

Abstract
We identify 2 homozygous mutations in the ε-subunit of the muscle acetylcholine receptor (AChR) in 3 patients with severe congenital myasthenia: εR218W in the pre-M1 region in 2 patients and εE184K in the β8-β9 linker in 1 patient. Arg218 is conserved in all eukaryotic members of the Cys-loop receptor superfamily, while Glu184 is conserved in the α-, δ-, and ε-subunits of AChRs from all species. εR218W reduces channel gating efficiency 338-fold and AChR expression on the cell surface 5-fold, whereas εE184K reduces channel gating efficiency 11-fold but does not alter AChR cell surface expression. Determinations of the effective channel gating rate constants, combined with mutant cycle analyses, demonstrate strong energetic coupling between εR218 and εE184, and between εR218 and εE45 from the β1-β2 linker, as also observed for equivalent residues in the principal coupling pathway of the α-subunit. Thus, efficient and rapid gating of the AChR channel is achieved not only by coupling between conserved residues within the principal coupling pathway of the α-subunit, but also between corresponding residues in the ε-subunit.
Keywords: Muscle Biology, Neuroscience
Keywords: Ion channels, Neuromuscular disease, Signal transduction
Efficient coupling of agonist binding to gating of the AChR ion channel requires interaction of conserved residues in both the α- and ε-subunit.
Introduction
Congenital myasthenic syndromes (CMSs) are disabling disorders in which the safety margin of neuromuscular transmission is compromised by one or more specific mechanisms. Although no fewer than 30 CMS disease genes have been identified to date, at least half of the CMSs stem from mutations of genes encoding subunits of the muscle acetylcholine receptor (AChR) (1). Mechanistic analyses of AChR disease-causing mutations have guided therapy (1), as well as yielded insights into structure-function relationships (2, 3).
The muscle AChR belongs to the cystine-loop superfamily of neurotransmitter receptors, and is a heteropentamer composed of homologous subunits with a stoichiometry of (α1)2β1δγ in fetal and (α1)2β1δε in adult muscle (4). The AChR aggregates at the tips of postsynaptic folds at a density of about 15,000/μm2, and is an approximately 250-kDa transmembrane protein in which the subunits align as barrel staves around a central ion channel. Each AChR subunit has a large extracellular domain comprising 6 inner β-strands linked by the signature Cys-loop to 4 outer β-strands (Figure 1A), a transmembrane domain composed of 4 α-helices that surround a central ion channel, and an intracellular domain (5, 6). The extracellular domains of specific subunit pairs form the agonist binding sites: a high-affinity site formed at the α/δ interface and a low-affinity site at the α/ε interface. Each binding site comprises 7 loops enriched in aromatic or electronegative residues; loops A, B, and C from the α-subunit form the principal face, and loops D, E, F, and G of the δ- and ε-subunits form the complementary face (Figure 1A) (7). High-resolution structures of several heteromeric and homomeric Cys-loop receptors have been determined: the Torpedo AChR (6), human α4β2 AChR (8), ELIC (9), GLIC (10), Caenorhabditis elegans glutamate-gated chloride channel (GluCl) (11), mouse 5-HT3 receptor (12), and human β3 GABAA receptor (13). Collectively, these structures reveal an abrupt transition zone where the predominantly β-sheet structure of the ligand-binding domain adjoins the α-helical structure of the transmembrane domains; this transition zone is crucial in conveying conformational changes from the agonist binding site to the ion channel.
Figure 1. Location of the mutations and single-channel patch-clamp recordings of wild-type and mutant acetylcholine receptors (AChRs) at low acetylcholine (ACh) concentrations.

(A) Side view of the α- and ε-subunits showing positions of the ACh binding pockets composed of loops A, B, and C in the α-subunit, of loops D, E, F, and G in ε-subunit, and the pre-M1 region and the Cys-loop, β1-2 loop, and β8-9 loops with equivalent residues indicated in both subunits. Mutations in the ε-subunit are shown in red. Based on the crystal structure of the ACh binding protein (PDB 1I9B). (B) Single-channel currents elicited by ACh from HEK cells expressing wild-type, εE184K, and εR218W ACHRs. Left: Channel openings are shown as upward deflections. Right: Logarithmically binned burst-duration histograms fitted to the sum of exponentials. Arrows indicate means of burst exponential components. The values for the longest components are indicated for each AChR.
Over the past decade, numerous studies addressed mechanisms by which the AChR transduces binding of ACh into opening of the ion channel (2, 3), and further, how mechanistic understanding can inform therapy (1). Analyses of energetic coupling between pairs of conserved residues in the α-subunit revealed a principal coupling pathway that functionally links the ligand-binding and pore domains (14). The center of this pathway is an invariant Arg residue (αR209) from the pre-M1 region that lodges within the hydrophobic interior of the subunit. The invariant Arg is strongly coupled to 3 nearby negatively charged residues (αE45, αE175, and αE138), each from a different loop structure that extends to the ligand binding site (15). By virtue of its location within the α-subunit that forms the principal face of the binding site, this pathway has been associated with communicating conformational changes at the agonist binding site to the channel gate. However, a central Arg and 3 negatively charged residues are also present at equivalent positions of the ε- and δ-subunits, which form the complementary face of the binding site, and the question arises of whether these residues contribute to signal transduction similarly to those in the α-subunit.
