

Summary
Aging is an inevitable outcome of life, characterized by progressive decline in tissue and organ function and increased risk of mortality. Accumulating evidence links aging to genetic and epigenetic alterations. Given the reversible nature of epigenetic mechanisms, these pathways provide promising avenues for therapeutics against age-related decline and disease. In this review, we provide a comprehensive overview of epigenetic studies from invertebrate organisms, vertebrate models, tissue and in vitro systems. We establish links between common operative aging pathways and hallmark chromatin signatures that can be used to identify “druggable” targets to counter human aging and age-related disease.
Introduction
“Aging” is the gradual loss of molecular fidelity after reaching sexual maturity, culminating in loss of function and ultimately in disease and death. In most organisms, the rate of aging is inversely proportional to mean lifespan. Increased age is also the largest risk factor for cancer, neurodegeneration and cardiovascular disease. On the other hand, “healthspan” is defined as the duration of time for which an organism remains free of chronic disease. Identifying ways to increase lifespan and healthspan are intriguing areas of biogerontology research.
Various animal models have been critical for uncovering key pathways related to aging. Genetically tractable models such as yeast have been used to investigate both replicative lifespan, measured by the maximum number of mitotic divisions a cell can undergo, and chronological lifespan, measured by the length of time a cell can survive in a post-mitotic state (Kaeberlein et al., 2007). Other studies have taken advantage of the short lifespans of worms and flies (Brandt and Vilcinskas, 2013; Tissenbaum, 2012). Studies in these models have contributed greatly to the field, but cannot fully recapitulate the complex nature of human aging, particularly with respect to age-related diseases and the decline of healthspan. Therefore, vertebrate models such as mice have been utilized, taking advantage of genetic proximity to humans and the availability of gene knockout and premature aging models (Quarrie and Riabowol, 2004). Unfortunately, mouse lifespan is too long for efficient laboratory studies of normal aging, creating the need for alternative short-lived vertebrate models such as the African turquoise killifish (Nothobranchius furzeri) (Harel et al., 2015) which lives for 4–6 months and recapitulates many of the age-related pathological changes found in humans.
Studies in various models have revealed that genetic differences and somatic mutations underlie longevity, but non-genetic contributions also play a major role (Cournil and Kirkwood, 2001). Calorie restriction (Bordone and Guarente, 2005), lowering of basal metabolic rate (Ruggiero et al., 2008), upregulated stress response (Migliaccio et al., 1999), restoration of mitonuclear protein balance (Houtkooper et al., 2013) and reduced fertility (Westendorp and Kirkwood, 1998) have all been shown to correlate with lifespan extension. These observations illuminate the role of “epi”-genetic mechanisms in modulating longevity pathways.
Chromatin is the polymer of nucleosomes composed of DNA wrapping the histone proteins. Chromatin and epigenetic factors influence gene expression dynamically or over long time scales - even through cell division - by regulating access of transcriptional machinery to DNA. These factors are enzymes that modify DNA directly or the core histones H2A, H2B, H3 and H4, as well as variant histones, including H3.3, macroH2A, and H2A.Z. A large number of epigenetic studies have begun to establish operational models in aging. It is clear that with age there is general loss of histones coupled with local and global chromatin remodeling, an imbalance of activating and repressive histone modifications, and global transcriptional change in all aging models. Additionally, particularly in mammalian systems there is global and local change in DNA methylation, site-specific loss and gain in heterochromatin, and significant nuclear reorganization (Figure 1).
Figure 1. The epigenetic hallmarks of senescence and aging.
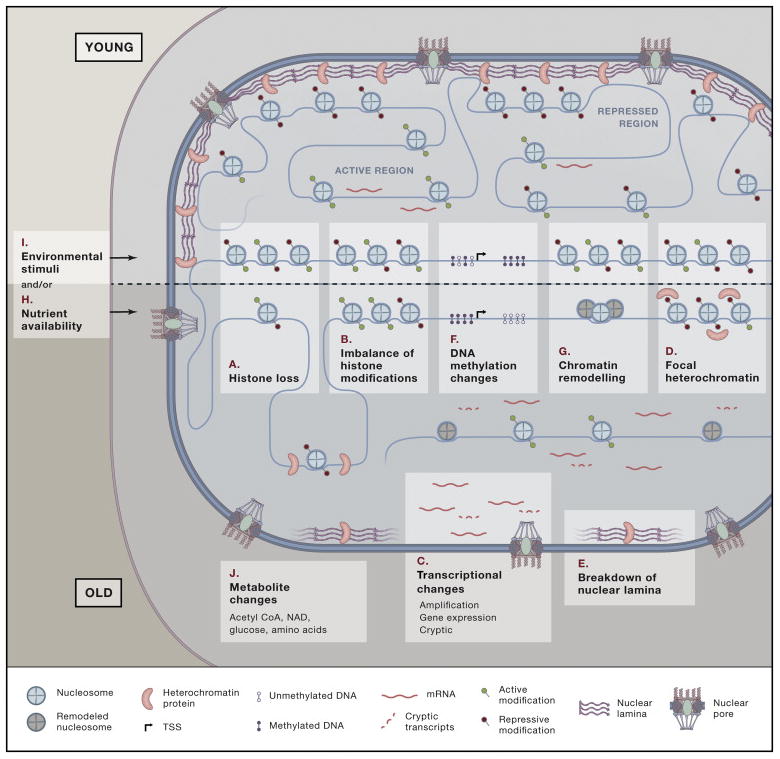
Senescence and aging are characterized by (A) loss of histones, (B) imbalance of activating and repressive modifications, (C) transcriptional changes, (D) losses and gains in heterochromatin, (E) breakdown of nuclear lamina, (F) global hypomethylation and focal hypermethylation and (G) chromatin remodeling. These changes are heavily dictated by (H) environmental stimuli (I) and nutrient availability that in turn (J) alter intracellular metabolite concentrations.
It is as yet unclear whether changes in the activity of epigenetic enzymes influence the expression of critical longevity genes or whether alterations in the longevity genes drive large scale epigenetic changes in the genome. In lower model organisms, single point mutations in epigenetic enzymes dramatically alter lifespan. For complex higher organisms that have many redundant enzymes, it is too simplistic to theorize that a few genes drive age-related changes. Rather, experimental evidence supports large-scale alterations in response to environmental stimuli or nutrient availability.
This review consolidates key findings in multiple systems that support an epigenetic role in aging. In addition to the text, epigenetic mechanisms are summarized for multiple systems in Supplemental Table 1 and general benefits and drawbacks of each model highlighted in Table 1 providing readers with a resource to quickly peruse key points for their own studies. Finally, readers are provided with lists of state-of-the-art methodologies, protocols, repositories tools and other resources that can be used to accelerate aging research (Table 2, Supplemental Table 2). Of particular interest are large-scale epigenomic studies and the recent CRISPR mutagenesis strategies.
Table 1.
Pros and cons of models used in aging and longevity studies
| Models | Pros | Cons |
|---|---|---|
| Yeast | Genetically tractable | Cannot model human aging since yeast expresses telomerase, lacks p53, no DNA methylation, no repressive histone methylation, and does not develop age-related diseases |
| Non-essential gene deletion collection and essential conditional deletions available | ||
| Short lifespan | ||
| Two lifespan arms to investigate mitotic and post-mitotic aging | ||
| Worm | Genetically tractable | Cannot model human aging, no DNA methylation and does not develop age-related diseases |
| RNAi | ||
| Short lifespan | ||
| Mostly post-mitotic cells to investigate senescent decline and organismal aging | ||
| Fly | Genetically tractable | Cannot model human aging and does not develop age-related diseases |
| RNAi | ||
| UAS-GAL4 system | ||
| Short lifespan | ||
| Mostly post-mitotic cells to investigate senescent decline and organismal aging | ||
| DNA is methylated (very low) | ||
| Killifish | Short-lived vertebrate model | Newly established model |
| Shows age-related human pathologies | ||
| Stable lines can be constructed by CRISPR | ||
| Normal mouse | Vertebrate model, highly orthologous to humans | Most strains used in aging research are inbred to a high degree that might not be representative of a genetically-diverse aging human population |
| Shorter lifespan compared to other vertebrates/mammals (2–3 years) | ||
| Multiple genetic tools, knockout mouse models, inbred strains | ||
| Stable lines can be constructed by CRISPR | ||
| Multiple premature aging and long-lived models available | ||
| Premature aging mouse models | Short lifespan | Epigenetic studies confounded by extreme consequences of genetic mutations |
| Long-lived dwarf mouse models | Useful models to study impaired insulin signaling | Most models show developmental delays rather than aging delays |
| Senescent cell cultures | Homogenous system | Senescent cells may not be relevant to in vivo tissue aging, unless the tissue accumulates senescent cells with age |
| Stable lines can be constructed by CRISPR and RNAi |
Table 2.
Repositories and tools for aging research
| Models | Description | Link/Reference |
|---|---|---|
| Yeast | Saccharomyces genome database | http://www.yeastgenome.org/ |
| Published lifespan data | http://lifespandb.sageweb.org/, McCormick et al, Cell Metabolism,2015 | |
| Wilcoxon rank sum test to test significance of lifespan differences | http://data.kaeberleinlab.org/scripts/ranksum.php | |
| Yeast outgrowth data analyzer (YODA) for chronological lifespan assays | http://yoda.sageweb.org/ | |
| Information about yeast gene deletion collection | http://www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html | |
| Library of histone mutants (plasmid) | (Nakanishi et al., 2008) | |
| Library of histone mutants (integrated) | (Dai et al., 2008) | |
| Screen for H3 and H4 residues affecting replicative lifespan | (Sen et al., 2015) | |
| Screen for yeast genes affecting replicative lifepsan | (McCormick et al., 2015) | |
| Screen for yeast genes affecting chronological lifespan | (Powers et al., 2006) | |
| Worm | C. elegans information resource | https://www.wormbase.org/species/c_elegans#041--10 |
| http://www.wormbook.org/ | ||
| Genome-wide data in C. elegans | http://www.modencode.org/ | |
| Published lifespan data | http://lifespandb.sageweb.org/ | |
| Worm transcriptome data | http://spell.caltech.edu:3000/ | |
| http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/index.html?worm | ||
| Fly | Database for Drosophila genetics and molecular biology | http://flybase.org/ |
| http://www.fruitfly.org/ | ||
| http://www.flymine.org/ | ||
| Genome wide data in Drosophila | http://www.modencode.org/ | |
| Published lifespan data | http://lifespandb.sageweb.org/ | |
| Killifish | Killifish genome browser | http://africanturquoisekillifishbrowser.org/ |
| Published lifespan data | http://lifespandb.sageweb.org/ | |
| Normal mouse | Mouse genome information | http://www.informatics.jax.org/ |
| Mouse ENCODE data | http://www.mouseencode.org/data | |
| Published lifespan data | http://lifespandb.sageweb.org/ | |
| Lifespan of commonly used inbred strains | https://www.jax.org/research-and-faculty/research-labs/the-harrison-lab/gerontology/available-data | |
| Premature aging mouse models | Targeted allele detail and overall phenotype | http://www.informatics.jax.org/marker/MGI:1890508https://techtransfer.universityofcalifornia.edu/NCD/20469.html |
| Long-lived dwarf mouse models | Targeted allele detail and overall phenotype | http://sageke.sciencemag.org/cgi/content/full/2001/1/tg11 |
| Cell cultures | Information about different cell types in the ENCODE project | http://genome.ucsc.edu/ENCODE/cellTypes.html |
| Genome wide data on multiple cell types (ENCODE project) | https://www.encodeproject.org/ | |
| Genome-wide epigenetic studies in human cells and tissues | http://www.roadmapepigenomics.org/ | |
| Resource for Progeria research | http://www.progeriaresearch.org/ | |
| https://catalog.coriell.org/ | ||
| National Institute on Aging’s (NIA) Aged Cell Bank at Coriell | https://catalog.coriell.org/ |
Evidence for epigenetic changes in aging
General loss of histones as a mechanism of aging
From yeast to humans, an emerging epigenetic trend detected in aging cells is the general loss of histones (Figure 1A, Supplemental Table 1) (Dang et al., 2009; O’Sullivan et al., 2010). Thus far, the majority of evidence suggests that histone loss is tightly linked to cell division. In replicatively aged yeast, micrococcal nuclease-DNA sequencing (MNase-seq) showed a global nucleosome loss of ~50% (Hu et al., 2014). The remaining nucleosomes had higher “fuzziness” in positioning or mapped to regions with stronger positioning sequences. That proper histone dosage is important in yeast is evident from the extended lifespan of strains overexpressing histones (Feser et al., 2010). In addition to transcriptional downregulation linked to cell cycle exit, loss of histones may also occur through post-translational mechanisms such as autophagy. Piecemeal microautophagy of the nucleus occurs in yeast but whether chromatin components are targeted for degradation by this process is unknown (Kvam and Goldfarb, 2007). In human senescent fibroblasts, nuclear blebs bud off cytoplasmic chromatin fragments (CCFs) that are positive for DNA, γ-H2AX and other repressive histone marks and targeted to lysosome for degradation (Dou et al., 2015; Ivanov et al., 2013). However, it is unclear whether such histone loss occurs in aged human tissues that accumulate senescent cells and tissues such as the brain containing non-renewable, differentiated neurons.
