

Summary
Arenaviruses cause fatal haemorrhagic disease in humans. Old-World arenavirus glycoproteins (GPs) mainly engage α-dystroglycan as a cell surface receptor, while New-World arenaviruses hijack transferrin receptor. However, the Lujo virus (LUJV) glycoprotein does not cluster with New-or Old-world arenaviruses. Using a recombinant vesicular stomatitis virus containing LUJV glycoprotein (LUJV GP) as its sole attachment and fusion protein (VSV-LUJV), we demonstrate that infection is independent of known arenavirus receptor genes. A genome-wide haploid genetic screen identified the transmembrane protein neuropilin 2 (NRP2) and tetraspanin CD63 as factors for LUJV GP-mediated infection. LUJV GP binds the N-terminal domain of NRP2, while CD63 stimulates pH-activated LUJV GP-mediated membrane fusion. Overexpression of NRP2 or its N-terminal domain enhances VSV-LUJV infection, and cells lacking NRP2 are deficient in wild-type LUJV infection. These findings uncover this distinct set of host cell entry factors in LUJV infection and are attractive focus points for therapeutic intervention.
Keywords: LUJV virus, Arenavirus, Haploid genetics, Entry receptor, NRP2, CD63
Introduction
Viruses in the family Arenaviridae often cause a persistent, asymptomatic infection in their natural rodent hosts. Transmission of such viruses to humans occurs through contact with infected rodents often through aerosolized urine and feces. Infection of humans can be associated with severe and sometimes fatal hemorrhagic disease (Moraz and Kunz, 2011). The geographic distribution of such viruses reflects that of the rodent hosts. The West African Lassa virus (LASV) and the South American Machupo virus (MACV), Guanarito virus (GTOV), Sabia virus (SABV) and Junin virus (JUNV) can instigate such severe hemorrhagic fever in humans (Moraz and Kunz, 2011).
Arenaviruses are enveloped viruses, with a bisegmented negative-strand RNA genome encoding for the expression of only four structural proteins, of which the envelope-embedded glycoprotein (GP) mediates cell entry. GP binds host-encoded receptors and enables membrane fusion that results in the release of the viral genome into the cytoplasm of the target cell (Rojek and Kunz, 2008). Based on Pairwise Sequence Comparison (PASC) of viral genomes, arenaviruses can be classified into two distinct groups (Radoshitzky et al., 2015): ‘Old-World arenaviruses’ that mainly utilize α-dystroglycan (α-DG) as a cell-surface receptor (Cao et al., 1998), and ‘New-World arenaviruses’, of which the pathogenic viruses (i.e. MACV, GTOV, SABV and JUNV) hijack TfR1 for entry into human cells (Radoshitzky et al., 2007). We recently identified lysosomal-associated membrane protein 1 (LAMP1) as an additional intracellular receptor for LASV (Jae et al., 2014), highlighting a receptor-switch from α-DG to LAMP1 during viral endocytosis, reminiscent of the multi-step entry strategy of Ebola virus (EBOV) (Jae and Brummelkamp, 2015).
LUJV virus (LUJV) was identified as the causative agent of an outbreak of lethal hemorrhagic fever disease in South Africa in 2008 (Briese et al., 2009). Genome sequence analysis demonstrates that the expected receptor binding region, GP1 of LUJV, does not cluster with other New-or Old-world arenaviruses, and therefore is postulated to exploit distinct entry receptors. (Fig.S1).
To explore the cell entry pathway(s) used by LUJV we constructed a recombinant vesicular stomatitis virus (VSV) containing as its sole attachment and fusion protein LUJV GP (VSV-LUJV) and employed this virus in a genome-wide haploid genetic screen. Interrogation of many independent genomic mutations in VSV-LUJV resistant haploid human cells identified a set of host factors, including the arenavirus receptor neuropilin-2 (NRP2) and CD63, a tetraspanin that aids LUJV GP-mediated membrane fusion.
Results
LUJV cell entry requires a specific set of host factors
Using haploid genetics, we previously identified host factors for the entry of EBOV, LASV and Rift Valley Fever Virus (Carette et al., 2011a; Jae et al., 2013, 2014; Riblett et al., 2016). To apply the same screening approach to search for host genes required for LUJV entry we generated a replication-competent recombinant vesicular stomatitis virus expressing LUJV GP as its sole attachment and fusion protein (VSV-LUJV). Haploid HAP1 cells supported amplification of VSV-LUJV resulting in a cytopathic effect. Consistent with the use of distinct host factors during LUJV entry, infection of HAP1 cells with VSV-LUJV was unaffected by genetic ablation of α-DG (DAG1), or by pre-treating cells with antibodies that block access to TfR1 (Fig.1A) (Tani et al., 2014).
Figure 1. LUJV GP-mediated cell entry requires a distinct set of host factors.
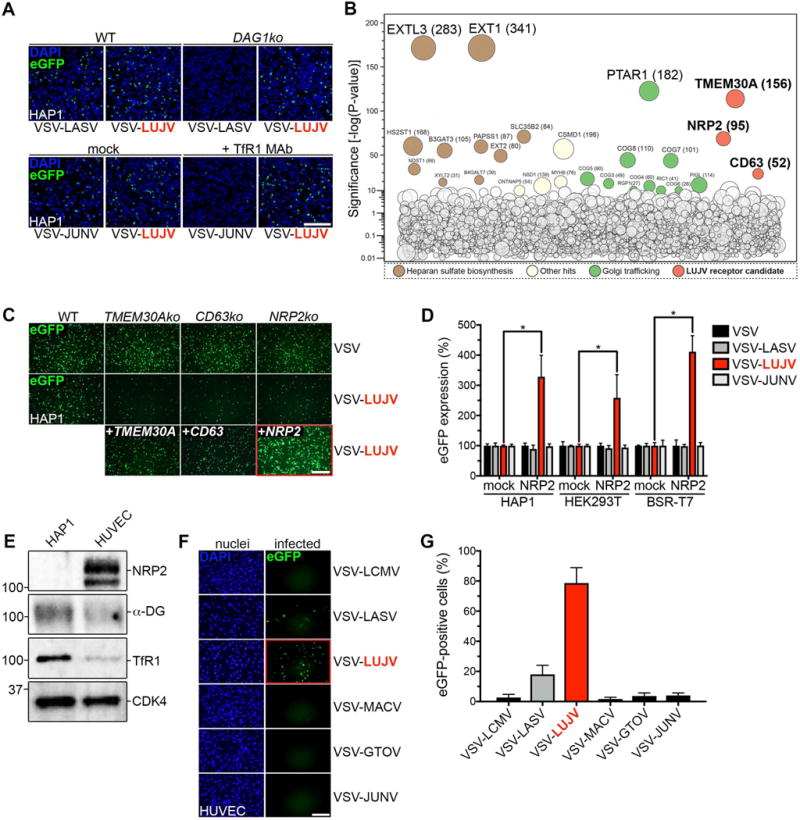
(A) Wild-type (WT) and α-DG knockout (DAG1ko) HAP1 cells were inoculated with the indicated VSVs (multiplicity of infection [MOI]=0.5). In parallel, HAP1 cells were mock-treated or pre-treated with TfR1 blocking antibodies for 1h before infection. Cells were fixed 8h post virus inoculation, stained with DAPI (to detect the nuclei) and infection (visible through virus-encoded eGFP expression) was detected by fluorescence microscopy. Scale bar: 10μm. (B) Data from the haploid screen with VSV-LUJV. The y-axis indicates the significance of enrichment of gene-trap insertions compared with unselected control cells. Circles represent individual genes and their size corresponds to the number of unique disruptive insertion sites identified in the virus-selected population. Genes with significance scores above 10 are labeled, colored and grouped according to function. (C) Wild-type HAP1 cells (WT), knockout clones (TMEM30Ako, CD63ko and NRP2ko) and reconstituted clones (+TMEM30A, +CD63 and +NRP2) were infected with VSV (control) or VSV-LUJV (MOI=3). Cells were fixed 6h post virus inoculation and infection was visualized by fluorescence microscopy. Scale bar: 10μm. (D) HAP1, HEK293T and BSR-T7 cells were transduced with empty (mock) retrovirus or with virus expressing NRP2-HA. Next, cells were inoculated with the indicated viruses (MOI=3). Cells were fixed 6h post inoculation and infection was measured by quantifying eGFP fluorescence. The data is presented as the percentage eGFP expression of each virus in cells overexpressing NRP2 compared to mock-transduced cells for each cell line. Significant differences (t-test; p-value <0.05) between each condition (n=3 experimental replicates) are indicated by the asterisks. (E) Western blot of total cell lysates from HAP1 and HUVEC cells. Membranes were probed with anti-NRP2, anti-α-DG or anti-TfR1 antibodies to detect endogenous expression in both cell types. An anti-CDK4 antibody was used as a loading control. (F) HUVEC cells were inoculated with the indicated viruses (MOI=1, as determined on HAP1 cells). Cells were fixed 6h post virus inoculation and infection was visualized by fluorescence microscopy. Cells were counter-stained with DAPI (shown as a separate panel). Scale bar: 10μm. (G) Quantification of the experiment shown in panel F. The data is presented as the percentage of infected (eGFP-positive) cells (n=3 experimental replicates).