Here we identify 2 potentially novel mutations in the AChR ε-subunit, εR218W at a position equivalent to αR209, and E184K at a position equivalent to αE175, in 3 congenital myasthenic patients. We first assess pathogenicity of each mutation by single-channel patch-clamp recording, and then use thermodynamic mutant cycle analyses to assess whether εR218 is functionally coupled to εE184, as well as whether εR218 is coupled to negatively charged residues at positions equivalent to αE45 and αD138. We find that both εR218W and εE184K severely reduce the efficiency and speed of channel opening, and that the 2 residues are strongly coupled in conferring efficient and rapid channel opening. In addition, we find that εR218 is strongly coupled to εE45. The overall results reveal striking parallels between equivalent structures in the α- and ε-subunits in conferring efficient and rapid channel gating.
Results
Characteristics of the patients.
Patient 1 is a 35-year-old woman born to healthy first-cousin parents. She had bilateral ptosis since age 2 months, learned to walk at 12 months, but was always weaker than her peers. Her symptoms worsened during infections and menses. Currently she has bilateral ptosis, markedly limited ocular ductions, and mild-moderate weakness in the upper and proximal lower extremity muscles. She responds very well to pyridostigmine and derives further marked benefit from salbutamol. Without treatment, she is often wheelchair dependent. Repetitive nerve stimulation shows a decrement of greater than 30% of the fifth compared with the first evoked muscle potential.
Patient 2, presently 34 years old, was born to a consanguineous, uncle-niece marriage. She had feeble cry since birth, fluctuating bilateral eyelid ptosis with limited ductions since age 5 months, and learned to walk at age 18 months. She had moderate generalized weakness, brisk tendon reflexes, and voice fatigue at age 2 years. Electromyographic studies revealed a 20% decrement of the fifth compared with the first evoked muscle action potential. She responded partially to pyridostigmine when also treated with of venlafaxine and fluoxetine for psychiatric symptoms, she developed complete ophthalmoplegia and her symptoms worsened during menses. Additional treatment with salbutamol at age 34 improved her fatigable weakness. There was no history of similarly affected family members.
Patient 3, currently 25 years old, was an adopted child. She had delayed motor milestones and walked at age 18 months, but fell frequently, could not climb stairs, complained of arm weakness, and had mild difficulty swallowing. Her symptoms worsened with infections. Presently, she has mild bilateral ptosis, mildly limited ocular ductions, and mild-moderate weakness of cervical, upper extremity, and proximal lower extremity muscles. She responds very well to pyridostigmine. Single-fiber electromyography (EMG) revealed increased jitter in all muscle fiber potential pairs studied.
None of the patients has a similarly affected family member. Each patient had negative tests for anti-AChR antibodies.
Mutation analysis.
The indicated codon numbers start from the first codon of the mature peptide (NP_000071.1). Nucleotide numbers start from the translational start site, with +1 corresponding to the A of the ATG translation initiation codon (NM_000080.3). Sequencing of each AChR subunit gene revealed homozygous mutations in the ε-subunit (CHRNE). Patients 1 and 2 harbor a homozygous c.712C>T/p.εR218W mutation located in the pre-M1 region near the junction of the extracellular and pore domains (Figure 1). The asymptomatic parents and sister of patient 1 are heterozygous for the same mutation. The mother of patient 2 is heterozygous for the same mutation. Patient 3 harbors a homozygous c.610G>A/p.εE184K mutation in the linker between β strands 8 and 9 of the major extracellular domain. Neither mutation was detected in 400 normal control alleles, and neither is listed in the Exome Variant Database (Exome Variant Server, NHLBI GO Exome Sequencing Project [ESP], Seattle, WA). Both mutations are predicted to be pathogenic by Mutation Taster and by Polyphen-2. εR218 is conserved at equivalent positions in all subunits of the Cys-loop receptor superfamily of all species, whereas εE184 is conserved at equivalent positions of α-, ε-, and δ-subunits of AChR. To date, no mutations were reported of residues at equivalent positions in non-ε AChR subunits or subunits of all other Cys-loop receptors. Thus, these naturally occurring mutations of conserved residues are strong candidates for disease causation, as well as key determinants of AChR function.
Expression of εR218W and εE184K in HEK cells.
To evaluate pathogenicity of the mutations, we engineered each mutation into the wild-type human ε-subunit, and cotransfected cDNAs encoding mutant or wild-type ε-subunits with complementary wild-type α-, β-, and δ-subunits into HEK cells. Expression of the εR218W-AChR on the surface of intact cells, measured by binding of [125I]α-bungarotoxin ([125I]α-bgt), was reduced to 19% of wild-type, whereas surface expression of the εE184K-AChR was normal. Previous studies showed that mutation of the equivalent Arg residue in the pre-M1 region of the α-subunit (αArg209) markedly reduced AChR expression on the cell surface (14, 16). The marked decrease in AChR expression by εR218W indicates that the conserved Arg in the pre-M1 region of the ε-subunit is similar in importance to the Arg in the pre-M1 region of the AChR α-subunit.