In addition to exploring whether histone loss is a feature of in vivo aging in higher organisms, future studies may be directed to reveal “pockets” of histone loss in the genome. This requires advanced single cell genomic analyses to acquire sufficient sequencing depth in a population of cells. Elucidating the signaling pathway (s) triggering histone loss will be a key conceptual advance. An additional area of future investigation is characterizing the alternative pool of canonical histones synthesized exclusively in senescence. These are formed via alternative splicing events from histone genes with two exons that eliminate the stem loop terminator, but allow polyadenylation for stabilization in non-proliferating cells. These alternative histones are then incorporated in the genome by replication independent chaperones to create a dynamic chromatin state (Rai et al., 2014), the further study of which may reveal key epigenetic differences between replicative and non-replicative states.
Imbalance of activating and repressive histone modifications
Histone methylation, specifically Histone 3 lysine 4 trimetylation (H3K4me3) (activating) and H3K27me3 (repressing), are epigenetic modifications with close ties to transcription, and have been directly linked to lifespan regulation in many organisms. In worms, the complex composed of ASH-2, WDR-5 and the H3K4me3 methyltransferase SET-2 affects lifespan. Knockdown of the methyltransferase subunits improves longevity, while knockdown of the demethylase subunit RBR2 shortens lifespan. Conversely, overexpression of RBR2 extends lifespan (Han and Brunet, 2012). Importantly, these complexes positively (methyltransferase) or negatively (demethylase) regulate global H3K4me3 levels, providing compelling evidence for an epigenetic role in lifespan regulation (Greer et al., 2010). These effects require an intact germline and longevity effects are transgenerational (Maures et al., 2011). Knockdown or mRNA suppression of the H3K4 mono- and di-demethylase LSD-1 extends lifespan (McColl et al., 2008). Although the exact mechanism by which H3K4 methylation regulates lifespan is not clear, the authors speculate a role for local and global transcriptional changes.
In contrast to H3K4me3, H3K27me3 is primarily a transcriptionally repressive modification catalyzed by the PRC2 complex and removed by UTX-1. During worm aging, there is global decrease in somatic H3K27me3 and increase in UTX-1 expression (Jin et al., 2011). RNAi depletion or heterozygous mutation of UTX-1 in worms increases global levels of H3K27me3 and extends lifespan in an insulin-dependent, but germline-independent manner (see below for insulin signaling pathway discussion)(Maures et al., 2011). The opposing effects of H3K4me3 and H3K27me3 on worm lifespan reflect an increasingly favored model in the aging field that proposes gain of activating marks and loss of repressive marks as hallmarks of aging. However, H3K27me3 provides confounding phenotypes in other organisms that limits enthusiasm in fully establishing such an encompassing model for aging; heterozygous mutations in the H3K27me3 methyltransferase MES-2 (E (z) in flies and EzH2 in mammals) lengthen lifespan (Ni et al., 2012), contrary to what might be expected. The longer lifespan may be due to altered regulation of specific genes that have a dominant lifespan extension effect, thereby overriding genome-wide effects of reduction of the repressive mark.
In flies, overexpression of Lid, the worm RBR-2 homolog, extends lifespan, while its knockdown shortens lifespan of male flies by 18% (Li et al., 2010). Lid affects lifespan by changing global levels of H3K4me3; however, knockdown of Trx, one of three H3K4 methylases, does not alter lifespan of male flies. The role of the other methylases and effect on female flies has not yet been tested. In flies, H3K27me3 seems to influence lifespan in a manner opposite to that in worms. Mutations in H3K27 methyltransferase (PRC2) subunits E (z) and ESC reduce global levels of H3K27me3 and extend lifespan of male flies by derepressing target genes Abd-B and Obc1. Interestingly, mutations in Trx increase H3K27me3 levels of E (z) mutants and suppress their longevity phenotype. Trx antagonizes polycomb-mediated silencing by interacting with CBP and promoting H3K27 acetylation (H3K27ac). Since acetylation and methylation at this site are mutually exclusive, increased acetylation results in both decreased methylation and loss of silencing at PRC2 target sites (Siebold et al., 2010).
Thus, histone modifications in flies influence lifespan; however, this effect appears to be less global, and more focused on targeted expression changes of specific genes influencing increased stress tolerance or modulating calorie restriction pathways (discussed later). In fact, the hypothetical working model for yeast and worms (increase in activating modifications and decrease in repressive modifications with age) appear to be oppositely regulated in flies. A ChIP-chip study of histone modification changes in young and old female fly heads showed a loss in activating marks like H3K4me3 and H3K36me3 and a gain of repressive marks like H3K9me3 with age (Wood et al., 2010). In aging killifish, there is global upregulation of H3K27me3 echoing a trend similar to that in flies (Baumgart et al., 2014). Additional studies will be required to determine if the observed opposite histone modification trends truly reflect major differences in age-related epigenetic changes within different aging models. Certainly, one technical explanation is the potential inability to measure global increases in activating modifications in some models due to the preferential loss of histones harboring activating marks, thus complicating the results. In the event that this is not a source of variance, it is also possible that some histone modifications are more dominant in some models than others, and as such, certain local modifications in one model may influence molecular aging pathways as strongly as global changes in other models.
Unlike histone methylation, the link between global histone acetylation and longevity is better understood. The polyamine spermidine, a small organic molecule derived from amino acids inhibits histone acetyltransferase (HAT) activity to generate hypoacetylated chromatin states. The level of spermidine declines from yeast to humans with age and spermidine supplementation increases lifespan in yeast, worms, flies and human cells. The longevity phenotype correlates with improved stress response and upregulated autophagy via increased histone acetylation at these genes (Eisenberg et al., 2009).
Evidence of global changes and imbalance of regional chromatin domains in humans come from in vitro studies in cultured cells. Senescence is a state of cell cycle arrest in response to stress, linked to age-associated tissue decline in vivo (van Deursen, 2014). During senescence, many cultured mammalian cells develop senescence-associated heterochromatin foci (SAHF), regions of highly condensed chromatin associated with heterochromatic histone modifications, heterochromatic proteins, histone variant macroH2A, high-mobility group A (HMGA) proteins and late replicating regions in the genome (Chandra et al., 2012; Zhang et al., 2007). A genome-wide study of senescent chromatin showed that more than 30% of chromatin is dramatically reorganized in senescent cells, including the formation of large scale domains (mesas) of H3K4me3 and H3K27me3 over lamin-associated domains (LADs), as well as large losses (canyons) of H3K27me3 outside of LADs(Shah et al., 2013). These changes are linked to the transcriptional downregulation of lamin B1 in senescence (Freund et al., 2012; Shah et al., 2013; Shimi et al., 2011). The profound destabilization of heterochromatic LADs leads to autophagic degradation of laminB1 and associated repressive histones (Dou et al., 2015). Additionally, senescent cells show strong enrichment of H4K16ac (acetylation at lysine 16 on H4) at promoter elements of active genes where it overlaps with HIRA and newly synthesized H3.3 peaks (Rai et al., 2014). HIRA, a DNA replication-independent histone chaperone that deposits variant histone H3.3 and H4 is required for the steady state maintenance of H4K16ac. These changes suggest that the senescent state hosts a dynamic and imbalanced chromatin environment that is markedly different from the proliferating state.
The role of ubiquitylation and sumoylation in senescence and aging are less understood due to the multitude of substrates targeted. These modifications involve the covalent addition of small peptides such as ubiquitin or SUMO (small ubiquitin related modifier) to lysine groups on histones and other proteins. In yeast, strains lacking subunits of the SAGA/SLIK deubiquitinase module such as Sgf73 are exceptionally long lived. Sgf73 impacts yeast replicative lifespan through multiple related pathways mediated by Sir2, a NAD+ dependent histone deacetylase (discussed later); (1) there is a direct physical interaction of Sgf73 with Sir2 in WT yeast that reduces Sir2 activity, (2) presence of Sgf73 results in asymmetric segregation of ribosomal DNA (rDNA) circles to mother cells and (3) sgf73Δ strains show increased silencing of telomere proximal genes (McCormick et al., 2014). Collectively, these pathways increase lifespan potentially by reinforcing a compact chromatin structure. The role of deubiquitinases in aging of higher organisms remains unknown. In senescent cells, the levels of many ubiquitylated and sumoylated proteins are upregulated including telomerase, p53, histone H2A and PCNA resulting in proteasomal degradation (Grillari et al., 2010). Ubiquitylation of DAF-16 (discussed later), an important longevity protein causes shorter lifespan in worms (Li et al., 2007). SUMO expression is increased in old rats and senescent fibroblasts and overexpression of SUMO causes premature aging in worms (Rytinki et al., 2011). SUMO-induced senescence acts through an interconnected network of pathways that involve sumoylation of proteins in promyelocytic leukemia (PML) nuclear bodies, p53/pRb and its interacting proteins to impact senescence-specific transcription programs (Ivanschitz et al., 2013). However, more work is required to identify the exact chromatin mechanisms.
Many chromatin-related senescence phenotypes occur in aging tissues from various organisms, underscoring the connection between senescence and longevity. High levels of histone macroH2A and heterochromatin protein HP1b accumulate with age in mouse and primate tissues (Herbig et al., 2006; Kreiling et al., 2011). Additionally, H3K9me3 is reduced in cultured cells from normal aged individuals and H4K20me3 is increased in the lysates of aged rats (Sarg et al., 2002; Scaffidi and Misteli, 2006). ChIP-seq in young and old hematopoietic stem cells (HSCs) show a modest increase in the number of H3K4me3 peaks with age, many of which broaden in coverage, especially over HSC identity and self-renewal genes. H3K27me3 peaks do not appreciably change in number but also broaden with age (Sun et al., 2014).
While many more studies will be required to ultimately determine if there is a key “signature” of aging chromatin, the data observed to date strongly suggest a massive alteration in histone modification patterns with aging in multiple models (Fig. 1B, Supplemental Table 1). An overarching theme of these studies is the loss of repressive marks and gain of activating marks that have profound effects on gene expression. In some cases, these global trends may not be evident due to the preferential loss of histones with active modifications. In other cases, the ultimate effects on lifespan may be driven by local and discrete chromatin changes at specific regulatory elements of the genome, resulting in altered transcription of key longevity genes. For example, increases in repressive marks during aging may lead to site specific gains in heterochromatin over genes whose expression are detrimental to lifespan. Together, these studies propose an altered histone modification profile in aging. The consequences of these regulatory changes are either destructive (as in tissue damage) or protective (as in genome stability), and distinguishing these is a major challenge in aging studies.
Transcriptional deregulation as a consequence of epigenetic change
Transcriptional amplification due to histone loss
A consequence of general histone loss in replicatively aged yeast is global transcriptional amplification (Fig. 1C, Supplemental Table 1). Genes most induced with age have specific chromatin signatures in young cells such as fuzzier nucleosome positioning, lack of a nucleosome depleted region (NDR) at the promoter, and weak nucleosome phasing. These genes also exhibit higher frequency of TATA elements and higher occupancy of repressive chromatin factors like Asf1 and Tup1. Interestingly, the same genes in old cells show a stronger nucleosome loss at the promoter and therefore the greatest induction in transcript levels. Many of these age-related changes can be ameliorated by increasing the levels of histones, strongly implicating direct transcriptional consequences of histone loss (Hu et al., 2014).