To identify those factors, we infected approximately 100 million mutagenized HAP1 cells with VSV-LUJV and expanded cells that were resistant to infection. Sites of retroviral insertion in those surviving cells were mapped using parallel deep sequencing and compared to mutation frequencies in an unselected control population (Fig.1B and Table S1). Those genes include 10 involved in the biosynthesis of heparan sulfate (EXT1, EXTL3, SLC35B2, HS2ST1, B3GAT3, PAPSS1, EXT2, NDST1, XYLT2 and B4GALT7) and several genes involved in Golgi-associated trafficking/glycosylation, including 6 out of 8 genes of the Conserved Oligomeric Golgi (COG) complex (COG3-8 and PTAR, RGP1, RIC1 and PIGL). A previous screen for cell-surface expression of heparan sulfate also identified PTAR1 and the Conserved Oligomeric Golgi (COG) complex genes identifying them as positive regulators (Jae et al., 2013). Consequently, the identification of those genes implicates heparan sulfate in LUJV GP-dependent infection.
Insertions were highly enriched in three other genes (TMEM30A, NRP2, and CD63), for which we identified 156, 95, and 52 independent mutations, respectively. TMEM30A (also called CDC50A) is required for phospholipid translocation from the outer to the inner plasma membrane leaflet (Paulusma et al., 2008). NRP2 encodes a transmembrane protein that serves as a receptor for several semaphorin proteins (Kolodkin et al., 1997) and vascular endothelial growth factor (Parker et al., 2015) and is thought to be involved in cardiovascular development (Takashima et al., 2002). CD63 encodes a protein that belongs to the tetraspanin family and is mainly associated with membranes of intracellular vesicles (Kobayashi et al., 2000).
NRP2 functions as a proteinaceous cell-surface receptor during LUJV GP-mediated infection
To validate the role of the genes identified in the haploid genetic screen, we enzymatically removed heparan sulfate from the cell surface of HAP1 cells before infecting them with VSV-LUJV or VSV. Infection of HAP1 cells with VSV was unaffected by heparinase treatment whereas VSV-LUJV infection was reduced approximatey 60% (Fig.S2A). This result is consistent with a role of heparan sulfate in LUJV infection likely at the level of viral attachment as has been described for several other viruses (Hilgard and Stockert, 2000; Riblett et al., 2016; Salvador et al., 2013).
The identification of three membrane protein genes (TMEM30A, NRP2 and CD63) prompted us to investigate their role in LUJV entry. To independently verify the importance of those genes in infection we generated knockout clones of TMEM30A, NRP2 and CD63 in HAP1 cells by CRISPR/Cas9 gene editing. All three clones showed a specific reduced susceptibility to VSV-LUJV infection compared with VSV (Fig.1C). In each case susceptibility of the knockout cells to VSV-LUJV infection was restored following expression of the corresponding cDNA. In contrast to NRP2 and CD63 (as described below), the function of TMEM30A could not be confirmed in primary human umbilical vein endothelial cells (HUVEC) cells (Fig.S2B) and was therefore excluded from further analysis.
Endothelial cells express high levels of NRP2 (Sulpice et al., 2008) and are targeted during viral infection. Overexpression of NRP2 enhanced infection of VSV-LUJV not only in HAP1 cells, but in a range of different cell lines (Fig.1D). A comparison of NRP2 levels demonstrates higher levels in HUVECs compared to HAP1 cells, in contrast to expression of α-DG and TfR1 (Fig.1E). Consistent with this data, VSV-LUJV infection was significantly enhanced in HUVEC’s when compared to VSV expressing other Old and New world arenavirus GPs (Fig.1F and G). Collectively, the above data implies that NRP2 is a LUJV specific entry factor and correlates expression levels with susceptibility to infection.
LUJV GP interacts with the N-terminal domain of NRP2 to facilitate cell entry and subsequent infection
Based on its membrane localization and function we hypothesized that NRP2 could serve as a LUJV receptor. To test this, we performed pull-down assays with immobilized Flag-tagged LUJV GP and cell lysates containing HA-tagged NRP2. Beads containing LUJV GP readily showed binding to NRP2, whereas control beads containing LASV GP failed to display an interaction (Fig.2A). To delineate the binding site on NRP2 we generated expression constructs containing different domains of NRP2 guided by the atomic structure (Appleton et al., 2007) (Fig.2B). This approach narrowed down the binding site of LUJV GP to the N-terminal region (domain A1) of NRP2. We did not detect such an association with the equivalent A1 domain of the neuropilin family member NRP1 (Fig.2C). Pull-down assays with soluble LUJV GP1 demonstrate that this predicted receptor binding subunit is able to interact with NRP2 (Fig.2D). Domain A1 of NRP2 is sufficient to mediate VSV-LUJV infection when overexpressed in HAP1 cells (Fig.2E). Using VSV-LUJV and VSV-LASV particles labeled with spectrally distinct fluorophores we observed a specific reduction in VSV-LUJV attachment to NRP2-deficient cells (Fig.2F and G). Taken together the above results define an interaction between NRP2 and LUJV GP provided by the N-terminal A1 domain of NRP2 and the receptor binding GP1 portion of LUJV GP, implicating NRP2 as a LUJV receptor.
Figure 2. LUJV GP binds the N-terminal domain of NRP2.
