

SUMMARY
The RNAi pathway provides both innate immunity and efficient gene-knockdown tools in many eukaryotic species, but curiously, not in zebrafish. We discovered that RNAi is less effective in zebrafish at least partly because Argonaute2-catalyzed mRNA slicing is impaired. This defect is due to two mutations that arose in an ancestor of most teleost fish, implying that most fish lack effective RNAi. Despite lacking efficient slicing activity, these fish have retained the ability to produce miR-451, a microRNA generated by a cleavage reaction analogous to slicing. This ability is due to a G–G mismatch within the fish miR-451 precursor, which substantially enhances its cleavage. An analogous G–G mismatch (or sometimes also a G–A mismatch) enhances target slicing, despite disrupting seed pairing important for target binding. These results provide a strategy for restoring RNAi to zebrafish and reveal unanticipated opposing effects of a seed mismatch with implications for mechanism and guide-RNA design.
Graphical abstract
INTRODUCTION
Diverse RNA-silencing pathways play important roles in transposon silencing, viral defense, heterochromatin formation, and posttranscriptional repression of cellular genes (Tomari and Zamore, 2005; Malone and Hannon, 2009). In the simplest of these pathways, RNA interference (RNAi), a Dicer endonuclease cleaves long, double-stranded RNA (dsRNA) into small interfering RNAs (siRNAs) that associate with an Argonaute (Ago) protein to guide the Ago-catalyzed slicing of transcripts with extensive pairing to the siRNA. The RNAi pathway arose early in eukaryotic evolution and has been retained by most eukaryotic lineages (Shabalina and Koonin, 2008). Some lineages also have derivative silencing pathways that are more elaborate and involve other types of guide RNAs, such as Piwi-interacting RNAs (piRNAs), which derive from single-stranded RNA rather than dsRNA (Weick and Miska, 2014; Iwasaki et al., 2015), or microRNAs (miRNAs), which derive from short hairpins rather than long dsRNA (Bartel, 2004). Despite their differences, the RNA-silencing pathways have each retained at their core a silencing complex that contains a short (20–32-nt) RNA associated with an Argonaute homolog. Within this complex, the RNA provides sequence specificity through direct pairing with target transcripts, and the Argonaute homolog either slices the target precisely between the nucleotides that pair to residues 10 and 11 of the guide RNA (Tuschl, 2001; Song et al., 2004) or recruits other proteins to promote other types of repression (Weick and Miska, 2014; Iwasaki et al., 2015; Jonas and Izaurralde, 2015).
The miRNA pathway is the dominant RNA-silencing pathway of mammalian somatic cells. Indeed, most cellular mRNAs are conserved regulatory targets of conserved mammalian miRNAs (Friedman et al., 2009). The miRNA silencing complex targets these mRNAs at sites that fall primarily in 3′ untranslated regions (3′ UTRs) and perfectly pair to nucleotides 2-7 of the miRNA, known as the miRNA seed (Bartel, 2009). Pairing to the seed region is insufficient to trigger slicing of the mRNA target, and repression is instead achieved primarily through the recruitment of factors that accelerate poly(A)-tail shortening (Jonas and Izaurralde, 2015). Thus, in this non-slicing mode of repression, the dominant effect of miRNAs depends on the consequences of the tail shortening, which change during development; i.e., in pre-gastrulation embryos, this tail shortening decreases translational efficiency, whereas later in development, it decreases mRNA stability (Subtelny et al., 2014).
Although the vast majority of miRNA regulatory sites in mammalian mRNAs have little more than seed pairing, some have the extensive complementarity required for Argonaute-catalyzed slicing. For example, in mouse embryos, miR-196 directs the cleavage of the HoxB8 3′ UTR at a site that has near-perfect complementarity to miR-196 and is conserved throughout most vertebrate species, including zebrafish (Yekta et al., 2004). To date, however, only 21 cleavage targets of mammalian miRNAs have been found (Davis et al., 2005; Shin et al., 2010; Hansen et al., 2011). Moreover, despite a high degree of homology among the four mammalian Argonaute proteins (Ago1, Ago2, Ago3, and Ago4), only Ago2 has retained slicing activity (Liu et al., 2004; Meister et al., 2004). Restoring activity to the other three paralogs requires a combination of changes that either restore the residues of the DEDH catalytic tetrad within the PIWI domain (Ago1 and Ago4), restore two structural elements (NTI and NTII) in the N domain (Ago1, Ago3, and Ago4), restore a short sequence cluster in the PIWI domain (Ago1), or remove a short insertion close to the glutamate of the catalytic center (Ago4) (Faehnle et al., 2013; Hauptmann et al., 2013; Nakanishi et al., 2013; Hauptmann et al., 2014).