Upregulation of intragenic cryptic transcripts due to reduced histone methylation over gene bodies
In addition to changes in canonical transcripts, a histone substitution screen in replicatively aged yeast uncovered a role for H3K36 methylation in lifespan regulation and an effect on cryptic transcription (Fig. 1C, Supplemental Table 1). The deletion of Set2, the only methyltransferase for H3K36 in yeast, shortened replicative lifespan (Sen et al., 2015). Conversely, deletion of the H3K36 di/tri demethylase Rph1, extended lifespan by ~30%, which was attributed to the ability of the demethylase mutant to suppress spurious cryptic transcripts originating from within gene bodies. Loss of the worm Set2 homolog, MET-1, is known to shorten lifespan whereas loss of the demethylase JMJD-2 has pro-longevity effects suggesting a conserved regulatory pathway (Hamilton et al., 2005). A subset of genes also shows upregulated cryptic transcription in old worms (Sen et al., 2015). H3K36me3 levels in young worms are negatively correlated with gene expression variation during aging (Pu et al., 2015). Overall, H3K36 methylation serves a protective role in preserving transcriptional precision and stability, and its loss in aging leads to an altered transcriptome detrimental to a longer life.
Transcriptional noise due to DNA damage
Age has been linked to increased transcriptional noise, defined as a stochastic deregulation of gene expression. Transcriptional profiling of single cardiomyocytes isolated from old mice showed considerable variation in transcript levels of a panel of heart-specific housekeeping genes compared to young controls. Mouse embryonic fibroblasts treated with the DNA damaging agent hydrogen peroxide also showed a similar increase in transcription noise, suggesting DNA damage as a key player (Bahar et al., 2006); however, the increase in noise after damage is a delayed effect when more permanent genomic changes such as mutations, rearrangements or epigenetic changes occur, rather than immediate changes such as protein or lipid oxidation.
Cardiomyocytes are post-mitotic, and interestingly, a similar experiment conducted using constantly renewing cells of the hematopoietic lineage (stem cells, granulocytes, naïve B cells and naïve T cells) failed to show significant increase in noise. Notably though, old cells exhibited a general trend toward higher transcript levels (Warren et al., 2007). One reason explaining this difference is the disparate somatic mutation rates between different tissues. Thus, age-related deregulation of transcription (among other changes) may contribute to cellular fitness and growth, but may differ vastly across differentiated cell types.
Altered transcriptional programs
The most obvious molecular consequence of age is an altered transcriptional program, a phenomenon that has been best studied in senescent human cells. In addition to upregulating alternative histone transcripts (Rai et al., 2014), senescent cells remodel the epigenetic landscape to induce a subset of cellular defense and inflammatory response genes, many of which are secreted from the cell and are characterized as part of the senescence-associated secretory phenotype (SASP)(Coppe et al., 2008). SASP has been theorized to promote immune clearance of pre-malignant senescent cells to facilitate tumor suppression; however, chronic SASP induction is also believed to promote tissue aging, underscoring the delicate balance between the “benefits” and “consequences” of senescence (Adams, 2009).
A strong epigenetic correlation was shown between loss of H3K27me3 and upregulation of SASP genes in senescent cells (Shah et al., 2013). Additionally, inhibition of the H3K4 methyltransferase MLL1 was shown to reduce SASP indirectly, via repression of pro-proliferative cell cycle genes and DNA damage response genes that in turn reduce SASP (Capell et al., 2016). Taken together, these lines of evidence indicate that transcriptional reprogramming in senescence occurs through altered activity of specific epigenetic proteins. Mimicking these trends, hematopoietic stem cells (HSCs) isolated from old mice show an upregulated inflammatory response and downregulated chromatin remodeling, DNA repair and transcriptional repression compared to young controls. These changes are concomitant with the inability of the older HSCs to rejuvenate the vascular system (Chambers et al., 2007). RNA-seq analyses of highly purified HSC populations showed upregulated gene ontology (GO) categories related to cell proliferation, cell adhesion and ribosomal protein genes while those related to cell cycle, DNA base excision repair and DNA replication were downregulated. Pathway analyses revealed that TGFβ signaling is reduced in aging HSCs, which could explain a myeloid bias that is established with age (Sun et al., 2014).
A final compelling genomic feature of senescent cells and aged mouse somatic tissue is upregulation of transcripts from repeat elements (De Cecco et al., 2013), much like in embryonic stem cells. Interestingly, HSCs also upregulate expression of repeat elements (such as LTRs, LINEs, SINEs and others) with age, although the consequence of this upregulation is unclear (Sun et al., 2014). Selective loss of heterochromatin in aged cells may contribute to the upregulation of these transcripts. Together, these studies implicate epigenetic mechanisms in regulation of transcription with age, and suggest that possible intervention resulting in transcriptional stabilization may result in increased lifespan and cellular fitness.
Site-specific losses and gains of heterochromatin
Heterochromatin domains are established early during embryonic development and were thought to be maintained throughout lifespan. However, loss of constitutive heterochromatin (commonly at telomeres, centromeres, pericentromeres) occurs in senescence and aging and is triggered by telomere shortening, transcription changes at boundaries and breakdown of the nuclear periphery. In contrast to a loss in constitutive heterochromatin, senescent fibroblast cells show focal increases in heterochromatin called SAHFs (discussed below) in otherwise euchromatic regions (Fig. 1D, Supplemental Table 1).
Yeast cells constitutively express telomerase and lack a well-defined nuclear periphery and deregulation of key heterochromatic proteins contributes to loss of silencing at heterochromatic regions such as the rDNA, MAT locus and subtelomeric regions. A yeast senescence model (tlc1Δ) shows relocalization of telomeric protein Rap1 to new positions in the genome, much like HP1 (Platt et al., 2013). Age-related chromatin deregulation at the rDNA locus results in the formation of extrachromosomal rDNA circles (ERCs) that are causal to replicative aging (Sinclair and Guarente, 1997). In addition, genes at the subtelomeric X-core and X-repeat elements are also derepressed with age (Dang et al., 2009). Yeast Sir2, the founding member of the Sirtuin family of proteins with NAD+ dependent histone deacetylation and ADP ribosylation activity, decreases in old yeast cells compromising silencing at rDNA, MAT, and subtelomeric loci, contributing to both aging (Dang et al., 2009) and onset of sterility (Smeal et al., 1996). Thus, loss of Sir2 shortens replicative lifespan while overexpression and small molecule Sir2 activators extend lifespan (Howitz et al., 2003; Kaeberlein et al., 1999). Similarly, a gain of function mutant of SIR4, another heterochromatic protein acting at the rDNA locus, extends yeast lifespan (Kennedy et al., 1995). Importantly, the Sir2 effect at subtelomeric genes is mediated through histone H4K16ac, which increases as cells age. It is unknown whether gene derepression at subtelomeric loci is causal to aging, but it is interesting to note that deletion of the H4K16 acetyltransferase Sas2 extends replicative lifespan in normal yeast (Dang et al., 2009) and the yeast senescence model mentioned above (Kozak et al., 2010).
Pathways affecting heterochromatin establishment and maintenance are tightly linked to senescence in mammalian cells, resulting in global and gene-specific changes linked to reduced cellular fitness. Constitutive heterochromatin structures are disorganized in senescence, as evidenced by reduced global DNA methylation (discussed below), H3K9me3 levels and HP1 (Scaffidi and Misteli, 2005, 2006). Senescence is also marked by a deficiency in the polycomb-repressive protein EZH2, a histone methyltransferase, and subsequent decrease of repression-associated H3K27me3, leading to rapid senescence. While likely resulting in broader genome wide effects, EZH2 and H3K27me3 loss also results in gene specific upregulation of p16INK4a (p16), a key upregulated SASP and ubiquitous marker for cellular senescence (Bracken et al., 2007). Conversely, overexpression of EZH2 results in increased cellular lifespan (Bracken et al., 2007), an effect attributed through repression of p16. It is interesting to speculate that global H3K27me3 and heterochromatin maintenance may also be stabilized with EZH2 overexpression.
In addition to a marked global loss of constitutive heterochromatin and general increases in transcription, it seems paradoxical that senescent cells form SAHFs that are foci of γ-H2AX, HP1γ, H3K9me3, macroH2A, HMGA proteins and hypoacetylated histones in otherwise euchromatic regions (Di Micco et al., 2011; Narita et al., 2003; Zhang et al., 2007). SAHF formation is induced by an initial chromatin condensation step mediated by the HUCA histone chaperone complex, composed of HIRA, ubinuclein-1 (UBN1), CABIN1 and histone chaperone ASF1a that deposits H3.3 into chromatin (Daniel Ricketts et al., 2015). Consistently, H3.3 expression is dramatically upregulated in senescence (Rai et al., 2014). In a subsequent step, other proteins are recruited to the condensed chromatin including phosphorylated HP1, macroH2A and HMGA proteins (Zhang et al., 2007). Loss of the nuclear lamina (Fig. 1E) triggers a breakdown of heterochromatin organization, relocalizing heterochromatic proteins to other regions in the genome where it may contribute to formation of region-specific foci like SAHFs. The location of SAHFs is not precisely known, but these regions have been correlated with late-replicating regions of the genome. Although the physiological explanation for the SAHFs is unclear, one speculation is that they contribute to the protective response discussed above, that is, as histones are lost and transcriptional noise increases (see below), then the histone chaperone system attempts to ameliorate this via H3.3 assembly to “plug the holes” in euchromatin to control gene expression.
Taken together, deregulation of chromatin organizational proteins and reorganization of heterochromatic domains are discernible features of senescence and aging. These changes encompass a broad decrease of heterochromatin, site-specific decrease of heterochromatin, as well as region-specific gains of heterochromatin. Broad changes may ultimately affect overall nuclear organization and cellular fitness, while region and site-specific heterochromatin changes may directly regulate expression-specific longevity genes that drive aging or affect lifespan.
Alteration in DNA methylation levels and patterns with age
In addition to histone modifications, epigenetic changes include chemical modifications directly on DNA. DNA methylation is a well-studied epigenetic modification and occurs mostly on the 5-carbon of cytosine residues in CpG dinucleotides. It is relatively easy to study due to simplicity of sample preparation and availability of cost-effective techniques that permit large-scale investigations, including bisulfite conversion coupled to PCR, pyrosequencing, microarrays and deep sequencing. DNA methylation is correlated with transcriptional repression and is important in X-chromosome silencing, centromeric/repetitive sequence repression and genomic imprinting.
Many commonly utilized aging models have no (such as yeast) or limited (worms and flies) DNA methylation although there are interesting links to aging. Worms lack DNA cytosine methylation (5mC), but methylation on N6 adenine (6mA) has been recently discovered (Greer et al., 2015). 5mC is also rare in flies (Capuano et al., 2014). Overexpression of dDnmt2, the non-redundant Drosophila DNA methyltransferase, increases fly lifespan, whereas dDnmt2 mutant flies are short-lived (Lin et al., 2005). DNA methylation in flies may function to repress transposition and 5mC has been localized to retrotransposons, although this finding is controversial (Oakes et al., 2003; Osorio et al., 2010). Like worms, 6mA was also recently identified in Drosophila and shown to mark and suppress transposons (Zhang et al., 2015). Given that transposons are also derepressed in senescent mammalian cells (De Cecco et al., 2013), it will be interesting to investigate this new epigenetic modification in higher organisms, and a possible role of 6mA in aging.