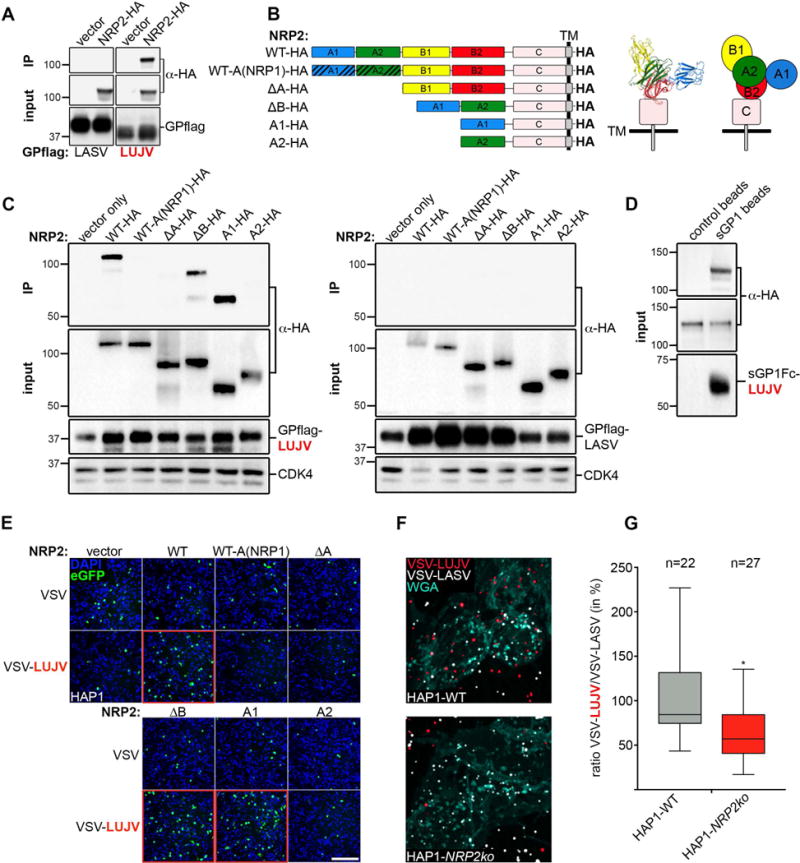
(A) Flag-tagged LASV GP and LUJV GP was immobilized on beads and incubated with cell lysates from HEK293T cells (transduced with control [vector] or NRP2-HA retrovirus). Bound proteins (IP: immunoprecipitation) and input samples were subjected to immunoblot analysis. (B) Schematic representation of all the NRP2 constructs used in the biochemistry and infection experiments described in panel C and D. The design of the constructs was based on the reported NRP2 crystal structure (Appleton et al., 2007). (C) Flag-tagged LASV GP and LUJV GP was immobilized on beads and incubated with cell lysates from HEK293T cells expressing vector control, HA-tagged full-length NRP2 or the individual mutants depicted in panel B. Bound proteins (IP: immunoprecipitation) and input samples were subjected to immunoblot analysis. An anti-CDK4 antibody was used as a loading control. (D) Soluble Fc-tagged LUJV GP1 (sGP1Fc-LUJV) was immobilized on beads and incubated with total cell lysate from HEK293T cells expressing NRP2-HA. Empty beads served as a control. Bound proteins (pulldown and beads) as wells as the input samples were subjected to immunoblot analysis. (E) HAP1 cells expressing vector control, HA-tagged full-length NRP2 or the individual NRP2 mutants were inoculated with VSV (control) or VSV-LUJV (MOI=0.2) for 6h at 37°C. Cells were fixed and stained with DAPI after which infection was visualized by fluorescence microscopy. Scale bar: 10μm. (F) HAP1 wild-type or NRP2ko cells were co-inoculated with fluorescently labeled VSV-LUJV (in red) and VSV-LASV (in white) for 15min at 37°C. Cells were subsequently washed and stained with fluorescently labeled WGA (in cyan) to visualize the cell boundaries and the viral particles attached to it. Representative images are shown. (G) Quantification of panel F (combined data of two independent experiments). Shown is the percentage of cell-associated VSV-LUJV particles over VSV-LASV particles in HAP1 wild-type (WT) or NRP2ko cells (n=22 and 27 cells, respectively). The significant difference (unpaired t-test; p-value=0.0023) is indicated by the asterisk.
CD63 is required for LUJV cell entry and stimulates membrane fusion
Following infection with VSV-LUJV we observed viral particles in CD63-positive compartments (Fig.3A and Movie S1). As expected, LUJV GP-mediated infection is sensitive to Bafilomycin A1 and thus pH-dependant (Fig.3B), highlighting the importance of the endo/lysosomal system in cell entry. To probe the role of CD63 in LUJV infection, we performed a cell-cell fusion experiment in cells overexpressing wild-type CD63 or a mutant (GY234AA) that localizes predominantly to the cell surface (Fig.S3) (Latysheva et al., 2006). Overexpression of CD63, but particularly the GY234AA mutant, together with LUJV GP led to syncytia formation only under acidic conditions, indicating the requirement of robust cell surface expression of CD63 in this assay (Fig.3C). In contrast, overexpression of NRP2 is insufficient to mediate membrane fusion following low pH treatment. The GP:NRP2 interaction is lost under such acidic conditions when measured by co-immunoprecipitation (Fig.S4A) suggesting that LUJV may employ a similar pH-dependent receptor mediated switch we previously described for LASV.
Figure 3. CD63 promotes LUJV GP-mediated membrane fusion under acidic conditions.
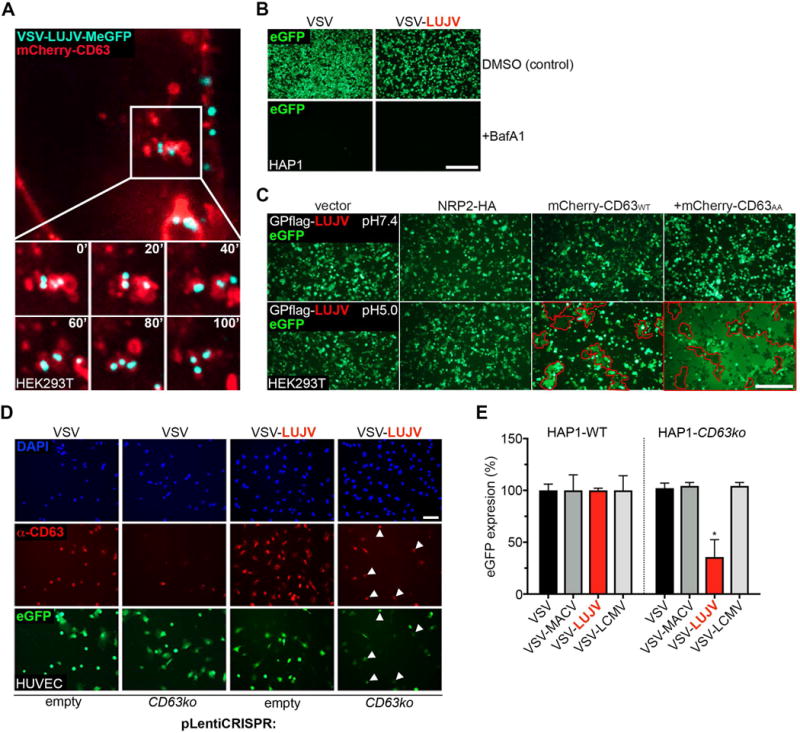
(A) HEK293T cells overexpressing mCherry-tagged CD63 (in red) were inoculated with VSV-LUJV-MeGFP (MOI=150; in cyan) and inoculum was removed 1h post infection. Virus entry was monitored by live-cell imaging 105min post virus inoculation (see Movie S1). Shown is an image of a representative cell for which still-frames are depicted with 20sec intervals. (B) HAP1 cells were pre-treated with DMSO (control) or with 100nM Bafilomycin A1 (BafA1) for 30min at 37°C. Cells were subsequently inoculated with VSV (control) or VSV-LUJV (MOI=3) for 6h and fixed. Infection was visualized by fluorescence microscopy. Scale bar: 10μm. (C) HEK293T cells transduced with control (empty) retrovirus or with virus expressing NRP2-HA, mCherry-tagged wild-type CD63 or a mutant with a disrupted lysosomal targeting motif (mCherry-CD63AA) were transfected with pCAGGS-LUJV GP-Flag and pCMV-eGFP. 48h post transfection, cells were washed with neutral or acidic culture medium for 5min at 37°C. Medium was replaced with normal medium and cells were recovered for 30min at 37°C. Subsequently, cells were fixed and syncytia (visible through extensive content [eGFP] mixing) were visualized by fluorescent microscopy (outlined in red). Scale bar: 10μm. (D) HUVEC cells were transduced with control (empty) lentivirus or with virus expressing a guide RNA targeting CD63. Cells were inoculated with VSV (control) or VSV-LUJV (MOI=3), fixed, stained with DAPI and CD63 antibodies (shown as separate panels) and analyzed for infection by fluorescence microscopy. White arrows indicate infected cells that are CD63-positive. Scale bar: 10μm. (E) Wild-type (WT) and CD63ko HAP1 cells were inoculated with the indicated viruses (MOI=3). Cells were fixed 6h post inoculation and infection was measured by quantifying eGFP fluorescence. The data is presented as the percentage eGFP expression of each virus in CD63ko cells compared to WT cells. A significant difference (Two-way ANOVA, p-value <0.05) between conditions is indicated by the asterisk (n=3 experimental replicates).