In addition its requirement for slicing of rare, extensively paired miRNA targets, the ability of Ago2 to cleave RNA is required for the unusual biogenesis of miR-451, a miRNA conserved among vertebrates (Cheloufi et al., 2010; Cifuentes et al., 2010; Yang et al., 2010). Most metazoan miRNAs are produced from the successive cleavage by Drosha and Dicer, two endonucleases with dual RNase III domains (Kim, 2005). Drosha first cleaves both strands near the base of the stem to liberate the pre-miRNA hairpin from the primary transcript, and then Dicer cleaves both strands near the loop to generate the miRNA duplex, which contains the mature miRNA paired with 2-nt 3′ overhangs to an RNA segment from the other arm of the hairpin. This duplex is then loaded into Ago such that the miRNA strand ultimately becomes the guide RNA, and the other strand is discarded. In mammals, fish, and presumably other vertebrate species, miR-451 biogenesis is unusual in that the pre-miR-451 hairpin, with a stem of only 17 bp, is too short to be cleaved by Dicer and is instead loaded into Ago, which cleaves the strand opposite the miRNA strand in an activity analogous to mRNA slicing (Cheloufi et al., 2010; Cifuentes et al., 2010; Yang et al., 2010). Following this cleavage, 3′ exonucleolytic resection generates the mature miR-451 miRNA (Yoda et al., 2013). Because miR-451 activity is required for proper erythropoiesis (Patrick et al., 2010; Rasmussen et al., 2010), mice with Ago2 mutations that abrogate slicing are anemic, as are fish lacking the full-length protein (Cheloufi et al., 2010; Cifuentes et al., 2010).
In some mammals, Ago2-catalzed slicing also plays a critical role in the RNAi pathway. Although this pathway is not typically found in somatic cells, endogenous siRNAs are observed in certain mouse cells, including oocytes, embryonic stem cells, and male germ cells (Babiarz et al., 2008; Tam et al., 2008; Watanabe et al., 2008; Song et al., 2011). The pathway is important in mouse oocytes, in that disrupting the Ago2 active site desilences transposon expression, causing meiotic defects and female sterility (Stein et al., 2015). However, whether RNAi plays such a critical role in other mammals is unclear, as the Dicer isoform primarily responsible for the production of murine siRNAs does not appear to be present outside of the Muridae family (Flemr et al., 2013).
Regardless of why Ago2 has retained slicing activity, be it to cleave a few miRNA targets, to enable miR-451 biogenesis, or to perform RNAi-mediated transposon control, the widespread presence of this activity in mammalian cells has greatly benefited biomedical research. Indeed, the ability of artificial siRNAs to direct mRNA slicing, discovered 16 years ago (Elbashir et al., 2001a), has transformed the way that biologists study mammalian gene function. The reason that these artificial siRNA duplexes are so effective is that they resemble endogenous miRNA duplexes and thereby become incorporated into the Ago2 silencing complex to direct the slicing of target mRNAs. For previously unknown reasons, however, RNAi is not generally an effective tool for gene-knockdown experiments in zebrafish (Oates et al., 2000; Mangos et al., 2001; Zhao et al., 2001; Gruber et al., 2005; Kelly and Hurlstone, 2011). Perhaps as a result, morpholino antisense reagents have been a much more popular choice for posttranslational gene-knockdown in fish.
We discovered one reason why RNAi is generally ineffective in zebrafish: two point substitutions that apparently occurred in a teleost ancestor ~0.3 billion years ago greatly diminish the slicing activity of zebrafish Ago2 (drAgo2). The crippling effect of these substitutions raised the question of how these fish are able to produce sufficient miR-451, which requires Ago2-catalyzed cleavage for its biogenesis. When answering this question, we found that a G–G mismatch involving position 6 of the miRNA substantially enhances both the cleavage of fish pre-miR-451 and the slicing of bound target transcripts. Our results indicate how RNAi might be restored to zebrafish and reveal an unanticipated feature of guide-RNA pairing, showing that non-Watson–Crick seed geometry is optimal for slicing bound target.