DNA methylation is more prominent in mammalian systems and is achieved by the action of three DNA methyltransferases: DNMT1, which has a maintenance role, and DNMT3a and 3b, which are de novo methylases. The removal of DNA methyl groups is mediated by the ten-eleven-translocation (TET) proteins (Du et al., 2014). With age, mammalian cells undergo global DNA hypomethylation and local DNA hypermethylation (Cruickshanks et al., 2013), a pattern which fits the aging model of global heterochromatin deregulation coupled with focal increase in repressive modifications (Fig. 1F, Supplemental Table 1). Loss of methylation occurs primarily in repetitive regions of the genome that correlate with constitutive heterochromatin while hypermethylation primarily occurs at promoter CpGs. In mice, progressive loss of 5mC in livers is evident as the animals age from 6 to 24 months (Singhal et al., 1987). Studies of livers from long-lived Ames mice (see below) compared to normal mice show lower basal levels of DNMT1 regulated by growth hormone deficiency. The differential expression of DNMT likely causes DNA methylation differences over repeat elements and key longevity genes impacting lifespan (Armstrong et al., 2014).
The DNA methylation status in a number of human tissues has been investigated at 26,486 autosomal CpGs using an arrayed format. The results revealed that promoter CpGs undergo hypermethylation while those outside undergo hypomethylation with age (Day et al., 2013). Other forms of DNA methylation such as 5-hydroxymethylcytosine, 6mA and non-CG methylation have also been investigated, but to a much lesser extent.
In a large scale study from UCLA, it was shown that DNA methylation can serve as a remarkable predictor of age in normal/non-cancerous tissues. This study analyzed ~8000 samples representing 51 healthy human tissue and cell types including liver, kidney, immune and brain cells and ~6000 cancer samples. It was convincingly identified that DNA methylation at 353 CpGs (termed clock CpGs) and not other epigenetic modifications accurately predicted age (Horvath, 2013, 2015). A parallel study investigated the rate of DNA methylation change by building a predictive model of the aging methylome from the blood of individuals aged 19–101 (Hannum et al., 2013). This study identified a smaller set of 71 CpGs that have a high accuracy of age prediction and occur near genes associated with aging. The prediction from blood and the smaller number of CpGs to probe increases the diagnostic value of this study. To understand the underlying mechanism of these epigenetic clocks, it is imperative to study it in model organisms where mutational analyses will reveal specific regulatory pathways. As of now, the clock has been identified in human cells and tissues and efforts to discover such a clock in model organisms such as mice are under way.
In summary, the overarching pattern of DNA methylation changes in aging encompasses global DNA hypomethylation and local hypermethylation that may activate specific transcriptional programs, and this is congruent with the histone modification changes that occur, as described above. It remains elusive what specific genes are directly affected by the altered DNA methylation.
ATP-dependent chromatin remodeling complexes in aging
ATP-dependent chromatin remodeling complexes use ATP hydrolysis to restructure chromatin either permitting or repressing transcription (Bartholomew, 2014). The role of remodelers in senescence and aging is limited, despite their function in large-scale chromatin organization; and it could be that there is an insufficient understanding of their roles due to functional complexity and redundancy of these enzymes in vivo. In line with the overall aging model presented above, the SWI/SNF class of remodelers associated with gene activation is predicted to promote aging, whereas the repressive ISWI class may promote longevity. However, we note that chromatin remodeling could also impact lifespan via regional or gene-specific effects that may not parallel global changes (Fig. 1G).
In support of gene-specific effects, deleting ISW2 in yeast robustly extends replicative lifespan. Increased resistance to stress is an emerging concept in lifespan extension, and deletion of ISW2 derepresses a cohort of stress response genes acting through the homologous recombination DNA repair pathway. In support, ISW2 deletion improved resistance to DNA damaging agents. Genome-wide analysis revealed that deletion of ISW2 partially mimics the transcriptome and nucleosome positioning profile of calorie restricted cells, also showing upregulation of stress response genes (Dang et al., 2014). Interestingly, reduction of the worm ortholog of the ISW2 complex extended lifespan while knockdown of BAZ1A (human homolog of Itc1) increased expression of many stress response genes mimicking findings in an isw2Δ yeast strain. These data argue in favor of gene-specific effects and suggest that ISW2 orthologous complexes may regulate longevity through stress response mechanisms.
The Nucleosome Remodeling and Deacetylase (NuRD) chromatin remodeling complex has been linked to aging in mammalian cells. The WD40 domain-containing protein, RBBP4, and the related RBBP7 histone chaperone proteins were identified as specific interactors of the lamin A C-terminal fragment. These subunits, along with two other NuRD-specific subunits, are downregulated in progeroid (HGPS) skin fibroblast cells. (Pegoraro et al., 2009). The same trends are recapitulated in primary fibroblast cells isolated from older patients, suggesting that loss of NuRD components is also relevant in physiological aging. The consequence of NuRD downregulation is loss of senescence heterochromatic features, fitting a model in which loss of global protective chromatin is predicted to promote aging. The exact mechanism of senescence induction through this pathway remains to be investigated, and it is particularly interesting to consider how nucleosome remodeling changes affect global and local changes in histone modifications and DNA methylation during aging.
Nutrient signaling and its effects on the chromatin
Nutrient availability is key to determining lifespan in several tested models (Bordone and Guarente, 2005). Nutrients in the form of growth factors, metabolites, amino acids and carbohydrates are sensed by a multitude of proteins including glucose sensors in yeast, the Tor (target of rapamycin) proteins conserved from yeast to humans, the insulin receptors conserved from worm to humans and sirtuins conserved from yeast to humans. One role of these factors is to relay signals to the nucleus leading to general epigenetic modulation of lifespan. In addition to nutrients, other environmental stimuli (such as circadian cycles, hormones, and exercise) also alter the epigenome to influence lifespan and healthspan. For additional discussion of these signals, we direct the readers to two recent reviews (Benayoun et al., 2015; Booth and Brunet, 2016). The best characterized pathway connecting the environment to the epigenome is nutrient signaling which we discuss in detail below.
Glucose sensing and the Ras/AC/PKA pathway
Glucose sensing in yeast involves the Ras/adenylate cyclase (AC)/protein kinase A (PKA) pathway. Deletion of Ras2 or adenylate cyclase has chronological lifespan benefits partially through the effects on oxidative stress protection (Fabrizio et al., 2003; Longo, 1999). The Ras and Tor (discussed below) pathways converge on the stress regulon SOD2 and involve the glucose-repressible kinase Rim15, and stress-response transcription factors Msn2/4 and Gis1 (Pedruzzi et al., 2000). Calorie restriction in the long-lived Ras2/Sch9 (of the Tor pathway) mutants further increases lifespan, for a combinatorial longevity increase of 10-fold, suggesting independent pathways in calorie restriction.
The Gis1 related protein Rph1, which is also a H3K36 demethylase, affects yeast replicative lifespan (Sen et al., 2015) and has a role in gene expression in the stationary phase (Orzechowski Westholm et al., 2012). Surprisingly, deletion of GIS1 shortens lifespan, and this phenotype may result from activation of Acs2 (nuclear acetyl-CoA synthetase), promoting histone acetylation, while deletion of RPH1 extends lifespan via a mitochondria-to-nucleus signaling via gene repression at subtelomeres (Orzechowski Westholm et al., 2012; Schroeder et al., 2013). All of the mutants mentioned in this section have markedly different gene expression profiles compared to wild type yeast, strongly indicative of large-scale epigenetic changes, although the exact changes driving longevity are undetermined.
Calorie restriction, the TOR pathway and histone acetylation
The yeast Tor protein (a serine/threonine kinase of the phosphatidyl inositol kinase related kinase (PIKK) family) assembles into two complexes: (a) rapamycin sensitive TORC1 comprising Tor1 or Tor2 kinases, Lst8, Kog1 and Tco89 and (b) rapamycin insensitive TORC2 comprising Tor2, Avo1, Avo2, Avo3 and Lst8 (Loewith and Hall, 2011). Unlike yeast, higher eukaryotes have only one Tor protein, although the assembly into two complexes is conserved. TORC1 signaling modulates ribosome biogenesis, translation, autophagy, cell growth and proliferation via signal transduction involving Sch9 kinase, whereas TORC2 signaling primarily regulates cytoskeletal changes required for cell growth. Deletion of TOR1 and SCH9, as well as administration of low doses of rapamycin, extend both replicative and chronological lifespan in yeast (Fabrizio et al., 2001; Kaeberlein et al., 2005; Powers et al., 2006). The broader question of interest here is whether TOR signaling modulates the epigenetic landscape to impact longevity.
There are several lines of evidence for TORC1 modulation of chromatin structure and function. Rapamycin treatment downregulates TORC1 signaling and increases genome-wide occupancy of Rsc9, a subunit of the RSC chromatin remodeling complex, ultimately resulting in gene expression changes (Damelin et al., 2002). Furthermore, rapamycin treatment or nutrient starvation reduces nucleoli size and displaces RNA polymerase I (pol I) from rDNA loci, thus inhibiting transcription at these sites. rDNA silencing is facilitated by increased binding of Rpd3-Sin3 HDAC complex (Rpd3(L)) and deacetylation of H4K5/K12 residues. At ribosomal protein (RP) gene promoters, TORC1 signaling favors the occupancy of Esa1-histone acetyltransferase complex, resulting in histone H4 acetylation and active transcription. In response to nutrient starvation or rapamycin treatment, TORC1 signaling is inhibited, releasing the Esa1-containing complex from RP gene promoters and returning chromatin to a condensed state by Rpd3 (L)(Rohde and Cardenas, 2003). These repressive chromatin effects of rapamycin all serve to lower transcription of protein synthesis apparatus, ultimately lowering growth.
A chemical genomics screen of H3/H4 residues sensitive to rapamycin identified H3K56 as crucial for TORC1 signaling (Chen et al., 2012a). In the presence of rapamycin, H3K56ac is reduced globally and locally at rDNA loci where this acetylation functions to make chromatin permissive for pol I transcription and is also required for rRNA processing. In H3K56 substitution mutants, deletion of the acetyltransferase Rtt109 and deacetylases Hst3/4 (both targeting H3K56) all decrease lifespan. Additionally, H3K56 and rtt109Δ mutants are sensitive to DNA damaging agents (Driscoll et al., 2007; Han et al., 2007). This implies that balanced and cycling H3K56ac is important to preserve genome integrity and promote longevity (Dang et al., 2009). Thus, in yeast, TORC1 signaling negatively regulates longevity via histone acetylation, affecting ribosome biogenesis, translation and rDNA regulation.
The worm homolog, CeTOR or let-363, is an amino acid sensor (McCormick et al., 2011); combining mutations of CeTOR substrate RSKS-1 (Sch9 of the Tor pathway in yeast) and DAF-2 (insulin signaling receptor in the insulin signaling pathway as discussed below) lead to a fivefold extension in worm lifespan (Chen et al., 2013). CeTOR associates with accessory proteins to form rapamycin sensitive TORC1 and rapamycin insensitive TORC2 complexes, as in yeast. TORC1 signaling increases protein translation, anabolic processes, and energy expenditure that is detrimental to a longer lifespan (Wullschleger et al., 2006). Depletion of many factors in this pathway leads to lifespan extension (Crawford et al., 2007; Hansen et al., 2007). From an epigenetic perspective, TOR-mediated expression of ribosomal proteins by pol II in worms may involve histone acetyltransferase activity as in yeast, although it has not been directly tested.
In flies, overexpression of TOR pathway proteins including dTsc1, dTsc2, PTEN, FOXO (homologous to worm DAF-16 discussed below), 4E-BP, or dominant negative versions of dTOR and dS6K, all extend lifespan by interrupting nutrient sensing and mimicking calorie restriction. Additionally, TORC1 inhibition by rapamycin also extends lifespan in some strains (Kapahi et al., 2004). The primary tissues where these pathways operate are the nervous system and fat body. Insulin signaling and TOR downregulation in flies improves cardiac function, locomotion and imparts neuroprotection against neurodegenerative diseases like Alzheimer’s and Parkinson’s (Hirth, 2010; Partridge et al., 2011), also suggesting major epigenetic changes underlying rapamycin treatment.