Single-channel currents from the εR218W and εE184K mutants elicited by low ACh concentrations.
To gain insight into the functional consequences of the mutations, we recorded single-channel currents elicited by low concentrations of ACh (50 nM to 1 μM), which appear as bursts of several channel openings in quick succession (Figure 1B). Compared with bursts from the wild-type AChR, bursts from both mutants are noticeably briefer and the number of openings per burst is reduced (Figure 1B). After defining bursts as successive openings separated by closings of a defined brief duration, the bursts were compiled into duration histograms, and the sum of exponentials was fitted to the data. The fitting analyses reveal 2 exponential components for εR218W and εE184K, with mean durations of 0.37 ms and 0.68 ms, and 3 components for the wild-type AChR, the longest of which has a mean duration of 3.3 ms (Figure 1B and Table 1). The briefer mean burst durations of the mutants predict that the synaptic response decays more rapidly at mutant compared with normal endplates, and thus the safety margin for neuromuscular transmission is curtailed.
Table 1. Open intervals and burst durations of wild-type and mutant AChRs on HEK cells.

Patch-clamp recordings from the εR218W and εE184K mutants elicited by increased ACh concentrations.
To gain further insight into the functional consequences of the εR218W and εE184K mutations, we recorded single-channel currents elicited by a range of intermediate and high ACh concentrations. By increasing the ACh concentration, channel openings appear in clusters of many successive openings, all from the same AChR channel, and the temporal distributions of openings and closings within clusters provide kinetic information on state transitions between closed and open states. Subsequently, fitting a kinetic scheme to the sequences of intracluster dwell times yields estimates of rate constants for state transitions in the scheme. For both the wild-type and mutant AChRs, the intracluster closings become progressively briefer as the ACh concentration increases, and the closed-duration histograms mirror the concentration-dependent change from long to brief intracluster closings (Figure 2). Notably, the change from long to brief intracluster closings occurs at a higher concentration of ACh for the mutants compared with the wild-type AChR. In addition, at intermediate ACh concentrations, the ratio of brief to long closings is reduced for the mutants compared with the wild-type AChR. These qualitative changes in intracluster closings suggest the mutations affect one or more rate constants that enable efficient and rapid channel opening.
Figure 2. Single-channel patch-clamp recordings of wild-type and mutant acetylcholine receptors (AChRs) at desensitizing concentrations of acetylcholine (ACh).

Activation kinetics of wild-type (A), εE184K (B), and εR218W (C). The left column shows representative single-channel currents at the indicated ACh concentrations recorded from HEK cells. Bandwidth was 10 kHz. The center and right columns show corresponding histograms of closed- and open-duration dwell times with superimposed global fits for Scheme 1. Fitted rate constants are shown in Table 2.
To identify rate constants altered by the mutation, we fitted the following kinetic scheme (Scheme 1) to sequences of open and closed dwell times obtained over a range of increased ACh concentrations:
(Scheme 1)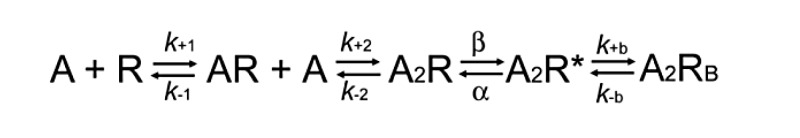
In this scheme, 2 agonists (A) bind to the receptor (R) with association rate constants k+1 and k+2, and dissociate with rate constants k–1 and k–2. The doubly occupied receptor channel opens with rate constant β and closes with rate constant α. In addition, ACh associates with the open channel with rate constant k+b, forming a nonconducting blocked state, and ACh dissociates from the blocked state with rate constant k–b. Owing to practical limitations in recording bandwidth, the previously identified closed state between A2R and A2R*, known as primed or flipped (17, 18), is not included; thus, the fitted rate constants β, α, and k–2 are considered effective rate constants. Also, a desensitized state is not included because each cluster begins just after a receptor recovered from the desensitized state, so that the openings and closings within each cluster represent sojourns in states specified in the scheme.
For both wild-type and mutant AChRs, the probability density functions computed from the fitted rate constants closely overlay the dwell-time histograms (Figure 2), indicating that the kinetic scheme well describes the overall process by which ACh activates the receptor. The fitting analyses reveal that compared with the wild-type AChR, the εR218W and εE184K mutants slow the effective channel opening rate constant β, while they slightly accelerate the channel closing rate constant α (Table 2). The ratio of the 2 effective gating rate constants, β/α, gives the effective channel gating equilibrium constant θ, which decreases 338- and 11-fold for the εR218W and εE184K mutants, respectively. For both mutants, the reductions in the effective channel opening rate constant mainly account for the lower effective gating equilibrium constant, while the ACh dissociation constant K2 decreases mildly (Table 2). Thus, the mutations predominately reduce the speed and efficiency with which the doubly occupied AChR channel opens.
Table 2. Kinetic analysis of wild-type and mutant AChRs.