RESULTS
Inefficient Slicing in Zebrafish
Although miR-196–directed slicing at the extensively paired site within the HoxB8 mRNA is readily detected in mouse embryos (Yekta et al., 2004), analogous efforts to detect slicing at the orthologous site within HoxB8a were unsuccessful in zebrafish embryos (S. Yekta & D.P.B., unpublished data). When considering this result together with the ineffectiveness of RNAi as a gene-knockdown tool in zebrafish, we decided to investigate the slicing ability of zebrafish Argonaute2 (drAgo2). We first assayed for miR-430–directed slicing in zebrafish embryos (Figure 1A). Capped RNA with a single site perfectly complementarity to the dominant isoform of miR-430 was injected into single-cell zebrafish embryos. Embryos were then harvested at 4 hours post-fertilization (4 hpf), a stage at which miR-430 dominates the endogenous miRNA pool, and total RNA was extracted and analyzed on RNA blots. No slicing was detected, even when an mRNA encoding additional drAgo2 was co-injected into the one-cell embryo (Figure 1B). In contrast, slicing was readily detected when mRNA for human Ago2 (hsAGO2) was co-injected, provided that the mRNA did not have mismatches at the cleavage site (mismatch), confirming that the conditions within the embryo were conducive to authentic slicing (Figure 1B). Additional experiments confirmed that another Ago paralog had not taken over slicing activity in zebrafish (Figure S1A).
Figure 1. Ineffective Slicing by drAgo2 in Zebrafish Embryos.
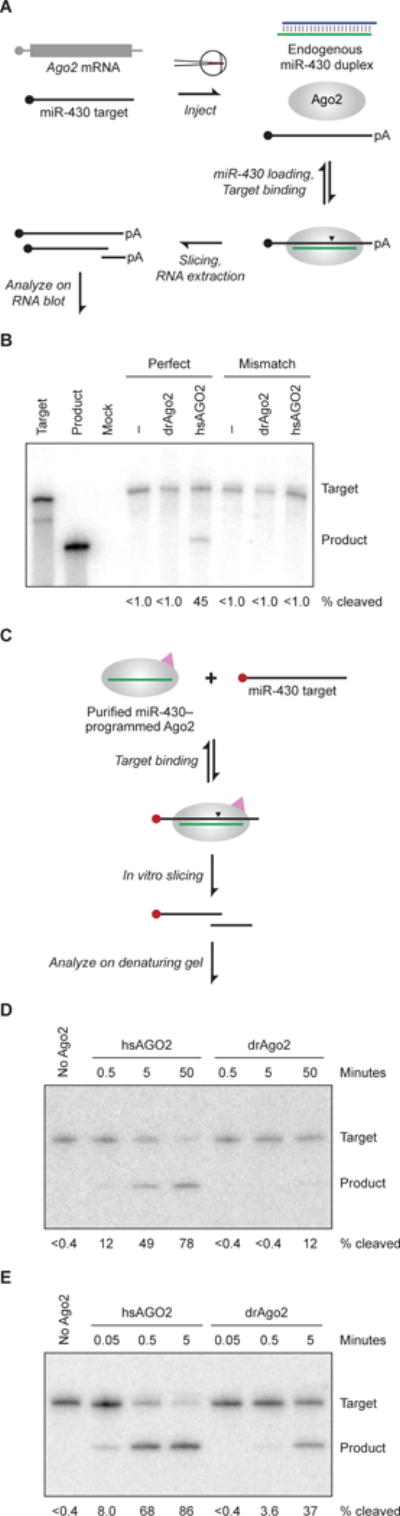
(A) Schematic of the in vivo miR-430–guided slicing assay. Capped target RNA is injected into zebrafish embryos at the one-cell stage, where it becomes polyadenylated (pA) and encounters endogenous miR-430 loaded into either endogenous Ago2 or protein translated from a co-injected Ago2 mRNA. To the extent that Ago2 is capable, it slices the target at the site indicted (arrowhead). Injected embryos develop until 4 hpf, at which point RNA is extracted and remaining target and sliced product are resolved and detected on an RNA blot.