Calorie restriction and the insulin signaling pathway
One of the most conserved and well-studied pathways in aging is the insulin and insulin-like growth factor 1 (IGF-1) signaling (IIS) pathway first identified in worms. Nutrient sensing by insulin/IGF-1 engages DAF-2, the only known member of the insulin receptor family in worms. This initiates a cascade of phosphorylation events via the activity of phosphatidyl inositol-3-kinase (PI3K) and Akt, ultimately inactivating and promoting the cytoplasmic sequestration of fork head (FOXO) transcription factor DAF-16. Under calorie restriction, DAF-16 is instead sequestered in the nucleus where it activates genes that promote longevity (Kenyon, 2005; Kenyon, 2010). In support of this pathway, it was shown that mutations in AGE-1, encoding PI3K, lengthen lifespan; these were the first long-lived mutants discovered in any organism (Friedman and Johnson, 1988). Subsequently, another group showed that mutations in DAF-2 double worm lifespan (Kenyon et al., 1993). It was further shown that DAF-16 mutations suppressed longevity phenotypes of AGE-1 and DAF-2, suggesting they operated in the same pathway (Lin et al., 1997; Ogg et al., 1997). Additionally, disabling DAF-2 arrests larval development in the dauer diapause stage that shows increased lifespan, inhibited reproductive maturity and delayed onset of senescence. Conversely, a high glucose diet decreases rate of dauer formation, shortens lifespan of DAF-2 knockdowns, decreases DAF-16 activity and shortens lifespan of WT worms (Kenyon et al., 1993; Kimura et al., 1997). DAF-16 may be phosphorylated at sites (not targeted by Akt) by AMPK, a pro-longevity kinase that is activated by low cellular energy states. DAF-16 phosphorylated by AMPK can influence gene expression patterns independent of its subcellular localization (Greer et al., 2007).
Of interest to this review, DAF-16/FOXO identification provided an exciting link to discovery of epigenetic mechanisms operating in the nucleus and ultimately affecting lifespan. DAF-16 co purifies with the ATP-dependent chromatin remodeling complex, SWI/SNF. Further, DAF-16 physically recruits SWI/SNF to coregulated target genes for local chromatin remodeling and activation of genes affecting dauer formation, increased stress resistance and longevity (Riedel et al., 2013). There is considerable cross-talk between SWI/SNF type remodelers and histone acetylation and this could be an interesting area of future study.
Reduced levels of Drosophila insulin like peptides (dilps), loss of insulin receptor homolog InR (homologous to worm DAF-2) or insulin receptor substrates such as CHICO and Lnk leads to considerable lifespan extension (Clancy et al., 2001; Gronke et al., 2010; Slack et al., 2010; Tatar et al., 2001). Some mutant “dwarf” mice, including the Ames (Bartke, 2000), Snell (Flurkey et al., 2001), Laron (Zhou et al., 1997) and Little mice (Eicher and Beamer, 1976), have impaired insulin signaling and increased lifespan. Their diminished growth is due to impaired development of the pituitary (thus decreasing growth hormone (GH) and other pituitary hormone levels) or mutations that make them GH resistant. A consequence of reduced GH is low circulating levels of IGF-1 and therefore impaired insulin signaling. In mammalian models, there is a strong correlation between growth retardation and duration of life (Sherman and Campbell, 1935), and there have been efforts to induce growth retardation to benefit lifespan, including reducing growth hormone (GH) levels, mutating GH receptors and calorie restriction (McCay et al., 1989). We note that aging studies in dwarf mice should be interpreted with caution since longevity is not necessarily a result of delayed aging but an effect of delayed attainment of sexual maturity. Regardless, these mice provide an excellent model to investigate aging delay due to reduced insulin signaling. A common theme to all the dwarf mice are low levels of IGF-1; it is likely that the downstream effectors of the IGF-1 pathway may in fact impact epigenetic changes much like in worms (Bartke and Brown-Borg, 2004).
In light of these studies, there has been renewed interest in characterizing the lifespan benefit of drugs that target the insulin pathway. Metformin has been prescribed as an anti-diabetic drug for decades and works by decreasing hepatic glucose production, sensitizing insulin, reducing mitochondrial respiratory chain function and activating AMPK (Viollet et al., 2012). Animals studies with metformin showed remarkable lifespan and healthspan benefits mimicking calorie restriction (Martin-Montalvo et al., 2013). Currently, the US Food and Drug Administration has approved a clinical trial (named Targeting Aging With Metformin or TAME) to repurpose the drug as an anti-aging intervention. Thus, it is of immense interest to continue characterizing the epigenetic outcomes of reduced insulin signaling in numerous models, possibly uncovering new pathways for targeted intervention strategies.
The link between metabolism and histone acetylation via sirtuins and other enzymes
The diverse environmental stimuli (including nutrients) alter the metabolic activity and supply of cofactors and substrates within cells. Cells can sense this altered energy status by, for example, assessing NAD+ levels that are reduced with age. Sirtuins are NAD-dependent deacetylases implicated in aging in multiple model systems (Longo and Kennedy, 2006). Calorie restriction induces an oxidative metabolic state manifested in high NAD+ levels and consequently high Sirtuin activity. Overexpression of yeast Sir2 and Sir2 homologs (SIR-2.1 in worms, dSir2 in flies and SIRT1 in mammals) extend lifespan (Herranz et al., 2010; Kaeberlein et al., 1999; Kim et al., 1999; Rogina and Helfand, 2004; Tissenbaum and Guarente, 2001); however, only yeast Sir2 has been strongly connected to calorie restriction pathways.
Importantly, Sir2 is a histone deacetylase and regulates lifespan in yeast through multiple epigenetic mechanisms impacting heterochromatin, extrachromosomal rDNA circles (ERCs), H4K16ac and subtelomeric gene expression as discussed above (Dang et al., 2009). Worms grown in presence of SirTuin Activating Compounds (STACs) like resveratrol (a plant polyphenol) show a slight (10%), but consistent, increase in lifespan (Wood et al., 2004). Boosting NAD+ levels by genetic or pharmacological means also extends worm and mouse lifespan (Mouchiroud et al., 2013; Zhang et al., 2016). Two models for Sir2 action have been found (1) in yeast, where Sir2 may stimulate heterochromatin formation or reduce H4K16 acetylation at subtelomeric loci to increase lifespan, (2) in mammals, where it may directly deacetylate FOXO transcription factors and regulate its target genes (Brunet et al., 2004; Daitoku et al., 2004; Frescas et al., 2005).
The effect of H4K16 deacetylation on fly lifespan is more complicated as different HDACs have disparate effects on lifespan. As in yeast and worms, dSir2 overexpression and STACs such as resveratrol and fisetin extend lifespan in flies acting via the calorie restriction pathway (Wood et al., 2004). However, haploinsufficiency of Rpd3, another HDAC targeting H4K16, also results in lifespan extension (Rogina et al., 2002). Although the Rpd3 findings are similar to yeast, Drosophila longevity is not mediated through heterochromatic gene silencing. Together, these data suggest that in flies, different deacetylases (dSir2 and Rpd3) function in opposite ways at euchromatin to affect longevity by influencing specific gene expression patterns (Frankel and Rogina, 2005). The disparity may stem from the multiple substrates targeted by these proteins but nevertheless underscore the importance of specific gene regulation in longevity, which, as discussed above, may be a more dominant mechanism regulating lifespan in some models.
Another metabolite, acetyl-CoA, increases in aged flies contributing to increased acetylation of H4K12 and key metabolic enzymes. Targeted reduction of ATP citrate lyase, an acetyl-CoA producing enzyme and the acetyltransferase for H4K12, Chameau, promotes longevity (Peleg et al., 2016). However, the exact role of acetylation in limiting lifespan in flies is still unclear. For one, different deacetylases, dSir2 and Rpd3, have different effects on lifespan. Second, increased H3K9ac achieved by mutation of Su (var) 2-1 has no effect on lifespan (Frankel and Rogina, 2005). Third, administration of different HDAC inhibitors like sodium butyrate, TSA, PBA and SAHA result in different changes in mean lifespan. The dosage, time of administration and genetic background of flies used are critical to observing lifespan changes (McDonald et al., 2013). Thus, more work will be required to determine the key epigenetic changes underlying fly lifespan modulation.
The hallmark study that first reported mean and maximum lifespan extension by calorie restriction in a vertebrate model was conducted in rats (McCay et al., 1989). In mice, calorie restriction intervention can be initiated at weaning (3–6 weeks), one year or even up to 19 months to see health benefits. Adult-initiated calorie restriction (~1 year) increases mean but not maximum lifespan (Weindruch and Walford, 1982). Calorie restriction initiated in older mice (19 months) has significant effects on mean and maximum lifespans in addition to reducing cancer incidence (Dhahbi et al., 2004). Late stage benefit occurs at least partially through the mTOR pathway; treatment with rapamycin at 600 days also extended mean and maximum lifespans of male and female mice (Harrison et al., 2009). For a more elaborate discussion of possible calorie restriction effects on the epigenome, we refer the readers to a specialized review (Vaquero and Reinberg, 2009).
The lifespan effect of altering mammalian sirtuins is complex. Mammals have seven Sirtuins (SIRT1-7) with SIRT1 being the closest mammalian homolog of yeast Sir2. SIRT1 localizes to the nucleus and deacetylates H1K26, H4K16, H3K9 and a number of non-histone targets including p53, p300, Suv39h1, FOXO transcription factors and PGC1α (Chen et al., 2012b). Unfortunately, altering systemic SIRT1 levels has not yet shown any lifespan changes in mice, although there are health benefits linked to SIRT1 expression (Libert and Guarente, 2013). Of note, there is one report of lifespan extension in mice by overexpression of neuronal SIRT1 (Satoh et al., 2013).
In 1997, resveratrol was found to be a potential chemotherapeutic agent in mice (Jang et al., 1997). Resveratrol reduced the Km for binding to both NAD and the acetylated substrate (Howitz et al., 2003; Mitchell et al., 2014). Later, it was shown that resveratrol had beneficial effects on health and survival of mice on a high calorie diet (Baur et al., 2006). SRT1720, another STAC, was capable of modestly extending mouse lifespan (Mitchell et al., 2014). One of the beneficial effects of SIRT1 activation is improved mitochondrial function. With age and declining NAD+ levels, mitochondrial (but not nuclear) OXPHOS subunits are downregulated resulting in a pseudohypoxic state. The effect is exaggerated by deleting SIRT1 and attenuated by increasing NAD+ levels, in this case by feeding mice NMN, a precursor of NAD+(Gomes et al., 2013).
Surprisingly, the less studied SIRT6 was recently shown to promote longevity in male (but not female) mice by ~16% when overexpressed (Kanfi et al., 2012). SIRT6 is nuclear, deacetylates H3K9 and H3K56 and acts as a PARP1 ADP-ribosylase under oxidative conditions to promote DNA repair.
These observations support the model whereby histone acetylation and regulation via sirtuins and other metabolic enzymes regulates lifespan. Overall, the epigenetic mechanisms of sirtuin action and their precise targets in aging are still unclear; however, the connection to histone acetylation is strong.
Diseases of epigenetics and aging – laminopathies
Laminopathies are heterogeneous diseases characterized by deleterious changes to nuclear organization, resulting from mutations in genes encoding nuclear lamina proteins (Dechat et al., 2008). Hutchinson-Gilford progeria syndrome (HGPS) is a rare premature aging laminopathy disease that has been the subject of intense study. HGPS is caused by mutations in lamin A, with most cases involving the expression of progerin, a truncated dominant negative lamin A protein (De Sandre-Giovannoli et al., 2003; Eriksson et al., 2003). HGPS is a complex disease with multiple phenotypes indicating possible defects in normal aging and underlying epigenetic regulation of aging. Of interest to this review, HGPS cells exhibit severe abnormalities in nuclear morphology (Goldman et al., 2004), compromised DNA damage repair, alterations in chromosome organization, abnormal heterochromatin and accelerated rates of cellular senescence (Burtner and Kennedy, 2010; Kudlow et al., 2007; Merideth et al., 2008; Norton et al., 2011; Prokocimer et al., 2013). Specifically, HGPS exhibits many alterations in heterochromatin factors, namely decreased levels of HP1, H3K9me3 and H3K27me3 and increased levels of H4K20me3 (McCord et al., 2013; Scaffidi and Misteli, 2005; Shumaker et al., 2006). Late passage HGPS cells exhibit loss of peripheral heterochromatin (Goldman et al., 2004), reduction in H3K9me3 and H3K27me3 levels (Scaffidi and Misteli, 2006; Shumaker et al., 2006), and the downregulation of H3K27me3 methylase EZH2 (Shumaker et al., 2006). A recent genome-wide analysis of H3K27me3 in HGPS cells suggested a redistribution of the remaining amount of this modification across the genome (McCord et al., 2013). In addition, pre-senescent H3K4me3 mesas have been observed in proliferating HGPS cells, indicative of major chromatin reorganization in progeroid cells prior to senescence (Shah et al., 2013).