The fitted rate constants give insight into how the mutations alter neuromuscular transmission. Following synaptic release of ACh, the probability that a doubly occupied AChR channel will open, given by the ratio β/(β + k–2), is predicted to be 0.22 for the εR218W, 0.53 for the εE184K, and 0.84 for the wild-type AChR. The predicted mean burst duration, given by (β/k–2 + 1)/α, is predicted to be 0.37 ms for the εR218W, 0.55 ms for the εE184K, and 2.6 ms for the wild-type AChR. These predicted mean burst durations are within 20% of the mean burst durations determined from recordings at low ACh concentrations, and indicate internal consistency between the estimated rate constants and the experimentally observed sequences of channel openings (Tables 1 and 2). Thus, both the εR218W and εE184K mutations are predicted to reduce the probability that a doubly occupied AChR channel will open, reduce the effective channel opening equilibrium constant, and shorten the burst duration. Moreover, the more than 300-fold reduction of the effective channel opening equilibrium constant by εR218W indicates that the invariant Arg in the ε-subunit is required for efficient and rapid channel opening.
Interaction between εR218 and εE184.
X-ray crystallographic data from several Cys-loop receptors show that within the transition zone between the extracellular and transmembrane domains, electron-rich residues from 3 extracellular loops surround an invariant Arg residue from the pre-M1 region (Figure 1A) (8, 11–13, 19). In the AChR, αR209 (aligned position equivalent to εR218) juxtaposes the conserved αE45 (aligned position equivalent to εE45) at the apex of the β1-2 loop (14), the conserved αE175 (aligned position equivalent to εE184) of the β8-9 loop (15), and the invariant αD138 (aligned position equivalent to εD138) of the Cys-loop (20) (Figure 1A). However, while these 3 negatively charged residues couple strongly to αR209 in promoting efficient and rapid channel opening, a key question is whether residues at equivalent aligned positions in the ε-subunit show similar functional coupling.
To assess whether εR218 and εE184 are functionally coupled in contributing to channel gating, we generated mutations of each residue, individually and as a pair, and used single-channel kinetic analysis to determine the functional consequences of the single and double mutants. Initially, we tested the double mutant εE184K/εR218W, but it did not generate detectable single-channel currents, suggesting a delicate structural interplay between εE184 and εR218 or between the mutants and the surrounding residues. Thus, we generated the double charge-reversal mutant, εE184R/εR218E, which maintained the net charge in this region (Figure 3A) and afforded readily detectable agonist-induced channel openings. Compared with the wild-type AChR, the single charge-reversal mutations, εE184R and εR218E, reduce the effective channel gating equilibrium constant, 53-fold and 104-fold, respectively (Table 2). However, the double mutant εE184R/εR218E reduces the effective channel gating equilibrium constant by 83-fold. Mutant cycle analysis (21), based on the effective channel gating equilibrium constants, reveals an inter-residue coupling free energy of –2.5 kcal/mol (Figure 3A), a value close to that obtained for the corresponding charge-reversal mutations in the α-subunit (–3.1 kcal/mol) (15). The substantial coupling free energy indicates that εR218 and εE184 are strongly interdependent in contributing to efficient and rapid channel gating (Figure 3C).
Figure 3. Mutant cycle analysis.

(A) A mutant cycle of energetic interaction among εE184 and εR218 and among εE45 and εR218. Single-channel currents correspond to each species of acetylcholine receptor (AChR) elicited by 300 μM acetylcholine (ACh). Numbers over the arrows indicate the difference in the apparent gating free energy between 2 different AChRs in kcal/mol. The standard error (SE) values were computed as described under Methods. The 95% confidence limit, or twice the SE, indicates a coupling energy significantly different from zero. The indicated diagonal arrows show the coupling free energy (ΔΔGint) for εE184R/εR218E and εE45R/εR218E. (B) A mutant cycle of energetic interaction among εD138 and εR218. (C) Stereo view of the interface between extracellular domains and transmembrane domains shows spatial disposition of interactions of Arg218 in pre-M1 with Glu184 in the β8-9 loop revealed by mutant cycle analysis in the current work, with Glu45 in the β1-2 loop and with Asp138 in the Cys-loop proposed in the Discussion.
Interactions between εR218 and εE45 and between εR218 and εD138.
To further investigate inter-residue energetic coupling in the ε-subunit, we evaluated coupling between εArg218 and either εGlu45 from the 1-2 loop or εAsp138 from the Cys-loop. In the α-subunit, negatively charged residues at aligned positions equivalent to εGlu45 and εAsp138 couple strongly to the invariant Arg residue equivalent to εR218 (14). The single charge-reversal mutation εE45R reduces the effective channel gating equilibrium constant by 1.7-fold compared with that determined for the wild-type AChR, while the εR218E mutation reduces it 104-fold, as described above (Table 2). However the double mutant εE45R/εR218E reduces the effective channel gating equilibrium constant by only 1.2-fold of the wild-type value, indicating the εE45R mutation almost completely rescues the deleterious effects of εR218E (Table 2). Thus, the functional consequences of the double mutation are much less than expected from additive effects of the single mutations. Mutant cycle analysis, based on the effective channel gating equilibrium constants, reveals a robust coupling free energy of –2.94 kcal/mol (Figure 3A), indicating that εR218 and εE45 are strongly interdependent in contributing to channel gating (Figure 3C).