To determine the importance of CD63 for LUJV GP-mediated infection of primary endothelial cells we transduced HUVEC cells with a lentivirus expressing Cas9 and a guide RNA targeting CD63. Such transduced cells displayed significant resistance to VSV-LUJV whereas VSV infection was unaffected (Fig.3D). Within the population of cells, those that were infected by VSV-LUJV expressed levels of CD63 that were detectable by immunofluorescence microscopy. Cell surface levels of NRP2 were unaffected in cells lacking CD63 (Fig.S4B), demonstrating that CD63 loss does not reduce infection by indirectly reducing available NRP2.
Infection of VSV recombinants containing other arenaviral GPs (LCMV GP or MACV GP) was unaffected in cells lacking CD63 (Fig.3E). These data suggest a model where NRP2 stimulates binding of viral particles to the cell surface, and CD63 facilitates acid pH-triggered LUJV GP-mediated fusion.
Productive infection of wild-type LUJV is dependent on NRP2
To validate the results of our findings with VSV-LUJV we next examined wild type LUJV infection in NRP2 knockout cells. For this purpose, we used primary HUVECs because they are known to support productive LUJV infection and represent a cell type that is frequently encountered by the virus in vivo. We generated pools of cells containing gene-disruptive mutations in NRP2 using CRISPR/Cas9 gene-editing. As expected, HUVECs lacking NRP2 exhibit reduced susceptibility to VSV-LUJV that was restored following transduction with NRP2-HA whereas VSV infection was unaffected. Infection of NRP2 knockout cells by wild-type LUJV under BSL4 conditions revealed a reduction in infection and production of LUJV progeny virions (Fig.4D and E). That defect in wild type LUJV infection is restored on reestablishing NRP2 expression. In addition, when HUVECs are incubated with an increasing dose of anti-NRP2 antibodies, cells become resistant to LUJV infection (Fig.4F). Overall, these findings validate the use of VSV-LUJV in entry studies and demonstrate a role of NRP2 in wild-type LUJV infection.
Figure 4. Wild-type LUJV requires NRP2 for infection.
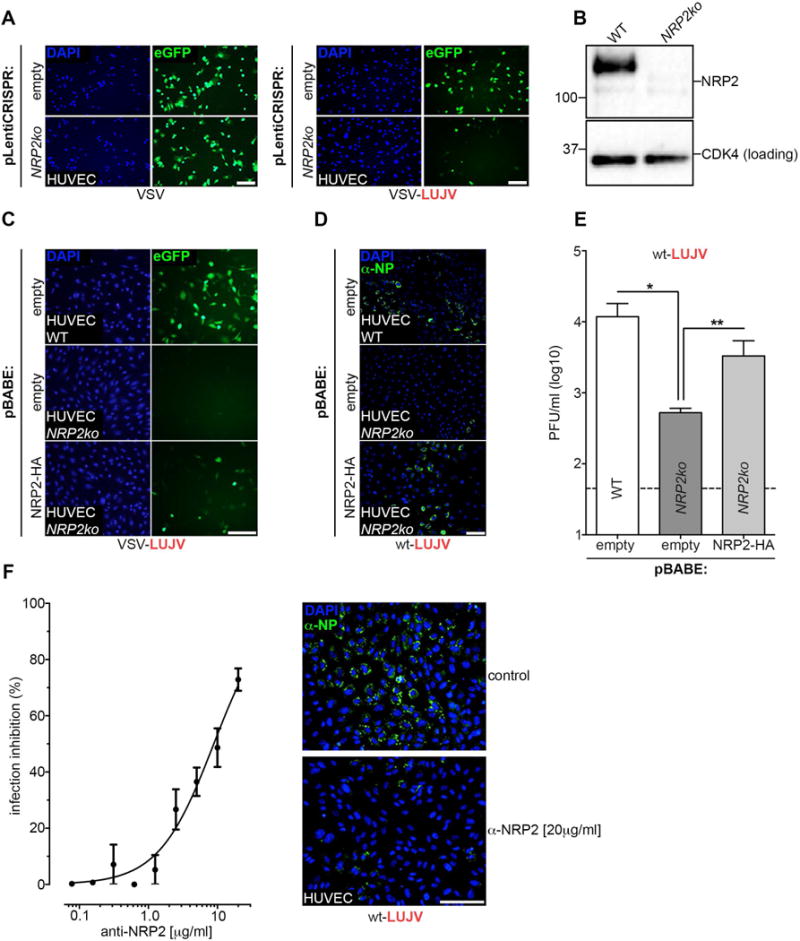
(A) HUVEC cells were transduced with control (empty) lentivirus or with virus expressing a guide RNA targeting NRP2. Next, cells were inoculated with VSV (control) or VSV-LUJV (MOI=3), fixed, stained with DAPI (shown as a separate panel) and analyzed for infection by fluorescence microscopy. Scale bar: 10μm. (B) Western blot analysis of the cells described in panel A. Membranes were probed with an anti-NRP2 antibody for detection of endogenous NRP2. An anti-CDK4 antibody was used as a loading control. (C) The pools of HUVECs mentioned in panel A were transduced with control (empty) retrovirus or with virus expressing NRP2-HA. Subsequently, cells were inoculated with VSV-LUJV and analyzed for infected cells as described above. Scale bar: 10μm. (D) Wild-type (WT), NRP2ko and NRP2-HA reconstituted HUVEC cells were infected with authentic LUJV (MOI=0.5). Cells were fixed 16h post virus inoculation, permeabilized and stained with DAPI. Infection was visualized by immunofluorescence microscopy using an α-NP antibody. Scale bar: 10μm. (E) In parallel to the immunofluorescence experiment in panel E, cells were inoculated with LUJV (MOI=0.2) and the production of viral progeny after 36h was monitored by plaque assays. The data is presented as plaque-forming units (PFU) per ml of culture medium. Significant differences (ANOVA test; p-value <0.05) between each condition are indicated by the asterisks (n=4 experimental replicates). (F) HUVECs were mock-treated or treated with serial dilutions of polyclonal NRP2 antibody. Following 30min incubation on ice, cells were exposed to LUJV Zambia R4356 (MOI=1) for 1h at 37°C in the presence or absence of NRP2 antibody and incubated at 37°C for an additional hour. Cells were then washed and incubated for 36h before fixation. Infected cells were visualized by immunostaining using an arenavirus NP-specific polyclonal guinea pig serum. The number of infected cells were quantified for all conditions and are expressed as percentage of inhibition (n=2 experimental replicates). The continuous line represents the nonlinear fit of all the data points (R2=0.93). Depicted are representative images of the control versus the highest NRP2 antibody concentration. All experiments involving wild-type LUJV infections were carried out under BSL4 conditions.
Discussion
In the present study, we used haploid genetics to identify host requirements for LUJV infection. We discovered several factors that facilitate LUJV GP-mediated cell entry including heparan sulfate, an interaction with NRP2 for cell attachment and CD63, which promotes GP-mediated membrane fusion under acidic conditions.
In addition to supporting robust viral replication, vascular endothelial cells have high expression of arenavirus entry receptors and attachment factors (Andrews et al., 1978; Radoshitzky et al., 2007; Rojek et al., 2007). Vascular dysfunction, exemplified by increased microvascular permeability, is frequently associated with fatal disease outcome of arenavirus-induced hemorrhagic fever (Kunz, 2009). The high levels of NRP2 expression in endothelial cells coupled with its identification as a LUJV entry factor underscores the important role of those cells of the vascular system during arenavirus infection. The mechanisms by which arenaviruses affect endothelial cell function remain unclear, but dysfunction may result from the cytopathic effects of virus replication or cytokine induction (de la Torre et al., 1991). Myeloid cells have also been implicated in the early stages of arenavirus infection and have been shown to abundantly express the identified LUJV host factors NRP2 and CD63 (Aung et al., 2016; Baize et al., 2004; Engering et al., 2003).