Laminopathies are well modeled in mice, including in the Zmpste24 and BubR1 models. Zmpste24 encodes a metalloproteinase involved in maturation of lamin A, one of the major components of the lamin network (Bergo et al., 2002; Corrigan et al., 2005; Pendas et al., 2002). BubR1 encodes a key protein involved in the mitotic checkpoint response, ensuring accurate mitotic chromosome segregation; levels of BubR1 are reduced in normally aged animals (Baker et al., 2004; Hartman et al., 2007; Matsumoto et al., 2007). Mutations in lamin A, Zmpste24, or BubR1 result in premature aging, including the acceleration onset of various age-related pathologies. BubR1 deficient mice also accumulate senescent cells, and in the BubR1 hypomorphic mouse, targeted clearance of senescent cells results in delay of onset for age-related phenotypes and BubR1 overexpression results in increased healthspan and lifespan (Baker et al., 2011), providing a strong link between onset of senescence and aging.
These models suggest other interesting links between epigenetic regulation and aging. Zmpste24 mice have been shown to have hypermethylation at rDNA (Osorio et al., 2010), a phenotype associated with normal rodent aging (Oakes et al., 2003). Zmpste24 mice have also been shown to have global hypoacetylation on core histones H4 and H2B, which has been attributed to changes in expression of key cell cycle and metabolic genes in pathways associated with age-related pathologies (Zhang et al., 2015). In addition, a recent study demonstrated that BubR1 is directly acetylated by the histone acetyltransferase CBP, and this modification targets the protein for degradation; however, acetylated BubR1 is as a novel deacetylation target of SIRT2 (North et al., 2014). Overexpression of SIRT2 or treatment with a NAD+ precursor in BubR1 hypomorphic mice results in increased BubR1 levels and increased median lifespan (Corrigan et al., 2005). However, SIRT2 does not restore BubR1 levels to WT and cannot reverse many of the age-related phenotypes.
We note that laminopathies are complex diseases and determining causality of aging is particularly challenging in these models. Specifically, it remains unclear whether reduction in lifespan and presence of age-related pathology directly results from the lamin or related mutations, or if the phenotypes are secondary to significant metabolic changes. Despite these challenges, it is of great interest to understand how the lifespan is extended in these premature aging models and uncover other epigenetic mechanisms that may underlie maintenance of normal levels of BubR1 or other key checkpoint factors whose reduction contributes to premature aging phenotypes or genetic instability.
Conclusions and Perspectives
This review provides a comprehensive overview of the compelling epigenetic evidence linked to aging from animal, tissue, and cell-based models that continue to be critical for identifying key longevity pathways. Studies of chromatin changes suggest two recurring themes in aging: (1) global upregulation of activating marks and downregulation of repressive marks and (2) gene-specific changes in chromatin states regulating expression of key longevity genes. These general themes are heavily influenced by environmental stimuli, nutrient signaling and metabolic state. While much has been uncovered in these studies, there remain many future challenges in epigenetic aging research, particularly with regard to characterizing key epigenetic changes that may be causal to aging. These challenges include linking specific chromatin modifications to changes in aging-specific signaling pathways using genetic manipulations, identifying causal epigenetic changes in age-related diseases, uncovering specific enzymes for targeted therapeutic strategies to improve lifespan and healthspan, linking metabolism to epigenetic outcomes in aging, particularly related to mitochondrial aging where a vast amount of literature is already available (Bratic and Larsson, 2013) and optimizing epigenomic modification strategies to become a viable option for potential therapy.
A number of recent technological advances provide promising leads towards addressing these challenges (Supplemental Table 2). Single cell/low cell number genomics (Nawy, 2014; Shankaranarayanan et al., 2011) may prove to be critically important in identifying mutations in key epigenetic enzymes and/or aberrant patterns of epigenetic modifications in specific disease cohorts or cell types. Continuing advances in CRISPR (Cong et al., 2013) technology, including use of CRISPR technology for enzyme targeting, will also provide new methods for genetic and epigenetic manipulation. In particular, genomics coupled with direct manipulation of epigenetic factors and targeted epigenetic reprogramming will greatly advance our understanding of the role of epigenetic changes that are causal to aging.
In addition to these advanced tools, new studies in emerging aging models, such as eusocial insects, and in yet-uncharacterized models will provide additional opportunities for insight into key epigenetic mechanisms in aging. In the case of the Indian jumping ant Harpegnathos saltator, a worker can replace a queen in the colony, resulting in a change in longevity, acquisition of reproductive function, and loss of worker behavior, all of which can ultimately be reversed. The epigenetic mechanisms that underlie this transition are of great interest, including characterization and manipulation of epigenetic patterning during development, which lead to key behavioral differences in these organisms (Simola et al, 2016). Particularly long lifespans have been observed in several types of deepwater fishes, various crustaceans, bow head whales, several turtles and naked mole rats (relative to other rodents) among others. While some may be unfeasible for creation of laboratory models, tissue and cellular studies of these or similar organisms may prove to be insightful. In addition, short-lived model organisms such as yeast, worms and killifish are useful for quick lifespan estimations (Table 1). Together with the technological advances highlighted above, new experimental avenues and models in aging research will provide key insight into the epigenetic pathways that underlie longevity and aging, and will likely identify factors and pathways that can be targeted to improve health and lifespan in humans.
Supplementary Material
Supplemental Table 1: Epigenetic pathways of aging and longevity in different model organisms
Supplemental Table 2- Protocols for aging research
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol Cell. 2009;36:2–14. doi: 10.1016/j.molcel.2009.09.021. [DOI] [PubMed] [Google Scholar]
- Armstrong VL, Rakoczy S, Rojanathammanee L, Brown-Borg HM. Expression of DNA methyltransferases is influenced by growth hormone in the long-living Ames dwarf mouse in vivo and in vitro. J Gerontol A Biol Sci Med Sci. 2014;69:923–933. doi: 10.1093/gerona/glt133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dolle ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, et al. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature. 2006;441:1011–1014. doi: 10.1038/nature04844. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomew B. Regulating the chromatin landscape: structural and mechanistic perspectives. Annu Rev Biochem. 2014;83:671–696. doi: 10.1146/annurev-biochem-051810-093157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartke A. Delayed aging in Ames dwarf mice. Relationships to endocrine function and body size. Results Probl Cell Differ. 2000;29:181–202. doi: 10.1007/978-3-540-48003-7_10. [DOI] [PubMed] [Google Scholar]
- Bartke A, Brown-Borg H. Life extension in the dwarf mouse. Curr Top Dev Biol. 2004;63:189–225. doi: 10.1016/S0070-2153(04)63006-7. [DOI] [PubMed] [Google Scholar]
- Baumgart M, Groth M, Priebe S, Savino A, Testa G, Dix A, Ripa R, Spallotta F, Gaetano C, Ori M, et al. RNA-seq of the aging brain in the short-lived fish N. furzeri - conserved pathways and novel genes associated with neurogenesis. Aging Cell. 2014;13:965–974. doi: 10.1111/acel.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol. 2015;16:593–610. doi: 10.1038/nrm4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A. 2002;99:13049–13054. doi: 10.1073/pnas.192460799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth LN, Brunet A. The Aging Epigenome. Mol Cell. 2016;62:728–744. doi: 10.1016/j.molcel.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005;6:298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
- Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, Theilgaard-Monch K, Minucci S, Porse BT, Marine JC, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525–530. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt A, Vilcinskas A. The Fruit Fly Drosophila melanogaster as a Model for Aging Research. Adv Biochem Eng Biotechnol. 2013;135:63–77. doi: 10.1007/10_2013_193. [DOI] [PubMed] [Google Scholar]
- Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013;123:951–957. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Burtner CR, Kennedy BK. Progeria syndromes and ageing: what is the connection? Nat Rev Mol Cell Biol. 2010;11:567–578. doi: 10.1038/nrm2944. [DOI] [PubMed] [Google Scholar]
- Capell BC, Drake AM, Zhu J, Shah PP, Dou Z, Dorsey J, Simola DF, Donahue G, Sammons M, Rai TS, et al. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev. 2016;30:321–336. doi: 10.1101/gad.271882.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuano F, Mulleder M, Kok R, Blom HJ, Ralser M. Cytosine DNA methylation is found in Drosophila melanogaster but absent in Saccharomyces cerevisiae, Schizosaccharomyces pombe, and other yeast species. Anal Chem. 2014;86:3697–3702. doi: 10.1021/ac500447w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5:e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra T, Kirschner K, Thuret JY, Pope BD, Ryba T, Newman S, Ahmed K, Samarajiwa SA, Salama R, Carroll T, et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell. 2012;47:203–214. doi: 10.1016/j.molcel.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Li PW, Goldstein BA, Cai W, Thomas EL, Chen F, Hubbard AE, Melov S, Kapahi P. Germline signaling mediates the synergistically prolonged longevity produced by double mutations in daf-2 and rsks-1 in C. elegans. Cell Rep. 2013;5:1600–1610. doi: 10.1016/j.celrep.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Fan M, Pfeffer LM, Laribee RN. The histone H3 lysine 56 acetylation pathway is regulated by target of rapamycin (TOR) signaling and functions directly in ribosomal RNA biogenesis. Nucleic Acids Res. 2012a;40:6534–6546. doi: 10.1093/nar/gks345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhao W, Yang JS, Cheng Z, Luo H, Lu Z, Tan M, Gu W, Zhao Y. Quantitative acetylome analysis reveals the roles of SIRT1 in regulating diverse substrates and cellular pathways. Mol Cell Proteomics. 2012b;11:1048–1062. doi: 10.1074/mcp.M112.019547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan DP, Kuszczak D, Rusinol AE, Thewke DP, Hrycyna CA, Michaelis S, Sinensky MS. Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem J. 2005;387:129–138. doi: 10.1042/BJ20041359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cournil A, Kirkwood TB. If you would live long, choose your parents well. Trends Genet. 2001;17:233–235. doi: 10.1016/s0168-9525(01)02306-x. [DOI] [PubMed] [Google Scholar]
- Crawford D, Libina N, Kenyon C. Caenorhabditis elegans integrates food and reproductive signals in lifespan determination. Aging Cell. 2007;6:715–721. doi: 10.1111/j.1474-9726.2007.00327.x. [DOI] [PubMed] [Google Scholar]
- Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, Singh Rai T, Brock C, Donahue G, Dunican DS, et al. Senescent cells harbour features of the cancer epigenome. Nat Cell Biol. 2013;15:1495–1506. doi: 10.1038/ncb2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Hyland EM, Yuan DS, Huang H, Bader JS, Boeke JD. Probing nucleosome function: a highly versatile library of synthetic histone H3 and H4 mutants. Cell. 2008;134:1066–1078. doi: 10.1016/j.cell.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daitoku H, Hatta M, Matsuzaki H, Aratani S, Ohshima T, Miyagishi M, Nakajima T, Fukamizu A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci U S A. 2004;101:10042–10047. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damelin M, Simon I, Moy TI, Wilson B, Komili S, Tempst P, Roth FP, Young RA, Cairns BR, Silver PA. The genome-wide localization of Rsc9, a component of the RSC chromatin-remodeling complex, changes in response to stress. Mol Cell. 2002;9:563–573. doi: 10.1016/s1097-2765(02)00475-6. [DOI] [PubMed] [Google Scholar]
- Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, Berger SL. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature. 2009;459:802–807. doi: 10.1038/nature08085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang W, Sutphin GL, Dorsey JA, Otte GL, Cao K, Perry RM, Wanat JJ, Saviolaki D, Murakami CJ, Tsuchiyama S, et al. Inactivation of yeast Isw2 chromatin remodeling enzyme mimics longevity effect of calorie restriction via induction of genotoxic stress response. Cell Metab. 2014;19:952–966. doi: 10.1016/j.cmet.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel Ricketts M, Frederick B, Hoff H, Tang Y, Schultz DC, Singh Rai T, Grazia Vizioli M, Adams PD, Marmorstein R. Ubinuclein-1 confers histone H3.3-specific-binding by the HIRA histone chaperone complex. Nat Commun. 2015;6:7711. doi: 10.1038/ncomms8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day K, Waite LL, Thalacker-Mercer A, West A, Bamman MM, Brooks JD, Myers RM, Absher D. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 2013;14:R102. doi: 10.1186/gb-2013-14-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Criscione SW, Peterson AL, Neretti N, Sedivy JM, Kreiling JA. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging (Albany NY) 2013;5:867–883. doi: 10.18632/aging.100621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22:832–853. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhahbi JM, Kim HJ, Mote PL, Beaver RJ, Spindler SR. Temporal linkage between the phenotypic and genomic responses to caloric restriction. Proc Natl Acad Sci U S A. 2004;101:5524–5529. doi: 10.1073/pnas.0305300101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G, dal Zuffo R, Matti V, d’Ario G, Montani E, et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol. 2011;13:292–302. doi: 10.1038/ncb2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015 doi: 10.1038/nature15548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll R, Hudson A, Jackson SP. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science. 2007;315:649–652. doi: 10.1126/science.1135862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Wang J, Jia J, Song N, Xiang C, Xu J, Hou Z, Su X, Liu B, Jiang T, et al. Human hepatocytes with drug metabolic function induced from fibroblasts by lineage reprogramming. Cell Stem Cell. 2014;14:394–403. doi: 10.1016/j.stem.2014.01.008. [DOI] [PubMed] [Google Scholar]
- Eicher EM, Beamer WG. Inherited ateliotic dwarfism in mice. Characteristics of the mutation, little, on chromosome 6. J Hered. 1976;67:87–91. doi: 10.1093/oxfordjournals.jhered.a108682. [DOI] [PubMed] [Google Scholar]
- Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11:1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Liou LL, Moy VN, Diaspro A, Valentine JS, Gralla EB, Longo VD. SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics. 2003;163:35–46. doi: 10.1093/genetics/163.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD. Regulation of longevity and stress resistance by Sch9 in yeast. Science. 2001;292:288–290. doi: 10.1126/science.1059497. [DOI] [PubMed] [Google Scholar]
- Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, Tyler JK. Elevated histone expression promotes life span extension. Mol Cell. 2010;39:724–735. doi: 10.1016/j.molcel.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flurkey K, Papaconstantinou J, Miller RA, Harrison DE. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci U S A. 2001;98:6736–6741. doi: 10.1073/pnas.111158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel S, Rogina B. Drosophila longevity is not affected by heterochromatin-mediated gene silencing. Aging Cell. 2005;4:53–56. doi: 10.1111/j.1474-9726.2005.00143.x. [DOI] [PubMed] [Google Scholar]
- Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem. 2005;280:20589–20595. doi: 10.1074/jbc.M412357200. [DOI] [PubMed] [Google Scholar]
- Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell. 2012;23:2066–2075. doi: 10.1091/mbc.E11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DB, Johnson TE. A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics. 1988;118:75–86. doi: 10.1093/genetics/118.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Blanco MA, Gu L, Sendinc E, Liu J, Aristizabal-Corrales D, Hsu CH, Aravind L, He C, Shi Y. DNA Methylation on N6-Adenine in C. elegans. Cell. 2015;161:868–878. doi: 10.1016/j.cell.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007;17:1646–1656. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Maures TJ, Hauswirth AG, Green EM, Leeman DS, Maro GS, Han S, Banko MR, Gozani O, Brunet A. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature. 2010;466:383–387. doi: 10.1038/nature09195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillari J, Grillari-Voglauer R, Jansen-Durr P. Post-translational modification of cellular proteins by ubiquitin and ubiquitin-like molecules: role in cellular senescence and aging. Adv Exp Med Biol. 2010;694:172–196. doi: 10.1007/978-1-4419-7002-2_13. [DOI] [PubMed] [Google Scholar]
- Gronke S, Clarke DF, Broughton S, Andrews TD, Partridge L. Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet. 2010;6:e1000857. doi: 10.1371/journal.pgen.1000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton B, Dong Y, Shindo M, Liu W, Odell I, Ruvkun G, Lee SS. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 2005;19:1544–1555. doi: 10.1101/gad.1308205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Zhou H, Horazdovsky B, Zhang K, Xu RM, Zhang Z. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science. 2007;315:653–655. doi: 10.1126/science.1133234. [DOI] [PubMed] [Google Scholar]
- Han S, Brunet A. Histone methylation makes its mark on longevity. Trends Cell Biol. 2012;22:42–49. doi: 10.1016/j.tcb.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007;6:95–110. doi: 10.1111/j.1474-9726.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- Harel I, Benayoun BA, Machado B, Singh PP, Hu CK, Pech MF, Valenzano DR, Zhang E, Sharp SC, Artandi SE, et al. A platform for rapid exploration of aging and diseases in a naturally short-lived vertebrate. Cell. 2015;160:1013–1026. doi: 10.1016/j.cell.2015.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman TK, Wengenack TM, Poduslo JF, van Deursen JM. Mutant mice with small amounts of BubR1 display accelerated age-related gliosis. Neurobiol Aging. 2007;28:921–927. doi: 10.1016/j.neurobiolaging.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- Herranz D, Munoz-Martin M, Canamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O, Serrano M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun. 2010;1:3. doi: 10.1038/ncomms1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirth F. Drosophila melanogaster in the study of human neurodegeneration. CNS Neurol Disord Drug Targets. 2010;9:504–523. doi: 10.2174/187152710791556104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S. Erratum to: DNA methylation age of human tissues and cell types. Genome Biol. 2015;16:96. doi: 10.1186/s13059-015-0649-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- Hu Z, Chen K, Xia Z, Chavez M, Pal S, Seol JH, Chen CC, Li W, Tyler JK. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 2014;28:396–408. doi: 10.1101/gad.233221.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF, et al. Lysosome-mediated processing of chromatin in senescence. J Cell Biol. 2013;202:129–143. doi: 10.1083/jcb.201212110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanschitz L, De The H, Le Bras M. PML, SUMOylation, and Senescence. Front Oncol. 2013;3:171. doi: 10.3389/fonc.2013.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- Jin C, Li J, Green CD, Yu X, Tang X, Han D, Xian B, Wang D, Huang X, Cao X, et al. Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab. 2011;14:161–172. doi: 10.1016/j.cmet.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, Burtner CR, Kennedy BK. Recent developments in yeast aging. PLoS Genet. 2007;3:e84. doi: 10.1371/journal.pgen.0030084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, Austriaco NR, Jr, Zhang J, Guarente L. Mutation in the silencing gene SIR4 can delay aging in S. cerevisiae. Cell. 1995;80:485–496. doi: 10.1016/0092-8674(95)90499-9. [DOI] [PubMed] [Google Scholar]
- Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Kim S, Benguria A, Lai CY, Jazwinski SM. Modulation of life-span by histone deacetylase genes in Saccharomyces cerevisiae. Mol Biol Cell. 1999;10:3125–3136. doi: 10.1091/mbc.10.10.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Kozak ML, Chavez A, Dang W, Berger SL, Ashok A, Guo X, Johnson FB. Inactivation of the Sas2 histone acetyltransferase delays senescence driven by telomere dysfunction. EMBO J. 2010;29:158–170. doi: 10.1038/emboj.2009.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiling JA, Tamamori-Adachi M, Sexton AN, Jeyapalan JC, Munoz-Najar U, Peterson AL, Manivannan J, Rogers ES, Pchelintsev NA, Adams PD, et al. Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell. 2011;10:292–304. doi: 10.1111/j.1474-9726.2010.00666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudlow BA, Kennedy BK, Monnat RJ., Jr Werner and Hutchinson-Gilford progeria syndromes: mechanistic basis of human progeroid diseases. Nat Rev Mol Cell Biol. 2007;8:394–404. doi: 10.1038/nrm2161. [DOI] [PubMed] [Google Scholar]
- Kvam E, Goldfarb DS. Nucleus-vacuole junctions and piecemeal microautophagy of the nucleus in S. cerevisiae. Autophagy. 2007;3:85–92. doi: 10.4161/auto.3586. [DOI] [PubMed] [Google Scholar]
- Li L, Greer C, Eisenman RN, Secombe J. Essential functions of the histone demethylase lid. PLoS Genet. 2010;6:e1001221. doi: 10.1371/journal.pgen.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Gao B, Lee SM, Bennett K, Fang D. RLE-1, an E3 ubiquitin ligase, regulates C. elegans aging by catalyzing DAF-16 polyubiquitination. Dev Cell. 2007;12:235–246. doi: 10.1016/j.devcel.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Libert S, Guarente L. Metabolic and neuropsychiatric effects of calorie restriction and sirtuins. Annu Rev Physiol. 2013;75:669–684. doi: 10.1146/annurev-physiol-030212-183800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- Lin MJ, Tang LY, Reddy MN, Shen CK. DNA methyltransferase gene dDnmt2 and longevity of Drosophila. J Biol Chem. 2005;280:861–864. doi: 10.1074/jbc.C400477200. [DOI] [PubMed] [Google Scholar]
- Loewith R, Hall MN. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics. 2011;189:1177–1201. doi: 10.1534/genetics.111.133363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD. Mutations in signal transduction proteins increase stress resistance and longevity in yeast, nematodes, fruit flies, and mammalian neuronal cells. Neurobiol Aging. 1999;20:479–486. doi: 10.1016/s0197-4580(99)00089-5. [DOI] [PubMed] [Google Scholar]
- Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006;126:257–268. doi: 10.1016/j.cell.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Baker DJ, d’Uscio LV, Mozammel G, Katusic ZS, van Deursen JM. Aging-associated vascular phenotype in mutant mice with low levels of BubR1. Stroke. 2007;38:1050–1056. doi: 10.1161/01.STR.0000257967.86132.01. [DOI] [PubMed] [Google Scholar]
- Maures TJ, Greer EL, Hauswirth AG, Brunet A. The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin-dependent manner. Aging Cell. 2011;10:980–990. doi: 10.1111/j.1474-9726.2011.00738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCay CM, Crowell MF, Maynard LA. The effect of retarded growth upon the length of life span and upon the ultimate body size. 1935. Nutrition. 1989;5:155–171. discussion 172. [PubMed] [Google Scholar]