Analogously, we tested for energetic coupling between εR218 and εD138. However, neither εD138R, εR218D nor εD138R/εR218D yielded detectable single-channel currents, suggesting these residues are structurally crucial. Thus, we made more conservative mutations that maintained the charge on both εD138 and εR218 (Figure 3B). The single-residue mutations εD138E and εR218K yielded agonist-induced single-channel currents, and kinetic analyses revealed modest to substantial decreases in the effective channel opening equilibrium constant of 2- and 16-fold, respectively (Table 2). The double mutation εD138E/εR218K showed a further reduction of the effective channel opening equilibrium constant of 24-fold, but mutant cycle analysis revealed an inter-residue coupling free energy of only –0.22 kcal/mol (Figure 3B), indicating negligible inter-residue coupling. However, because the charge on both residues could not be reversed simultaneously, the question of whether εR218 and εD138 are functionally coupled could not be fully addressed (Figure 3C).
Discussion
We identify 2 potentially novel homozygous mutations in the AChR ε-subunit: εR218W in the pre-M1 region in 2 patients, and εE184K in the β8-9 linker in 1 patient. εR218W reduces the effective channel gating efficiency 338-fold, and reduces cell surface expression 5-fold, while εE184K reduces the effective channel gating efficiency 11-fold but does not alter cell surface expression. To our knowledge, εR218W is the first naturally occurring mutation of the invariant pre-M1 Arg residue in the eponymous Cys-loop receptor superfamily. The dual effects of εR218W on AChR expression and activation kinetics are expected to reduce the safety margin of neuromuscular transmission and thus account for the disease phenotype. The more severe in vitro consequences of εR218W than of εE184K do not explain why the clinical consequences of the 2 mutations are similar. A plausible explanation is that reduced expression of the εR218W mutation elicits compensatory expression of the fetal γ-subunit, as documented in previous cases of fast CMS (22–24). Why the 2 patients harboring the same εR218W mutation responded to pyridostigmine to a different extent remains unexplained.
The essential function of the AChR and its Cys-loop receptor family relatives is to transduce binding of agonist into opening of an intrinsic ion channel, but the structural mechanism behind this chemo-electrical transduction remains a topic of vigorous investigation. In particular, the overall studies provide evidence for structural changes both within and between the subunits. There is a general consensus that upon entry of agonist into the binding pocket, loop structures from both the principal and complementary faces close in upon and trap the agonist (25–28). Whether this trapping of agonist is due to rigid body or local bending motions is not resolved. In addition, upon agonist binding, the M2 α-helices lining the ion channel change from being nearly normal to the plasma membrane to having their extracellular ends tilted outward, while also rotating about their long axes (29, 30). These motions of α-helices, which ultimately allow ions to pass through the channel, arise in response to changes from the structurally remote ligand binding site. Previous studies of inter-residue coupling showed that conserved residues within the structural transition zone between the extracellular and pore domains of the α-subunit were required for this binding to gating signal transduction (15, 31), and suggested a minimal structural mechanism in which inter-residue interactions among key residues within the 2 α-subunits were required. It remained possible, however, that given the high sequence homology among the subunits, structural changes in the adjoining non–α-subunits were also required; in addition, whether the non–α-subunits were active or passive participants remained unknown.
Here we show that the ε-subunit is not only required for signal transduction, but also that it is an active rather than a passive participant. In particular, ε-subunit residues equivalent to those required for signal transduction in the α-subunit are strongly coupled, and decoupling them by mutation prevents efficient and rapid channel opening in response to agonist. However, in the α-subunit the pre-M1 region bearing the central Arg connects directly to loop C at the principal face of the agonist binding site, suggesting a structural basis through which the α-subunit mediates binding-to-gating signal transduction. The equivalent Arg in the pre-M1 region of the ε-subunit also connects to an intrasubunit structure analogous to loop C, but that structure does not form part of an agonist binding site. Instead, in the ε-subunit, the trio of negatively charged residues that surrounds εR218 stem from structures that extend to the complementary face of the agonist binding site: the β8-9 linker that harbors εE184, β-strand 2 that harbors εE45, and β-strand 6 that harbors εD138 (Figure 1A and Figure 3C). While agonist binding alters the principal face of the binding site formed by the α-subunit, it is possible that the same binding event alters the complementary face formed by the ε-subunit. Thus, AChR signal transduction may arise from structural changes within both the principal and complementary subunits that form the ligand binding sites. Our overall approach highlights the dual benefits of studying naturally occurring pathogenic mutations — they allow mechanism-based therapy and bring to light potentially novel structure-function relationships of the AChR.
Methods
Mutation analysis.
We directly sequenced muscle AChR α-, β-, δ-, and ε-subunit genes using the patient’s genomic DNA. Using allele-specific PCR, we determined whether the patient’s mutation was present in family members and 400 normal alleles of 200 unrelated controls.
Construction and expression of wild-type and mutant AChR.