(B) Slicing activities of zebrafish and human proteins in zebrafish embryos. Shown is an RNA blot probing for an injected target with or without a perfectly complementary miR-430 site (perfect and mismatch, respectively) and with or without co-injection of mRNA for the indicated protein. The left two lanes are uninjected standards representing the full-length target and sliced product. Mock is an uninjected negative control. Below each lane is the percentage of total signal (target plus product bands) represented by product (% cleaved).
(C) Schematic of the in vitro slicing assay, which uses Ago2 that was affinity-purified based on both its association with a specific miRNA (miR-430 in this example, green line) and its 3X-FLAG tag (pink triangle). After binding to the purified complex, cap-labeled (red filled circle) target is sliced at the site indicated (arrowhead), and labeled product is resolved from target on a denaturing gel.
(D) In vitro slicing activities of human and zebrafish Ago2 proteins programed with miR-430. Shown is a denaturing gel resolving cap-labeled target and product after incubation of limiting target (0.1 nM) with the indicated Ago2 protein (1.0 nM) for the indicated time. Below each lane is the percentage of target converted to product, reporting the mean from two experiments.
(E) In vitro slicing activities of human and zebrafish Ago2 proteins programmed with miR-1. Below each lane is the percentage of target converted to product. Otherwise, this panel is as in (D).
See also Figure S1.
Although our assay within zebrafish embryos had the advantage of examining slicing under physiological conditions, the in vivo setting of the developing embryo, with its large number of seed-matched miR-430 targets, its unknown amount of loaded Ago2 that varied over time, and its dynamic nuclease activities, prevented quantitative analysis of slicing kinetics. Therefore, to supplement the in vivo analyses, we adapted the protocol of Flores-Jasso et al. (2013) to purify the different Ago2 proteins loaded with miR-430 and then measured their ability to slice a cap-labeled substrate in vitro (Figure 1C). To isolate the slicing step from the substrate-binding and product-release steps, we monitored single-turnover reactions in which miR-430–programed human or zebrafish Ago2 was in 10-fold excess over the slicing substrate (1.0 and 0.1 nM, respectively) and in even greater excess over the enzyme–substrate dissociation constant (KD), expected to be in the low-picomolar range (Wee et al., 2012). In this more sensitive assay, drAgo2-catalyzed slicing was detectable but only ~1% as rapid as hsAGO2-catalyzed slicing (Figure 1D).
These results mirrored our earlier, unsuccessful efforts to detect endogenous miR-196–directed slicing at the extensively paired HoxB8a mRNA in zebrafish, mentioned above. However, they seemed at odds with previous reports of slicing activity observed in zebrafish embryos injected with a miR-1 duplex and complementary substrate (Giraldez et al., 2005; Cifuentes et al., 2010). To explore this potential discrepancy, we replicated the previous miR-1 experiment using the same reagents and approach. Capped RNA with three perfectly paired miR-1 sites was co-injected with miR-1 duplex into one-cell embryos, and then at 4 hpf, RNA was extracted and analyzed on RNA blots. Our results were consistent with those previously reported, in that a miR-1–dependent product appeared, which migrated at a size expected for cleavage at one of the three sites (Figure S1B). This product accumulation substantially increased when hsAGO2 mRNA was also co-injected, which confirmed that the product was of the size expected for Ago2-catalyzed slicing.
We do not know which features of this previous experimental regime enabled detection of endogenous drAgo2-catalyzed slicing in embryos. Perhaps it helped to have three miRNA sites rather than one, or relatively large quantities of injected miRNA and target, or perhaps inherent differences between the two miRNAs, miR-430 and miR-1, made this setup less sensitized to differences in slicing activity. To test whether the large difference between the activities of the human and zebrafish proteins was still observed when using the miR-1 guide, we purified miR-1–programed hsAGO2 and miR-1–programed drAgo2 and compared their activities in the single-turnover in vitro slicing assay (Figure 1C). With the miR-1 guide, drAgo2-catalyzed slicing was ~3% as rapid as hsAGO2-catalyzed slicing (Figure 1E, Figure S1C). Thus, regardless of the guide RNA, zebrafish Ago2 slicing activity was substantially reduced compared to that of the human protein. In addition to revealing this difference between to two proteins, the quantitative in vitro assays revealed a difference between the two miRNA–target pairs, showing that miR-1 directed slicing of its target was ≥ 10-fold more rapid than miR-430 directed slicing of its target. This type of difference helps to explain why drAgo2-catalyzed slicing of a miR-1 target was readily detected in fish embryos, whereas drAgo2-catalyzed slicing of a miR-430 target was not.
