/S0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 26.Yasuda H, et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139(3):1329–1337. doi: 10.1210/endo.139.3.5837. [DOI] [PubMed] [Google Scholar]
- 27.Lemos MC, et al. A novel EXT1 splice site mutation in a kindred with hereditary multiple exostosis and osteoporosis. J Clin Endocrinol Metab. 2005;90(9):5386–5392. doi: 10.1210/jc.2004-2520. [DOI] [PubMed] [Google Scholar]
- 28.Orimo H, et al. Diagnostic criteria for primary osteoporosis: year 2000 revision. J Bone Miner Metab. 2001;19(6):331–337. doi: 10.1007/s007740170001. [DOI] [PubMed] [Google Scholar]
- 29.[No authors listed] Prevention and management of osteoporosis. World Health Organ Tech Rep Ser. 2003;921:1–164. [PubMed] [Google Scholar]
- 30.Theoleyre S, Wittrant Y, Tat SK, Fortun Y, Redini F, Heymann D. The molecular triad OPG/RANK/RANKL: involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004;15(6):457–475. doi: 10.1016/j.cytogfr.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 31.Baud’huin M, et al. Osteoprotegerin: multiple partners for multiple functions. Cytokine Growth Factor Rev. 2013;24(5):401–409. doi: 10.1016/j.cytogfr.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 32.Pino AM, et al. Concentration of adipogenic and proinflammatory cytokines in the bone marrow supernatant fluid of osteoporotic women. J Bone Miner Res. 2010;25(3):492–498. doi: 10.1359/jbmr.090802. [DOI] [PubMed] [Google Scholar]
- 33.Nakamura H, Tsuji T, Hirata A, Yamamoto T. Localization of osteoprotegerin (OPG) on bone surfaces and cement lines in rat tibia. J Histochem Cytochem. 2002;50(7):945–953. doi: 10.1177/002215540205000708. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura H, Ozawa H. Immunohistochemical localization of heparan sulfate proteoglycan in rat tibiae. J Bone Miner Res. 1994;9(8):1289–1299. doi: 10.1002/jbmr.5650090819. [DOI] [PubMed] [Google Scholar]
- 35.Ariyoshi W, et al. Heparin inhibits osteoclastic differentiation and function. J Cell Biochem. 2008;103(6):1707–1717. doi: 10.1002/jcb.21559. [DOI] [PubMed] [Google Scholar]
- 36.Ling L, Murali S, Stein GS, van Wijnen AJ, Cool SM. Glycosaminoglycans modulate RANKL-induced osteoclastogenesis. J Cell Biochem. 2010;109(6):1222–1231. doi: 10.1002/jcb.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Théoleyre S, et al. Characterization of osteoprotegerin binding to glycosaminoglycans by surface plasmon resonance: role in the interactions with receptor activator of nuclear factor kappaB ligand (RANKL) and RANK. Biochem Biophys Res Commun. 2006;347(2):460–467. doi: 10.1016/j.bbrc.2006.06.120. [DOI] [PubMed] [Google Scholar]
- 38.Irie A, Takami M, Kubo H, Sekino-Suzuki N, Kasahara K, Sanai Y. Heparin enhances osteoclastic bone resorption by inhibiting osteoprotegerin activity. Bone. 2007;41(2):165–174. doi: 10.1016/j.bone.2007.04.190. [DOI] [PubMed] [Google Scholar]
- 39.Fuller K, Chambers TJ, Gallagher AC. Heparin augments osteoclast resorption-stimulating activity in serum. J Cell Physiol. 1991;147(2):208–214. doi: 10.1002/jcp.1041470204. [DOI] [PubMed] [Google Scholar]
- 40.Fannon M, Forsten KE, Nugent MA. Potentiation and inhibition of bFGF binding by heparin: a model for regulation of cellular response. Biochemistry. 2000;39(6):1434–1445. doi: 10.1021/bi991895z. [DOI] [PubMed] [Google Scholar]
- 41.Yu K, et al. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130(13):3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- 42.Miao D, et al. Impaired endochondral bone development and osteopenia in Gli2-deficient mice. Exp Cell Res. 2004;294(1):210–222. doi: 10.1016/j.yexcr.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 43.Mishina Y, et al. Bone morphogenetic protein type IA receptor signaling regulates postnatal osteoblast function and bone remodeling. J Biol Chem. 2004;279(26):27560–27566. doi: 10.1074/jbc.M404222200. [DOI] [PubMed] [Google Scholar]
- 44.Kamiya N, Kaartinen VM, Mishina Y. Loss-of-function of ACVR1 in osteoblasts increases bone mass and activates canonical Wnt signaling through suppression of Wnt inhibitors SOST and DKK1. Biochem Biophys Res Commun. 2011;414(2):326–330. doi: 10.1016/j.bbrc.2011.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kamiya N, et al. Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J Bone Miner Res. 2008;23(12):2007–2017. doi: 10.1359/jbmr.080809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sgariglia F, et al. Epiphyseal abnormalities, trabecular bone loss and articular chondrocyte hypertrophy develop in the long bones of postnatal Ext1-deficient mice. Bone. 2013;57(1):220–231. doi: 10.1016/j.bone.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ono N, Ono W, Nagasawa T, Kronenberg HM. A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat Cell Biol. 2014;16(12):1157–1167. doi: 10.1038/ncb3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seeliger C, et al. Five freely circulating miRNAs and bone tissue miRNAs are associated with osteoporotic fractures. J Bone Miner Res. 2014;29(8):1718–1728. doi: 10.1002/jbmr.2175. [DOI] [PubMed] [Google Scholar]