Sources of human α-, β-, δ-, and ε-subunit cDNAs and subcloning of the cDNAs into the CMV-based mammalian expression vector pRBG4 were as previously described (32). The mutations were engineered into wild-type AChR subunit cDNAs in pRBG4 using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies). The presence of each mutation and absence of unwanted mutations were confirmed by sequencing the entire inserts. HEK293 cells (ATTC) were transfected with plasmid DNA encoding α-, β-, δ- and ε-subunits, and pEGFP-N1 at a ratio of 2:1:1:1:1, using the FuGENE 6 transfection reagent (Roche) (24).
α-Bungarotoxin binding measurements.
The total number of 125I-α-bgt sites on the surface of transfected HEK cells was determined as previously described (32). Briefly, intact cells were harvested 3 days after transfection by gentle agitation in PBS with 5 mM EDTA. Cells were briefly centrifuged, resuspended in high-potassium Ringer’s solution, and divided into aliquots for measurements of α-bgt binding. The total number of α-bgt binding sites was determined by incubation for 1 hour in the presence of 5 nM 125I-α-bgt. Unbound toxin was removed by washing with high-potassium Ringer’s solution containing 300 μM d-tubocurarine, followed by filtration using a cell harvester (Brandel Incorporated). Radioactivity retained by glass fiber filters (Whatman GF-B, 1-μm cutoff) was measured with a gamma counter. Nonspecific binding was determined in the presence of 300 μM d-tubocurarine.
Patch-clamp recordings and single-channel kinetic analysis.
Recordings were obtained in the cell-attached patch configuration at a membrane potential of –80 mV at 22°C and with bath and pipette solutions containing (in mM): 142 KCl, 5.4 NaCl, 1.8 CaCl2, 1.7 MgCl2, 10 HEPES, pH 7.4. Single-channel currents were recorded using an Axopatch 200A amplifier (Axon Instruments) at a bandwidth of 50 kHz, digitized at 5-μs intervals using a Digidata 1322A (Axon Instruments) and recorded to a hard disk using the program Clampex 8.2 (Axon Instruments). Recordings obtained with ACh concentrations of 1 μM or lower were analyzed at a uniform bandwidth of 11.7 kHz with an imposed dead time of 15.3 μs. Recordings obtained with ACh concentrations of 10 μM or higher were analyzed at a bandwidth of 10 kHz and an imposed dead time of 25 μs using TACx4.0.9 software (Bruxton). Dwell-time histograms were plotted on a logarithmic abscissa and fitted with the sum of exponentials by maximum likelihood (33).
To estimate rate constants underlying AChR activation, we employed desensitizing concentrations of ACh that cause events from a single receptor channel to cluster into sequences of identifiable activation episodes (34). Clusters were identified as a series of closely spaced openings preceded and followed by closed intervals greater than a defined critical time. The critical time was determined by a method that misclassifies an equal number of events between 2 adjacent closed-time components (35). For each receptor, the critical time that provided the best fit for the closed-time histogram was chosen for the final analysis. Clusters with fewer than 5 openings were excluded from analysis. Individual clusters were examined for homogeneity based on the mean open probability (Popen) and open duration for each cluster, and clusters within 2 SDs of the mean were accepted for further analysis (36, 37). The resulting global set of open and closed dwell times from wild-type and mutant AChRs were analyzed using the program MIL (QuB suite; http://medicine.buffalo.edu/research/service.html), which uses an interval-based maximum likelihood method that also corrects for missed events (36) to yield fitted rate constants in a kinetic scheme for receptor activation.
For each wild-type or mutant AChR, single-channel dwell times obtained over a range of ACh concentrations (10 to 1,000 μM) were fitted simultaneously. The final set of rate constants was checked by comparing probability density functions calculated from the rate constants and the experimental dwell-time histograms and by the ability of the rate constants to predict the mean burst duration at limiting low concentrations of ACh (38, 39).
Thermodynamic mutant cycle analysis.
We performed mutant cycle analysis to determine whether the functional contributions of 2 residues, X and Y, are independent or interdependent. The functional consequences of mutating residue X, with and without the mutation of residue Y, is computed from the free energy change associated with a given reaction step for the wild-type AChR and the mutants AChRX, AChRY, and AChRXY. In the case of the AChR, the relevant reaction step was the apparent channel gating equilibrium constant of the di-liganded receptor, θ, which has an associated free energy change ΔG = –RTlnθ. Although θ is a composite of closed-state priming and channel gating steps (17), and thus is an apparent equilibrium constant, it remains a highly sensitive measure of inter-residue coupling. The changes in gating free energy due to the mutations X, Y, and XY relative to the wild type are designated ΔΔGX, ΔΔGY, and ΔΔGXY. These terms are related to the free energy of inter-residue interaction, ΔΔGint, as follows: ΔΔGXY= ΔΔGX + ΔΔGY + ΔΔGint (21, 40). Given this relationship and noting that ΔΔGs for the individual mutant AChRs are ΔGX – ΔGW, ΔGY – ΔGW, and ΔGXY – ΔGW, ΔΔGint can be calculated from –RTln[(θW × θXY)/(θX × θY)], where θ is the apparent channel gating equilibrium constant for wild-type (W) and mutant (X, Y, XY) AChRs (14, 41).
Statistics.