Our previous work on LASV and EBOV highlighted the importance of intracellular receptors that facilitate fusion of the virus envelope with the endo/lysosomal membrane (Jae and Brummelkamp, 2015). EBOV binds to several different molecules at the cell surface, but requires the lysosomal protein, Niemann-Pick C1 (NPC1), for membrane fusion (Carette et al., 2011a; Côté et al., 2011; Miller et al., 2012). LASV binds α-DG at the plasma membrane, but switches to LAMP1 during virus entry to establish fusion of its envelope in a low pH environment (Jae et al., 2014). Earlier work by others suggested an additional cue, besides low pH, is necessary for LUJV GP to mediate membrane fusion (Tani et al., 2014). In this work, we identify NRP2 as a binding partner for LUJV GP and demonstrate that CD63 promotes LUJV GP mediated membrane fusion. CD63 is a tetraspanin that mainly resides in intracellular vesicles, including endosomes and lysosomes (Kobayashi et al., 2000) consistent with the possibility that it serves as an endolysosomal receptor for LUJV. It is tempting to speculate that LUJV also employs a receptor switching strategy during cell entry where surface NRP2 binding followed by endosomal CD63 engagement lead to productive infection. Our efforts to detect binding between CD63 and LUJV GP have thus far been unsuccessful suggesting that if a similar receptor switching operates during LUJV infection the interaction between CD63 and LUJV GP may be weak and/or transient.
The 2014–2016 outbreak of EBOV in West Africa underscores the challenges of controlling hemorrhagic fever viruses with profound impacts on human health (Simulundu et al., 2016). Deciphering the interplay between virus and host is not only paramount to understanding mechanisms of pathogenesis but may aid the development of antivirals. Virus-receptor interactions, such as the LUJV GP:NRP2 interaction described here, are attractive focus points for therapeutic intervention.
STAR ★ Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents may be directed to the Lead Contact, Thijn R. Brummelkamp (t.brummelkamp@nki.nl).
Experimental Models and Subject details
Cell lines
HAP1 cells (obtained from Horizon Discovery, gender: male) and derivatives were maintained in IMDM supplemented with 10% FCS, L-glutamine, and penicillin–streptomycin. BSRT-7 (authenticity was not confirmed), HEK293T (obtained from ATCC, gender: unknown) and Vero-E6 (obtained from ATCC, species: African green monkey, gender: unknown) cells were maintained in DMEM supplemented with 10% FCS, L-glutamine, and penicillin– streptomycin. Vero cells were used for amplification of all recombinant VSV viruses. HEK293T cells were used for making gene-trap retrovirus for mutagenesis of HAP1 cells. Human umbilical vein endothelial cells (HUVECs) were obtained from Lonza (Walkersville, MD) and maintained in endothelial grown medium (EGM) supplemented with the appropriate growth factors according to the manufacturer’s instructions (Lonza). Note that all human material utilized for Lonza’s products is ethically obtained and meets global guidelines. All cell lines were propagated under the following conditions: 37°C and 5% CO2 atmosphere.
Methods details
Generation of recombinant viruses
A recombinant VSV expressing enhanced green fluorescent protein (eGFP) and LUJV GP (VSV-LUJV) was generated by replacement of the coding region for VSV G with a codon-optimized version of LUJV GP using restriction sites MluI and NotI in cDNA clone pVSV(+)-eGFP (Wong et al., 2010). The amino acid sequence of LUJV GP (454 aa) is identical to that of the field isolate (NCBI Reference Sequence: YP_002929490.1). A functional fluorescent VSV M protein (MeGFP) was introduced into VSV-LUJV as reported. Fluorescent VSV-LUJV-MeGFP was amplified and purified as described previously (Soh and Whelan, 2015). VSV-LCMV, VSV-JUNV, VSV-GTOV and VSV-MACV were generated in a similar manner by using gBlocks Gene Fragments (from Integrated DNA Technologies) of the corresponding GP precursor coding sequences (GenBank accession number: AY847350.1 [for LCMV GP], GenBank accession number: D10072.2 [for JUNV GP], GenBank accession number: AF485258.1 [for GTOV GP] and GenBank accession number: KM198592.1 [for MACV GP]). Generation of VSV-LASV has been described previously (Jae et al., 2013). All recombinant viruses were recovered, amplified, and purified as described (Whelan et al., 1995), except that a plasmid expressing VSV G was transfected into cells at early passages to boost viral titers. Where indicated, VSV (also encoding eGFP; recovered from cDNA clone pVSV(+)-eGFP) was used as a control in most experiments. Newly generated rVSVs were plaque-purified twice and infectivity was subsequently measured by exposing HAP1 cell monolayers to serial dilutions of virus and counting fluorescent forming units (FFU) at 24-48h post infection. The MOIs used in all experiments are based on the infectivity of the individual viruses in HAP1 cells. A low MOI (ranging from 0.2 to 3) was used in order to maximize the detection of conditions that inhibit/enhance viral infectivity. A high MOI (±150) was used in the fluorescent particle imaging experiments in order to get sufficient events for imaging and subsequent quantification.
Haploid genetic screen and analysis of gene-trap insertion sites
Production of gene-trap retrovirus and mutagenesis of HAP1 cells have been described previously (Jae et al., 2013). Approximately 100 million mutagenized cells were selected with VSV-LUJV (MOI ≈ 0.5) in the presence of 3mM ammonium chloride. Following selection, the surviving colonies were expanded to ±30 million total cells and their genomic DNA was isolated using a QIAamp DNA mini kit (Qiagen). Recovery by LAM-PCR and subsequent mapping of gene-trap insertion sites from cells selected with VSV-LASV was carried out as previously reported (Carette et al., 2011b). Briefly, reads were aligned to unique protein-coding genomic regions (hg19) allowing a single mismatch. For every gene, enrichment of disruptive gene-trap insertions in the VSV-LUJV-selected population over an unselected HAP1 control dataset (available at the NCBI sequence read archive: SRX223544) was calculated by applying a one-sided Fisher’s exact test. Genes with P-value ≤ 10−4 were considered enriched (see Table S1). All P-values were adjusted for false discovery rate (FDR).
Generation of knockout clones using the CRISPR/Cas9 system
Guide RNAs were designed targeting TMEM30A (exon1; 5′-TGGGCCCCCGTGTGCTCCGG-3′), NRP2 (exon3; 5′-AGGAGATTCGATGGTCCCGT-3′), and CD63 (exon5; 5′-ACACTGCTTCGATCCTGGAC-3′) and cloned into the pX330 expression vector. HAP1 cells were co-transfected with the gene-specific vectors and a plasmid containing an expression cassette for a guide RNA targeting the zebrafish TIA gene (5′-GGTATGTCGGGAACCTCTCC-3′) followed by a CMV promotor sequence driving expression of a blasticidin resistance gene flanked by two TIA target sites (Lackner et al., 2015). Co-transfection of these plasmids occasionally results in the incorporation of the blasticidin resistance cassette at the site of the targeted genomic locus by non-homologous end joining, rendering cells resistant to blasticidin while also providing a genomic tag at the site of mutation. Four days after DNA transfection, the culture medium was supplemented with blasticidin (20 μg/mL). Surviving colonies were clonally expanded and their mutations were verified by Sanger sequencing. For editing of NRP2 in primary HUVECs, the guide RNA was cloned into transduction vector pLentiCRISPR. After virus production and transduction, HUVECs were selected with puromycin for 2 days. Cells were expanded and polyclonal pools were used for further experiments. As a control, empty pLentiCRISPR vector was used for generating virus and transduction of cells. The generation of DAG1-deficient cells has been previously described (Jae et al., 2013).