- 49.Hashimoto Y, Orellana A, Gil G, Hirschberg CB. Molecular cloning and expression of rat liver N-heparan sulfate sulfotransferase. J Biol Chem. 1992;267(22):15744–15750. [PubMed] [Google Scholar]
- 50.Kasza Z, et al. MicroRNA-24 suppression of N-deacetylase/N-sulfotransferase-1 (NDST1) reduces endothelial cell responsiveness to vascular endothelial growth factor A (VEGFA) J Biol Chem. 2013;288(36):25956–25963. doi: 10.1074/jbc.M113.484360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grzesik WJ, Frazier CR, Shapiro JR, Sponseller PD, Robey PG, Fedarko NS. Age-related changes in human bone proteoglycan structure. Impact of osteogenesis imperfecta. J Biol Chem. 2002;277(46):43638–43647. doi: 10.1074/jbc.M202124200. [DOI] [PubMed] [Google Scholar]
- 52.Inatani M, Irie F, Plump AS, Tessier-Lavigne M, Yamaguchi Y. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 2003;302(5647):1044–1046. doi: 10.1126/science.1090497. [DOI] [PubMed] [Google Scholar]
- 53.Tallquist MD, Soriano P. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis. 2000;26(2):113–115. doi: 10.1002/(SICI)1526-968X(200002)26:2<113::AID-GENE3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 54.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 55.Matsumoto K, Irie F, Mackem S, Yamaguchi Y. A mouse model of chondrocyte-specific somatic mutation reveals a role for Ext1 loss of heterozygosity in multiple hereditary exostoses. Proc Natl Acad Sci USA. 2010;107(24):10932–10937. doi: 10.1073/pnas.0914642107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kawamoto T. Use of a new adhesive film for the preparation of multi-purpose fresh-frozen sections from hard tissues, whole-animals, insects and plants. Arch Histol Cytol. 2003;66(2):123–143. doi: 10.1679/aohc.66.123. [DOI] [PubMed] [Google Scholar]
- 57.Arima Y, et al. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell. 2012;148(3):447–457. doi: 10.1016/j.cell.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 58.Harada S, et al. Daily administration of eldecalcitol (ED-71), an active vitamin D analog, increases bone mineral density by suppressing RANKL expression in mouse trabecular bone. J Bone Miner Res. 2012;27(2):461–473. doi: 10.1002/jbmr.555. [DOI] [PubMed] [Google Scholar]
- 59.Inubushi T, Nozawa S, Matsumoto K, Irie F, Yamaguchi Y. Aberrant perichondrial BMP signaling mediates multiple osteochondromagenesis in mice. JCI Insight. 2017;2(15):e90049. doi: 10.1172/jci.insight.90049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vignery A, Baron R. Dynamic histomorphometry of alveolar bone remodeling in the adult rat. Anat Rec. 1980;196(2):191–200. doi: 10.1002/ar.1091960210. [DOI] [PubMed] [Google Scholar]
- 61.Hessle L, et al. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci USA. 2002;99(14):9445–9449. doi: 10.1073/pnas.142063399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Narisawa S, Harmey D, Yadav MC, O’Neill WC, Hoylaerts MF, Millán JL. Novel inhibitors of alkaline phosphatase suppress vascular smooth muscle cell calcification. J Bone Miner Res. 2007;22(11):1700–1710. doi: 10.1359/jbmr.070714. [DOI] [PubMed] [Google Scholar]
- 63.Hessle L, et al. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci USA. 2002;99(14):9445–9449. doi: 10.1073/pnas.142063399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hikita A, et al. Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-kappaB ligand. J Biol Chem. 2006;281(48):36846–36855. doi: 10.1074/jbc.M606656200. [DOI] [PubMed] [Google Scholar]
- 65.Lidholt K, et al. A single mutation affects both N-acetylglucosaminyltransferase and glucuronosyltransferase activities in a Chinese hamster ovary cell mutant defective in heparan sulfate biosynthesis. Proc Natl Acad Sci USA. 1992;89(6):2267–2271. doi: 10.1073/pnas.89.6.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Irie F, Okuno M, Matsumoto K, Pasquale EB, Yamaguchi Y. Heparan sulfate regulates ephrin-A3/EphA receptor signaling. Proc Natl Acad Sci USA. 2008;105(34):12307–12312. doi: 10.1073/pnas.0801302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Metcalf JA, Zhang Y, Hilton MJ, Long F, Ponder KP. Mechanism of shortened bones in mucopolysaccharidosis VII. Mol Genet Metab. 2009;97(3):202–211. doi: 10.1016/j.ymgme.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bono F, Rigon P, Lamarche I, Savi P, Salel V, Herbert JM. Heparin inhibits the binding of basic fibroblast growth factor to cultured human aortic smooth-muscle cells. Biochem J. 1997;326(Pt 3):661–668. doi: 10.1042/bj3260661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.