Error estimates of the scheme rate constants for wild-type and mutant receptors in Table 2 were computed by the MIL program from the curvature of the likelihood function at its maximum (36). To compute the standard error (SE) for ΔΔG, the likelihood surfaces for each fitted β or α are assumed to be symmetrical about the maximum. The SE of ΔΔG is then computed from the SE for each rate constant (i.e., SE βN) as follows (15):
SE = RTsqrt[var(log βW) + var(log αW) + var(log βX) + var(log αX) + var(log βY) + var(log αY) + var(log βXY) + var(log αXY)], where var(log βN) = (SE βN/ βN)2; var(log αN) = (SE αN/αN)2.
Study approval.
All human studies were in accordance with the guidelines of, and approved by, the Institutional Review Board of the Mayo Clinic. The patients reported in this study provided written informed consent to participate.
Author contributions
XMS, SMS, and AGE designed the study and wrote the paper. Clinical phenotyping and EMG were performed by HD, NY, VPK, SV, AN, and FD. JMB performed Sanger sequencing and the binding assays. XMS and SS prepared the plasmids. XMS performed and analyzed the patch-clamp experiments.
Acknowledgments
This work was supported in part by NIH grants NS 6277 to AGE and NS031744 to SMS.
Version 1. 01/25/2018
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2018;3(2):e97826. https://doi.org/10.1172/jci.insight.97826.
Contributor Information
Xin-Ming Shen, Email: shen.xinming@mayo.edu.
Shelley Shen, Email: shen.shelley55@gmail.com.
Hacer Durmus, Email: durmushacer@yahoo.com.
Veeramani Preethish-Kumar, Email: prthshkumar@gmail.com.
Nur Yuceyar, Email: nuryuceyar@gmail.com.
Seena Vengalil, Email: seenavengalil@gmail.com.
Atchayaram Nalini, Email: atchayaramnalini@yahoo.co.in.
Feza Deymeer, Email: fezadeymeer@gmail.com.
Andrew G. Engel, Email: age@mayo.edu.
References
- 1.Engel AG, Shen XM, Selcen D, Sine SM. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015;14(4):420–434. doi: 10.1016/S1474-4422(14)70201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sine SM, Engel AG. Recent advances in Cys-loop receptor structure and function. Nature. 2006;440(7083):448–455. doi: 10.1038/nature04708. [DOI] [PubMed] [Google Scholar]
- 3.Sine SM. End-plate acetylcholine receptor: structure, mechanism, pharmacology, and disease. Physiol Rev. 2012;92(3):1189–1234. doi: 10.1152/physrev.00015.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karlin A, Holtzman E, Yodh N, Lobel P, Wall J, Hainfeld J. The arrangement of the subunits of the acetylcholine receptor of Torpedo californica. J Biol Chem. 1983;258(11):6678–6681. [PubMed] [Google Scholar]
- 5.Galzi JL, Revah F, Bessis A, Changeux JP. Functional architecture of the nicotinic acetylcholine receptor: from electric organ to brain. Annu Rev Pharmacol Toxicol. 1991;31:37–72. doi: 10.1146/annurev.pa.31.040191.000345. [DOI] [PubMed] [Google Scholar]
- 6.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346(4):967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 7.Sine SM. The nicotinic receptor ligand binding domain. J Neurobiol. 2002;53(4):431–446. doi: 10.1002/neu.10139. [DOI] [PubMed] [Google Scholar]
- 8.Morales-Perez CL, Noviello CM, Hibbs RE. X-ray structure of the human α4β2 nicotinic receptor. Nature. 2016;538(7625):411–415. doi: 10.1038/nature19785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hilf RJ, Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452(7185):375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- 10.Bocquet N, et al. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457(7225):111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- 11.Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474(7349):54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hassaine G, et al. X-ray structure of the mouse serotonin 5-HT3 receptor. Nature. 2014;512(7514):276–281. doi: 10.1038/nature13552. [DOI] [PubMed] [Google Scholar]
- 13.Miller PS, Aricescu AR. Crystal structure of a human GABAA receptor. Nature. 2014;512(7514):270–275. doi: 10.1038/nature13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee WY, Sine SM. Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature. 2005;438(7065):243–247. doi: 10.1038/nature04156. [DOI] [PubMed] [Google Scholar]
- 15.Mukhtasimova N, Sine SM. Nicotinic receptor transduction zone: invariant arginine couples to multiple electron-rich residues. Biophys J. 2013;104(2):355–367. doi: 10.1016/j.bpj.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Purohit P, Auerbach A. Acetylcholine receptor gating at extracellular transmembrane domain interface: the “pre-M1” linker. J Gen Physiol. 2007;130(6):559–568. doi: 10.1085/jgp.200709857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukhtasimova N, Lee WY, Wang HL, Sine SM. Detection and trapping of intermediate states priming nicotinic receptor channel opening. Nature. 2009;459(7245):451–454. doi: 10.1038/nature07923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lape R, Colquhoun D, Sivilotti LG. On the nature of partial agonism in the nicotinic receptor superfamily. Nature. 2008;454(7205):722–727. doi: 10.1038/nature07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zouridakis M, Giastas P, Zarkadas E, Chroni-Tzartou D, Bregestovski P, Tzartos SJ. Crystal structures of free and antagonist-bound states of human α9 nicotinic receptor extracellular domain. Nat Struct Mol Biol. 2014;21(11):976–980. doi: 10.1038/nsmb.2900. [DOI] [PubMed] [Google Scholar]