Generation of expression plasmids
The LUJV GP precursor codon-optimized coding sequence was cloned into vector pCAGGS-LASV-GP-Flag replacing LASV GP by using the restriction enzymes NotI and XhoI creating pCAGGS-LUJV-GP-Flag. A plasmid encoding soluble LUJV GP1 (phCMV-LUJV-GP1-Fc; amino acid 59-271 of LUJV GP fused to the rabbit Fc-tail from IgG1) was kindly provided by the Saphire lab. An expression plasmid encoding TMEM30A was obtained from C.C. Paulusma (Tytgat Institute for Liver and Intestinal Research, the Netherlands). The coding sequences for NRP2 was cut from vector pCR4-NRP2 (from the Harvard PlasmidID Database; Clone ID: HsCD00341194) and cloned into pBABE-PURO using the restriction enzymes NaeI and SalI resulting in vector pBABE-NRP2-HA. HA-tagged NRP2 deletion and chimeric constructs were designed and generated using gBlocks Gene Fragments (from Integrated DNA Technologies) and cloned into pBABE-PURO (see Figure 3B for details on NRP2 domain boundaries) using the restriction enzymes BamHI and SalI, with the exception of pBABE-NRP2-dB1B2-HA for which we used NaeI and SalI). Expression vectors encoding mCherry-CD63 (wild-type and GY234AA mutant) were constructed and designed using gBlocks encoding CD63 flanked by EcoRI and SalI restriction sites. The DNA fragments were digested and cloned into pmCherry-C1 (Clontech). The resulting plasmid was used as a template for a PCR reaction using primers 5′-gcaacgGGATCCACCATGGTGAGCAAGGGCGAGGAGG-3′ and 5′-gcaacgGTCGACCTACATCACCTCGTAGCCACTTCTGATACTCTTC-3′. The PCR product was digested with BamHI and SalI and subsequently cloned into pBABE-PURO. All constructs were sequence-verified by Sanger sequencing using vector-specific primers.
Antibodies
The following antibodies were used for immunoblotting and/or immunofluorescent imaging: anti-HA and anti-Flag M2 (both from Sigma-Aldrich), anti-NRP2 (from R&D Systems), anti-CD63 (clone NKI-C3, a kind gift from J. Neefjes), anti-TfR1 (clone 66Ig10, obtained from J. Hilkens), anti-α-DG (clone IIH6C4 from Millipore) and anti-CDK4 (clone C-22, from Santa Cruz Biotechnology). The corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Bio-Rad Laboratories. Alexa Fluor-conjugated secondary antibodies were used for all immunofluorescent experiments (from Thermo Fisher Scientific).
Enzymatic removal of cell-surface glycosaminoglycans
Glycosaminoglycans, including heparan sulfate, were stripped from the cell surface of HAP1 cells using an enzymatic cocktail of heparinases (i.e. heparinase I-III, purchased from Sigma-Aldrich). Enzymes were resuspended in HEPES buffer (20mM HEPES [pH 7.5], 50mM NaCl, 4mM CaCl2 and 0.01% BSA), after which dilutions were prepared in digestion buffer (20mM HEPES [pH 7.5], 150mM NaCl, 4mM CaCl2, 0.1% BSA). HAP1 cells were treated for 1h at 37°C with the enzymatic cocktail at the indicated concentrations. The cells were washed three times with culture medium and then incubated with VSV-LUJV or VSV control virus for 30min at 37°C. The cells were washed twice with culture medium and further incubated in culture medium at 37°C for 6h, after which the cells were fixed with 4% PFA. Infected cells (i.e. eGFP-positive cells) were visualized by fluorescence microscopy and counted for all conditions.
Immunoprecipitations
Flag-tagged LUJV GP was produced in HEK293T cells. Cells were transiently transfected with pCAGGS-LUJV GP-Flag and lysates were prepared 48h post transfection in NETN buffer (50mM Tris-HCl, 150mM NaCl, 1mM EDTA, 0.5% NP-40, pH7.4) and supplemented with cOmplete protease inhibitor cocktail (Roche, Basel, Switzerland). Lysates were briefly sonicated and LUJV GP was immobilized on anti-Flag M2 agarose beads (Sigma-Aldrich). Beads were washed and incubated with whole cell lysates prepared in NETN buffer from cells overexpressing HA-tagged NRP2. After incubation, beads were washed extensively with NETN buffer to remove non-specific interactions. Bound proteins were eluted and subjected to SDS polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore) by Western blotting. Subsequently, membranes were blocked (PBS containing 0.1% Tween-20 and 5% non-fat milk powder) and incubated with primary antibodies in the same buffer.
Fc pull-down assay
HEK293T cells were transiently transfected with phCMV-LUJV GP1-Fc. 48h post transfection the supernatant was harvested and filtered through a 0.45μm filter to remove cell debris. Cleared supernatant was incubated with Protein A Sepharose (GE Healthcare Life Sciences) for 2h at 4°C under slow rotation. Beads were washed four times with NETN buffer and incubated with whole cell lysates from HEK293T cells overexpressing HA-tagged NRP2 for 1h at 4°C. Beads were washed extensively with NETN buffer after which protein complexes were taken up directly in sample buffer and subjected to SDS-PAGE and immunoblot analysis (see above).
Cell-cell fusion assay
HEK293T cells stably expressing HA-tagged NRP2, mCherry-tagged wild-type CD63 or a cell-surface localized mutant containing a mutated C-terminal lysosomal targeting motif (GY234AA) were transiently co-transfected with an eGFP expression vector (pCMV-eGFP) and pCAGGS-LUJV-GP-Flag or empty pCAGGS vector. 48h post transfection, the medium was briefly replaced with acidic medium (pH4.5) for 3min. Cells were recovered in normal medium for 1h after which cells were fixed and examined for membrane fusion events by fluorescence microscopy. Fused (polykaryotic) cells showed enlarged regions with homogenous eGFP signal as a result of cell content mixing.
Fluorescent microscopy
Images of infected cells (i.e. eGFP-positive cells) were obtained with an FV1000 confocal laser scanning microscope (Olympus) equipped with an oil immersion objective (U Plan S Apo 100×, N.A. 1.40) and Olympus Fluoview Software (3.0a) and a Leica-Microsystems microscope. Representative pictures were acquired with LCS software (Leica-Microsystems, Vienna, Austria).
Virus labeling, imaging and quantification
Gradient purified VSV-LUJV particles were labeled with AlexaFluor 594 and VSV-LASV particles were labeled with AlexaFluor 647 as previously described (Cureton et al., 2009). Viruses were added together to the indicated cells and incubated at 37°C for 15min. Samples were stained with 5μg/ml AlexaFlour 488 labeled wheat germ agglutinin (AF488-WGA), washed two times with PBS, fixed with 2% PFA and mounted with ProLong Gold (Molecular Probes). Samples were imaged using a Zeiss Axiovert 200M inverted fluorescence microscope, equipped with a spinning disc head (Yokogawa), a cooled electron multiplication CCD camera (model QuantEM, Photometics) and a computer controlled piezo Z stage (Applied Scientific Instruments). Each image was acquired as a z-stack (0.25μm interval) with a 100×, 1.4 NA objective (Zeiss), and at least ten images were taken per sample. Excitation wavelengths were 491, 561 and 660nm. Image analysis was performed using Fiji. The processing started by creating a maximum intensity Z projection for each channel image followed by background subtraction, threshold segmentation and generation of binary masks for each image. The binary masks corresponding to the VSV-LUJV particles were multiplied with that of the AF488-WGA, resulting in a new binary mask representing the cell associated VSV-LUJV particles. The same procedure was performed to generate binary masks showing the cell associated VSV-LASV particles. From these newly generated binary masks, the number of cell associated VSV-LUJV, VSV-LASV particles were counted using the Fiji Analyze Particle plugin. The surface areas of the cells were measured from the AF488-WGA masks using the build in Measure function in Fiji. Particle binding measurements were calculated by dividing the particle count by the measured cell area.