- McColl G, Killilea DW, Hubbard AE, Vantipalli MC, Melov S, Lithgow GJ. Pharmacogenetic analysis of lithium-induced delayed aging in Caenorhabditis elegans. J Biol Chem. 2008;283:350–357. doi: 10.1074/jbc.M705028200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord RP, Nazario-Toole A, Zhang H, Chines PS, Zhan Y, Erdos MR, Collins FS, Dekker J, Cao K. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013;23:260–269. doi: 10.1101/gr.138032.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick MA, Delaney JR, Tsuchiya M, Tsuchiyama S, Shemorry A, Sim S, Chou AC, Ahmed U, Carr D, Murakami CJ, et al. A Comprehensive Analysis of Replicative Lifespan in 4,698 Single-Gene Deletion Strains Uncovers Conserved Mechanisms of Aging. Cell Metab. 2015;22:895–906. doi: 10.1016/j.cmet.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick MA, Mason AG, Guyenet SJ, Dang W, Garza RM, Ting MK, Moller RM, Berger SL, Kaeberlein M, Pillus L, et al. The SAGA histone deubiquitinase module controls yeast replicative lifespan via Sir2 interaction. Cell Rep. 2014;8:477–486. doi: 10.1016/j.celrep.2014.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick MA, Tsai SY, Kennedy BK. TOR and ageing: a complex pathway for a complex process. Philos Trans R Soc Lond B Biol Sci. 2011;366:17–27. doi: 10.1098/rstb.2010.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald P, Maizi BM, Arking R. Chemical regulation of mid- and late-life longevities in Drosophila. Exp Gerontol. 2013;48:240–249. doi: 10.1016/j.exger.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358:592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Martin-Montalvo A, Mercken EM, Palacios HH, Ward TM, Abulwerdi G, Minor RK, Vlasuk GP, Ellis JL, Sinclair DA, et al. The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep. 2014;6:836–843. doi: 10.1016/j.celrep.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell. 2013;154:430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi S, Sanderson BW, Delventhal KM, Bradford WD, Staehling-Hampton K, Shilatifard A. A comprehensive library of histone mutants identifies nucleosomal residues required for H3K4 methylation. Nat Struct Mol Biol. 2008;15:881–888. doi: 10.1038/nsmb.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- Nawy T. Single-cell sequencing. Nat Methods. 2014;11:18. doi: 10.1038/nmeth.2771. [DOI] [PubMed] [Google Scholar]
- Ni Z, Ebata A, Alipanahiramandi E, Lee SS. Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell. 2012;11:315–325. doi: 10.1111/j.1474-9726.2011.00785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North BJ, Rosenberg MA, Jeganathan KB, Hafner AV, Michan S, Dai J, Baker DJ, Cen Y, Wu LE, Sauve AA, et al. SIRT2 induces the checkpoint kinase BubR1 to increase lifespan. EMBO J. 2014;33:1438–1453. doi: 10.15252/embj.201386907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton L, Fourcaudot M, Abdul-Ghani MA, Winnier D, Mehta FF, Jenkinson CP, Defronzo RA. Chromatin occupancy of transcription factor 7-like 2 (TCF7L2) and its role in hepatic glucose metabolism. Diabetologia. 2011;54:3132–3142. doi: 10.1007/s00125-011-2289-z. [DOI] [PubMed] [Google Scholar]
- O’Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol. 2010;17:1218–1225. doi: 10.1038/nsmb.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes CC, Smiraglia DJ, Plass C, Trasler JM, Robaire B. Aging results in hypermethylation of ribosomal DNA in sperm and liver of male rats. Proc Natl Acad Sci U S A. 2003;100:1775–1780. doi: 10.1073/pnas.0437971100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- Orzechowski Westholm J, Tronnersjo S, Nordberg N, Olsson I, Komorowski J, Ronne H. Gis1 and Rph1 regulate glycerol and acetate metabolism in glucose depleted yeast cells. PLoS One. 2012;7:e31577. doi: 10.1371/journal.pone.0031577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio FG, Varela I, Lara E, Puente XS, Espada J, Santoro R, Freije JM, Fraga MF, Lopez-Otin C. Nuclear envelope alterations generate an aging-like epigenetic pattern in mice deficient in Zmpste24 metalloprotease. Aging Cell. 2010;9:947–957. doi: 10.1111/j.1474-9726.2010.00621.x. [DOI] [PubMed] [Google Scholar]
- Partridge L, Alic N, Bjedov I, Piper MD. Ageing in Drosophila: the role of the insulin/Igf and TOR signalling network. Exp Gerontol. 2011;46:376–381. doi: 10.1016/j.exger.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedruzzi I, Burckert N, Egger P, De Virgilio C. Saccharomyces cerevisiae Ras/cAMP pathway controls post-diauxic shift element-dependent transcription through the zinc finger protein Gis1. EMBO J. 2000;19:2569–2579. doi: 10.1093/emboj/19.11.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegoraro G, Kubben N, Wickert U, Gohler H, Hoffmann K, Misteli T. Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol. 2009;11:1261–1267. doi: 10.1038/ncb1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleg S, Feller C, Forne I, Schiller E, Sevin DC, Schauer T, Regnard C, Straub T, Prestel M, Klima C, et al. Life span extension by targeting a link between metabolism and histone acetylation in Drosophila. EMBO Rep. 2016;17:455–469. doi: 10.15252/embr.201541132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendas AM, Zhou Z, Cadinanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodriguez F, Tryggvason K, et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet. 2002;31:94–99. doi: 10.1038/ng871. [DOI] [PubMed] [Google Scholar]
- Platt JM, Ryvkin P, Wanat JJ, Donahue G, Ricketts MD, Barrett SP, Waters HJ, Song S, Chavez A, Abdallah KO, et al. Rap1 relocalization contributes to the chromatin-mediated gene expression profile and pace of cell senescence. Genes Dev. 2013;27:1406–1420. doi: 10.1101/gad.218776.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers RW, 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–184. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokocimer M, Barkan R, Gruenbaum Y. Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell. 2013;12:533–543. doi: 10.1111/acel.12070. [DOI] [PubMed] [Google Scholar]
- Pu M, Ni Z, Wang M, Wang X, Wood JG, Helfand SL, Yu H, Lee SS. Trimethylation of Lys36 on H3 restricts gene expression change during aging and impacts life span. Genes Dev. 2015;29:718–731. doi: 10.1101/gad.254144.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarrie JK, Riabowol KT. Murine models of life span extension. Sci Aging Knowledge Environ. 2004;2004:re5. doi: 10.1126/sageke.2004.31.re5. [DOI] [PubMed] [Google Scholar]
- Rai TS, Cole JJ, Nelson DM, Dikovskaya D, Faller WJ, Vizioli MG, Hewitt RN, Anannya O, McBryan T, Manoharan I, et al. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014;28:2712–2725. doi: 10.1101/gad.247528.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel CG, Dowen RH, Lourenco GF, Kirienko NV, Heimbucher T, West JA, Bowman SK, Kingston RE, Dillin A, Asara JM, et al. DAF-16 employs the chromatin remodeller SWI/SNF to promote stress resistance and longevity. Nat Cell Biol. 2013;15:491–501. doi: 10.1038/ncb2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogina B, Helfand SL, Frankel S. Longevity regulation by Drosophila Rpd3 deacetylase and caloric restriction. Science. 2002;298:1745. doi: 10.1126/science.1078986. [DOI] [PubMed] [Google Scholar]
- Rohde JR, Cardenas ME. The tor pathway regulates gene expression by linking nutrient sensing to histone acetylation. Mol Cell Biol. 2003;23:629–635. doi: 10.1128/MCB.23.2.629-635.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero C, Metter EJ, Melenovsky V, Cherubini A, Najjar SS, Ble A, Senin U, Longo DL, Ferrucci L. High basal metabolic rate is a risk factor for mortality: the Baltimore Longitudinal Study of Aging. J Gerontol A Biol Sci Med Sci. 2008;63:698–706. doi: 10.1093/gerona/63.7.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rytinki MM, Lakso M, Pehkonen P, Aarnio V, Reisner K, Perakyla M, Wong G, Palvimo JJ. Overexpression of SUMO perturbs the growth and development of Caenorhabditis elegans. Cell Mol Life Sci. 2011;68:3219–3232. doi: 10.1007/s00018-011-0627-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarg B, Koutzamani E, Helliger W, Rundquist I, Lindner HH. Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J Biol Chem. 2002;277:39195–39201. doi: 10.1074/jbc.M205166200. [DOI] [PubMed] [Google Scholar]
- Satoh A, Brace CS, Rensing N, Cliften P, Wozniak DF, Herzog ED, Yamada KA, Imai S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013;18:416–430. doi: 10.1016/j.cmet.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005;11:440–445. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder EA, Raimundo N, Shadel GS. Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab. 2013;17:954–964. doi: 10.1016/j.cmet.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Dang W, Donahue G, Dai J, Dorsey J, Cao X, Liu W, Cao K, Perry R, Lee JY, et al. H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev. 2015;29:1362–1376. doi: 10.1101/gad.263707.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27:1787–1799. doi: 10.1101/gad.223834.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaranarayanan P, Mendoza-Parra MA, Walia M, Wang L, Li N, Trindade LM, Gronemeyer H. Single-tube linear DNA amplification (LinDA) for robust ChIP-seq. Nat Methods. 2011;8:565–567. doi: 10.1038/nmeth.1626. [DOI] [PubMed] [Google Scholar]
- Sherman HC, Campbell HL. Rate of Growth and Length of Life. Proc Natl Acad Sci U S A. 1935;21:235–239. doi: 10.1073/pnas.21.5.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimi T, Butin-Israeli V, Adam SA, Hamanaka RB, Goldman AE, Lucas CA, Shumaker DK, Kosak ST, Chandel NS, Goldman RD. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011;25:2579–2593. doi: 10.1101/gad.179515.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006;103:8703–8708. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebold AP, Banerjee R, Tie F, Kiss DL, Moskowitz J, Harte PJ. Polycomb Repressive Complex 2 and Trithorax modulate Drosophila longevity and stress resistance. Proc Natl Acad Sci U S A. 2010;107:169–174. doi: 10.1073/pnas.0907739107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Singhal RP, Mays-Hoopes LL, Eichhorn GL. DNA methylation in aging of mice. Mech Ageing Dev. 1987;41:199–210. doi: 10.1016/0047-6374(87)90040-6. [DOI] [PubMed] [Google Scholar]
- Slack C, Werz C, Wieser D, Alic N, Foley A, Stocker H, Withers DJ, Thornton JM, Hafen E, Partridge L. Regulation of lifespan, metabolism, and stress responses by the Drosophila SH2B protein, Lnk. PLoS Genet. 2010;6:e1000881. doi: 10.1371/journal.pgen.1000881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeal T, Claus J, Kennedy B, Cole F, Guarente L. Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell. 1996;84:633–642. doi: 10.1016/s0092-8674(00)81038-7. [DOI] [PubMed] [Google Scholar]
- Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, Wang H, Le T, Faull KF, Chen R, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014;14:673–688. doi: 10.1016/j.stem.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science. 2001;292:107–110. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- Tissenbaum HA. Genetics, life span, health span, and the aging process in Caenorhabditis elegans. J Gerontol A Biol Sci Med Sci. 2012;67:503–510. doi: 10.1093/gerona/gls088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquero A, Reinberg D. Calorie restriction and the exercise of chromatin. Genes Dev. 2009;23:1849–1869. doi: 10.1101/gad.1807009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012;122:253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren LA, Rossi DJ, Schiebinger GR, Weissman IL, Kim SK, Quake SR. Transcriptional instability is not a universal attribute of aging. Aging Cell. 2007;6:775–782. doi: 10.1111/j.1474-9726.2007.00337.x. [DOI] [PubMed] [Google Scholar]
- Weindruch R, Walford RL. Dietary restriction in mice beginning at 1 year of age: effect on life-span and spontaneous cancer incidence. Science. 1982;215:1415–1418. doi: 10.1126/science.7063854. [DOI] [PubMed] [Google Scholar]
- Westendorp RG, Kirkwood TB. Human longevity at the cost of reproductive success. Nature. 1998;396:743–746. doi: 10.1038/25519. [DOI] [PubMed] [Google Scholar]
- Wood JG, Hillenmeyer S, Lawrence C, Chang C, Hosier S, Lightfoot W, Mukherjee E, Jiang N, Schorl C, Brodsky AS, et al. Chromatin remodeling in the aging genome of Drosophila. Aging Cell. 2010;9:971–978. doi: 10.1111/j.1474-9726.2010.00624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Zhang G, Huang H, Liu D, Cheng Y, Liu X, Zhang W, Yin R, Zhang D, Zhang P, Liu J, et al. N6-methyladenine DNA modification in Drosophila. Cell. 2015;161:893–906. doi: 10.1016/j.cell.2015.04.018. [DOI] [PubMed] [Google Scholar]
- Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D’Amico D, Ropelle ER, Lutolf MP, Aebersold R, et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352:1436–1443. doi: 10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]
- Zhang R, Chen W, Adams PD. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol. 2007;27:2343–2358. doi: 10.1128/MCB.02019-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Xu BC, Maheshwari HG, He L, Reed M, Lozykowski M, Okada S, Cataldo L, Coschigamo K, Wagner TE, et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse) Proc Natl Acad Sci U S A. 1997;94:13215–13220. doi: 10.1073/pnas.94.24.13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: Epigenetic pathways of aging and longevity in different model organisms
Supplemental Table 2- Protocols for aging research