- 20.Shen XM, Brengman J, Neubauer D, Sine SM, Engel AG. Investigation of congenital myasthenia reveals functional asymmetry of invariant acetylcholine receptor (AChR) Cys-loop aspartates. J Biol Chem. 2016;291(7):3291–3301. doi: 10.1074/jbc.M115.683995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horovitz A, Fersht AR. Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. J Mol Biol. 1990;214(3):613–617. doi: 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
- 22.Engel AG, Ohno K, Bouzat C, Sine SM, Griggs RC. End-plate acetylcholine receptor deficiency due to nonsense mutations in the epsilon subunit. Ann Neurol. 1996;40(5):810–817. doi: 10.1002/ana.410400521. [DOI] [PubMed] [Google Scholar]
- 23.Ohno K, et al. Congenital myasthenic syndromes due to heteroallelic nonsense/missense mutations in the acetylcholine receptor epsilon subunit gene: identification and functional characterization of six new mutations. Hum Mol Genet. 1997;6(5):753–766. doi: 10.1093/hmg/6.5.753. [DOI] [PubMed] [Google Scholar]
- 24.Shen XM, Ohno K, Sine SM, Engel AG. Subunit-specific contribution to agonist binding and channel gating revealed by inherited mutation in muscle acetylcholine receptor M3-M4 linker. Brain. 2005;128(Pt 2):345–355. doi: 10.1093/brain/awh364. [DOI] [PubMed] [Google Scholar]
- 25.Gao F, et al. Agonist-mediated conformational changes in acetylcholine-binding protein revealed by simulation and intrinsic tryptophan fluorescence. J Biol Chem. 2005;280(9):8443–8451. doi: 10.1074/jbc.M412389200. [DOI] [PubMed] [Google Scholar]
- 26.Hansen SB, Sulzenbacher G, Huxford T, Marchot P, Taylor P, Bourne Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005;24(20):3635–3646. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang HL, et al. Single-channel current through nicotinic receptor produced by closure of binding site C-loop. Biophys J. 2009;96(9):3582–3590. doi: 10.1016/j.bpj.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li SX, et al. Ligand-binding domain of an α7-nicotinic receptor chimera and its complex with agonist. Nat Neurosci. 2011;14(10):1253–1259. doi: 10.1038/nn.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Althoff T, Hibbs RE, Banerjee S, Gouaux E. X-ray structures of GluCl in apo states reveal a gating mechanism of Cys-loop receptors. Nature. 2014;512(7514):333–337. doi: 10.1038/nature13669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Du J, Wu S, Cheng Y, Gouaux E. Glycine receptor mechanism elucidated by electron cryo-microscopy. Nature. 2015;526(7572):224–229. doi: 10.1038/nature14853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukhtasimova N, Free C, Sine SM. Initial coupling of binding to gating mediated by conserved residues in the muscle nicotinic receptor. J Gen Physiol. 2005;126(1):23–39. doi: 10.1085/jgp.200509283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohno K, et al. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor epsilon subunit. Neuron. 1996;17(1):157–170. doi: 10.1016/S0896-6273(00)80289-5. [DOI] [PubMed] [Google Scholar]
- 33.Sigworth FJ, Sine SM. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J. 1987;52(6):1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakmann B, Patlak J, Neher E. Single acetylcholine-activated channels show burst-kinetics in presence of desensitizing concentrations of agonist. Nature. 1980;286(5768):71–73. doi: 10.1038/286071a0. [DOI] [PubMed] [Google Scholar]
- 35.Colquhoun D, Sakmann B. Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J Physiol (Lond) 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qin F, Auerbach A, Sachs F. Maximum likelihood estimation of aggregated Markov processes. Proc Biol Sci. 1997;264(1380):375–383. doi: 10.1098/rspb.1997.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen XM, et al. Mutation causing severe myasthenia reveals functional asymmetry of AChR signature cystine loops in agonist binding and gating. J Clin Invest. 2003;111(4):497–505. doi: 10.1172/JCI16997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Colquhoun D, Hawkes AG. The Principles of the Stochastic Interpretation of Ion-Channel Mechanisms. In: Sakmann B, Neher E, eds. Single-Channel Recording. Boston, MA: Springer; 1995: 397–482. [Google Scholar]
- 39. Colquhoun D, Hawkes AG. A Q-Matrix Cookbook: How to Write Only One Program to Calculate the Single-Channel and Macroscopic Predictions for Any Kinetic Mechanism. In: Sakmann B, Neher E, eds. Single-Channel Recording. Boston, MA: Springer; 1995: 589–633. [Google Scholar]
- 40.Wells JA. Additivity of mutational effects in proteins. Biochemistry. 1990;29(37):8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- 41.Shen XM, Fukuda T, Ohno K, Sine SM, Engel AG. Congenital myasthenia-related AChR delta subunit mutation interferes with intersubunit communication essential for channel gating. J Clin Invest. 2008;118(5):1867–1876. doi: 10.1172/JCI34527. [DOI] [PMC free article] [PubMed] [Google Scholar]