Flow cytometry
HEK293T cells (transduced with the different expression cassettes described in Fig.3C) or HUVEC cells (transduced with a lentivirus expressing a guide RNA targeting CD63) were expanded in 10cm dishes, harvested with TripLE Express (Thermo Fisher Scientific) and stained with an anti-CD63 antibody (clone NKI-C3) together with a goat isotype control antibody or with an anti-NRP2 antibody (AF2215, R&D Systems) in PBS supplemented with 2% FCS. For the polyclonal HUVECs, two cell populations were gated from the living cell pool (i.e. CD63+ and CD63−) and were analyzed for NRP2 cell surface expression using a flow cytometry (LSR Fortessa cell analyzer, BD Biosciences) and FACSDiva Software 8.0.1 (BD Biosciences). 20,000 events were measured per experimental condition.
LUJV infections
Experiments with infectious wild-type LUJV were carried out under BSL-4 containment at the United States Army Medical Institute of Infectious Diseases (USAMRIID, Fort Detrick, MD). For immunofluorescence assays, HUVEC cells were exposed to wild-type LUJV Zambia R4356 at a MOI of 1 for 1 hour at 37°C. Cells were then washed and incubated with growth medium at 37°C for 36 hours before fixing with 10% formalin. Cells were then permeabilized with 0.02% Triton-X and incubated sequentially with arenavirus NP-specific polyclonal guinea pig serum, then Alexa Flour488-conjugated secondary antibody. Cells were imaged using the Operetta high-content imaging system (PerkinElmer, Waltham, MA) and Harmony high-content imaging and analysis software (PerkinElmer). For titration assays, HUVECs were infected with LUJV at an MOI of 0.2. Supernatants were harvested 36h post infection and virus titers were determined by plaque assay.
Anti-NRP2 antibody inhibition experiment
HUVECs, seeded in 96-well plates (Greiner BioOne), were placed on ice for 15min prior to treating with serial dilutions of chilled polyclonal NRP2 antibody (AF2215, R&D Systems) or media alone. Following 30min incubation on ice, cells were exposed to LUJV Zambia R4356 at a MOI of 1 for 1 hour at 37°C, 5% CO2 in the presence of NRP2 antibody or media alone and incubated at 37°C, 5% CO2 for 1 hour. Cells were then washed and incubated with growth medium at 37°C for 36 hours before fixing with 10% formalin. Cells were then permeabilized with 0.02% Triton-X and incubated sequentially with arenavirus NP-specific polyclonal guinea pig serum, then Alexa Flour488-conjugated secondary antibody. Cells were imaged using the Operetta high-content imaging system (PerkinElmer, Waltham, MA) and Harmony high-content imaging and analysis software (PerkinElmer).
Quantification and Statistical Analysis
All data is expressed as arithmetic means ± standard deviation (SD). For the comparison of continuous variables from independent groups we used Student’s t-test for two groups and one-way ANOVA for more than two groups. All values were normally distributed. The statistical details (n-numbers, mean ± SD and tests) are given in the figure legends and were calculated by Prism version 7.0b software (GraphPad).
Data and Software Availability
The deep sequencing data from the VSV-LUJV selection screen in HAP1 cells is available at the NCBI sequence read archive: SRR5947468. The control dataset can also be found at the NCBI sequence read archive: SRX223544.
Supplementary Material
Acknowledgments
We thank E.O. Saphire (Scripps Research Institute, La Jolla, CA, USA) for providing cDNA encoding the LUJV GP precursor as well as the expression construct encoding Fc-tagged LUJV GP1, and the members of the Brummelkamp and Whelan laboratories for helpful discussions. We also thank Kathleen Cashman (USAMRIID, Fort Detrick, MD, USA) for providing the arenavirus NP-specific polyclonal guinea pig serum and John Hilkens (Netherlands Cancer Institute, Amsterdam, the Netherlands) for sharing antihuman TfR antibodies (clone 66Ig10). This work was supported by a Marie Sklodowska-Curie Action fellowship (H2020-MSCA-IF-2014 660417) to M.R., by Defense Threat Reduction Agency funding (CB4088) to J.M.D., by National Institutes of Health funding (AI109740) to S.P.W. and T.K. and by a European Research Council (ERC) Starting Grant (ERC-2012-StG 309634) to T.R.B. Opinions, conclusions, interpretations, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army. The mention of trade names or commercial products does not constitute endorsement or recommendation for use by the Department of the Army or the Department of Defense.
Footnotes
Author contributions
M.R., L.T.J., A.S.H., T.K., J.M.D., S.P.W. and T.R.B. were responsible for the overall design of the study, recombinant viruses were generated and characterized by M.R. in the lab of S.P.W., the haploid genetic screen was carried out and analyzed by M.R., L.T.J. and V.A.B. in the lab of T.R.B., A.I.K., A.S.H. and J.M.D. planned and performed the experiments with authentic LUJV under BSL4 conditions, S.H.S. and Y.Y.C. carried out virus binding and live-cell imaging experiments in the lab of S.P.W and T.K., M.R., T.R.B. and S.P.W. wrote the manuscript, every author commented on the manuscript.
References
- Andrews BS, Theofilopoulos AN, Peters CJ, Loskutoff DJ, Brandt WE, Dixon FJ. Replication of dengue and junin viruses in cultured rabbit and human endothelial cells. Infect Immun. 1978;20:776–781. doi: 10.1128/iai.20.3.776-781.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleton BA, Wu P, Maloney J, Yin J, Liang WC, Stawicki S, Mortara K, Bowman KK, Elliott JM, Desmarais W, et al. Structural studies of neuropilin/antibody complexes provide insights into semaphorin and VEGF binding. EMBO J. 2007;26:4902–4912. doi: 10.1038/sj.emboj.7601906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aung NY, Ohe R, Meng H, Kabasawa T, Yang S, Kato T, Yamakawa M. Specific Neuropilins Expression in Alveolar Macrophages among Tissue-Specific Macrophages. PLoS One. 2016;11:e0147358. doi: 10.1371/journal.pone.0147358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baize S, Kaplon J, Faure C, Pannetier D, Georges-Courbot MC, Deubel V. Lassa virus infection of human dendritic cells and macrophages is productive but fails to activate cells. J Immunol. 2004;172:2861–2869. doi: 10.4049/jimmunol.172.5.2861. [DOI] [PubMed] [Google Scholar]
- Briese T, Paweska JT, McMullan LK, Hutchison SK, Street C, Palacios G, Khristova ML, Weyer J, Swanepoel R, Egholm M, et al. Genetic Detection and Characterization of Lujo Virus, a New Hemorrhagic Fever–Associated Arenavirus from Southern Africa. PLoS Pathog. 2009;5:e1000455. doi: 10.1371/journal.ppat.1000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Henry MD, Borrow P, Yamada H, Elder JH, Ravkov EV, Nichol ST, Compans RW, Campbell KP, Oldstone MB. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science. 1998;282:2079–2081. doi: 10.1126/science.282.5396.2079. [DOI] [PubMed] [Google Scholar]
- Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, et al. Ebola virus entry requires the cholesterol transporter Niemann–Pick C1. Nature. 2011a;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette JE, Guimaraes CP, Wuethrich I, Blomen VA, Varadarajan M, Sun C, Bell G, Yuan B, Muellner MK, Nijman SM, et al. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nat Biotechnol. 2011b;29:542–546. doi: 10.1038/nbt.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, et al. Small molecule inhibitors reveal Niemann–Pick C1 is essential for Ebola virus infection. Nature. 2011;477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, Whelan SPJ. Vesicular Stomatitis Virus Enters Cells through Vesicles Incompletely Coated with Clathrin That Depend upon Actin for Internalization. PLoS Pathog. 2009;5:e1000394. doi: 10.1371/journal.ppat.1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engering A, Kuhn L, Fluitsma D, Hoefsmit E, Pieters J. Differential post-translational modification of CD63 molecules during maturation of human dendritic cells. Eur J Biochem. 2003;270:2412–2420. doi: 10.1046/j.1432-1033.2003.03609.x. [DOI] [PubMed] [Google Scholar]
- Hilgard P, Stockert R. Heparan sulfate proteoglycans initiate dengue virus infection of hepatocytes. Hepatology. 2000;32:1069–1077. doi: 10.1053/jhep.2000.18713. [DOI] [PubMed] [Google Scholar]
- Jae LT, Brummelkamp TR. Emerging intracellular receptors for hemorrhagic fever viruses. Trends Microbiol. 2015;23:392–400. doi: 10.1016/j.tim.2015.04.006. [DOI] [PubMed] [Google Scholar]
- Jae LT, Raaben M, Riemersma M, van Beusekom E, Blomen VA, Velds A, Kerkhoven RM, Carette JE, Topaloglu H, Meinecke P, et al. Deciphering the Glycosylome of Dystroglycanopathies Using Haploid Screens for Lassa Virus Entry. Science (80-) 2013;340:479–483. doi: 10.1126/science.1233675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jae LT, Raaben M, Herbert AS, Kuehne AI, Wirchnianski AS, Soh TK, Stubbs SH, Janssen H, Damme M, Saftig P, et al. Lassa virus entry requires a trigger-induced receptor switch. Science (80-) 2014;344:1506–1510. doi: 10.1126/science.1252480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Vischer UM, Rosnoblet C, Lebrand C, Lindsay M, Parton RG, Kruithof EK, Gruenberg J. The tetraspanin CD63/lamp3 cycles between endocytic and secretory compartments in human endothelial cells. Mol Biol Cell. 2000;11:1829–1843. doi: 10.1091/mbc.11.5.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodkin AL, Levengood DV, Rowe EG, Tai YT, Giger RJ, Ginty DD. Neuropilin is a semaphorin III receptor. Cell. 1997;90:753–762. doi: 10.1016/s0092-8674(00)80535-8. [DOI] [PubMed] [Google Scholar]
- Kunz S. The role of the vascular endothelium in arenavirus haemorrhagic fevers. Thromb Haemost. 2009 doi: 10.1160/TH09-06-0357. [DOI] [PubMed] [Google Scholar]
- de la Torre JC, Borrow P, Oldstone MB. Viral persistence and disease: cytopathology in the absence of cytolysis. Br Med Bull. 1991;47:838–851. doi: 10.1093/oxfordjournals.bmb.a072515. [DOI] [PubMed] [Google Scholar]
- Lackner DH, Carré A, Guzzardo PM, Banning C, Mangena R, Henley T, Oberndorfer S, Gapp BV, Nijman SMB, Brummelkamp TR, et al. A generic strategy for CRISPR-Cas9-mediated gene tagging. Nat Commun. 2015;6:10237. doi: 10.1038/ncomms10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latysheva N, Muratov G, Rajesh S, Padgett M, Hotchin NA, Overduin M, Berditchevski F. Syntenin-1 is a new component of tetraspanin-enriched microdomains: mechanisms and consequences of the interaction of syntenin-1 with CD63. Mol Cell Biol. 2006;26:7707–7718. doi: 10.1128/MCB.00849-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, et al. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 2012;31:1947–1960. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraz ML, Kunz S. Pathogenesis of arenavirus hemorrhagic fevers. Expert Rev Anti Infect Ther. 2011;9:49–59. doi: 10.1586/eri.10.142. [DOI] [PubMed] [Google Scholar]
- Parker MW, Linkugel AD, Goel HL, Wu T, Mercurio AM, Vander Kooi CW. Structural Basis for VEGF-C Binding to Neuropilin-2 and Sequestration by a Soluble Splice Form. Structure. 2015;23:677–687. doi: 10.1016/j.str.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulusma CC, Folmer DE, Ho-Mok KS, de Waart DR, Hilarius PM, Verhoeven AJ, Oude Elferink RPJ. ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology. 2008;47:268–278. doi: 10.1002/hep.21950. [DOI] [PubMed] [Google Scholar]
- Radoshitzky SR, Abraham J, Spiropoulou CF, Kuhn JH, Nguyen D, Li W, Nagel J, Schmidt PJ, Nunberg JH, Andrews NC, et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature. 2007;446:92–96. doi: 10.1038/nature05539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radoshitzky SR, Bào Y, Buchmeier MJ, Charrel RN, Clawson AN, Clegg CS, DeRisi JL, Emonet S, Gonzalez JP, Kuhn JH, et al. Past, present, and future of arenavirus taxonomy. Arch Virol. 2015;160:1851–1874. doi: 10.1007/s00705-015-2418-y. [DOI] [PubMed] [Google Scholar]
- Riblett AM, Blomen VA, Jae LT, Altamura LA, Doms RW, Brummelkamp TR, Wojcechowskyj JA. A Haploid Genetic Screen Identifies Heparan Sulfate Proteoglycans Supporting Rift Valley Fever Virus Infection. J Virol. 2016;90:1414–1423. doi: 10.1128/JVI.02055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojek JM, Kunz S. Cell entry by human pathogenic arenaviruses. Cell Microbiol. 2008;10:828–835. doi: 10.1111/j.1462-5822.2007.01113.x. [DOI] [PubMed] [Google Scholar]
- Rojek JM, Spiropoulou CF, Campbell KP, Kunz S. Old World and clade C New World arenaviruses mimic the molecular mechanism of receptor recognition used by alpha-dystroglycan’s host-derived ligands. J Virol. 2007;81:5685–5695. doi: 10.1128/JVI.02574-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador B, Sexton NR, Carrion R, Nunneley J, Patterson JL, Steffen I, Lu K, Muench MO, Lembo D, Simmons G. Filoviruses Utilize Glycosaminoglycans for Their Attachment to Target Cells. J Virol. 2013;87:3295–3304. doi: 10.1128/JVI.01621-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simulundu E, Mweene AS, Changula K, Monze M, Chizema E, Mwaba P, Takada A, Ippolito G, Kasolo F, Zumla A, et al. Lujo viral hemorrhagic fever: considering diagnostic capacity and preparedness in the wake of recent Ebola and Zika virus outbreaks. Rev Med Virol. 2016;26:446–454. doi: 10.1002/rmv.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh TK, Whelan SPJ. Tracking the Fate of Genetically Distinct Vesicular Stomatitis Virus Matrix Proteins Highlights the Role for Late Domains in Assembly. J Virol. 2015;89:11750–11760. doi: 10.1128/JVI.01371-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulpice E, Plouët J, Bergé M, Allanic D, Tobelem G, Merkulova-Rainon T. Neuropilin-1 and neuropilin-2 act as coreceptors, potentiating proangiogenic activity. Blood. 2008;111:2036–2045. doi: 10.1182/blood-2007-04-084269. [DOI] [PubMed] [Google Scholar]
- Takashima S, Kitakaze M, Asakura M, Asanuma H, Sanada S, Tashiro F, Niwa H, Miyazaki Ji J, Hirota S, Kitamura Y, et al. Targeting of both mouse neuropilin-1 and neuropilin-2 genes severely impairs developmental yolk sac and embryonic angiogenesis. Proc Natl Acad Sci U S A. 2002;99:3657–3662. doi: 10.1073/pnas.022017899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani H, Iha K, Shimojima M, Fukushi S, Taniguchi S, Yoshikawa T, Kawaoka Y, Nakasone N, Ninomiya H, Saijo M, et al. Analysis of Lujo Virus Cell Entry using Pseudotype Vesicular Stomatitis Virus. J Virol. 2014;88:7317–7330. doi: 10.1128/JVI.00512-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan SP, Ball LA, Barr JN, Wertz GT. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc Natl Acad Sci U S A. 1995;92:8388–8392. doi: 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AC, Sandesara RG, Mulherkar N, Whelan SP, Chandran K. A forward genetic strategy reveals destabilizing mutations in the Ebolavirus glycoprotein that alter its protease dependence during cell entry. J Virol. 2010;84:163–175. doi: 10.1128/JVI.01832-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.