Two Substitutions in a Teleost Ancestor Explain the Loss of Efficient Slicing
A search for differences that might explain the loss of efficient drAgo2-catalyzed slicing started with the observation that drAgo2 and hsAGO2 differ mainly in their amino-terminal (N) domains and the knowledge that changes in the N domains explained the loss of slicing activity of hsAGO1, hsAGO3, and hsAGO4 (Faehnle et al., 2013; Hauptmann et al., 2013; Hauptmann et al., 2014). However, when we swapped the N domains of drAgo2 and hsAGO2 and examined the activity of these chimeric proteins using our assay for miR-430–guided slicing, drAgo2 with a human N domain was not substantially better at slicing, and hsAGO2 with the zebrafish N domain had no worse activity, indicating that differences outside the drAgo2 N domain were inhibiting slicing activity (Figure S2A–B).
To search for differences outside the N domain that might explain the loss of efficient slicing, we compared Ago2 sequences from 11 vertebrate species. Assuming the most parsimonious evolutionary scenario, in which 1) efficient slicing was lost only once in the vertebrate clade, and 2) mammals have retained the ancestral slicing activity that is present in invertebrates and throughout most eukaryotes, we surmised that efficient slicing must have been lost at some point in the jawed-fish lineage that gave rise to zebrafish, after the common ancestor of humans and zebrafish (Figure 2A, highlighted lineage of cladogram). By this reasoning, any substitution that compromised slicing in zebrafish would be at a residue that is identical in lamprey and the Sarcopterygii (ceolacanth and tetrapods) and different in zebrafish. These criteria narrowed the number of candidate substitutions to 20, all of which imparted conservative amino acid changes, and most of which were at residues on the surface of the protein. Of the three at interior residues, the two best candidates for explaining the loss of efficient slicing were near the active site (Figure 2B). Indeed, one changed the active-site glutamate (E), previously found to complete a DEDH catalytic tetrad (Nakanishi et al., 2012); in zebrafish and other representatives of the teleost clade, this E changed to an aspartate (D). The second changed a nearby phenylalanine (F) to tyrosine (Y), and this change also occurred in all representatives of the teleost clade.
Figure 2. Two Point Substitutions that Explain the Ineffective Slicing by drAgo2.

(A) Comparative analysis of drAgo2 and its orthologs in 10 other vertebrate species. The cladogram shows the evolutionary relationships between the species (left), and the sequence alignment highlights differences within a short region of the Ago2 PIWI domain (right). Segments of the jawed-fish lineage originally under consideration for the loss of drAgo2 slicing activity are highlighted (red) in the cladogram. All residues that vary among these species are in bold, and those identical in Sarcopterygii and lamprey but different in zebrafish are in blue. The two substitutions that were the primary candidates for conferring the loss of slicing are shaded in red in the alignment, with the most parsimonious timing of their occurrence indicated with a red asterisk in the cladogram.
(B) Structure of the Ago2 active-site residues and selected neighboring residues, modeling the amino acid changes (E-to-D and F-to-Y) that explain the loss of efficient slicing in zebrafish. Residues were modeled within the context of the hsAGO2 structure (Schirle and MacRae, 2012), substituting the ancestral residues of the human protein (top) with those of zebrafish (bottom). The residues of the catalytic tetrad, including the active-site E that changes to a D, are in green. Also shown is part of the hydrogen-bond network (dashed lines) that positions the E of the ancestral active-site and involves residues shown in blue (Nakanishi et al., 2012). The co-varying F-to-Y residue is in purple, with a potential additional hydrogen bond also shown, which might perturb the hydrogen-bond network in zebrafish.
(C) The effect of restoring the ancestral residues on the slicing activity of drAgo2 in zebrafish embryos. Otherwise, this panel is as in Figure 1B. The graph plots mean values from three experiments (error bars, standard deviation).
(D) The effect of restoring the ancestral residues on the slicing activity of drAgo2 in vitro. Below each lane is the percentage of target converted to product, reporting the mean from three experiments. Otherwise, this panel is as in Figure 1D. Results for hsAGO2 (grey), drAgo2 (blue), drAgo2D–E (red), drAgo2Y–F (green), and drAgo2DY–EF (orange) are shown for three replicates (circle, squares, and triangles). The line for each substrate represents the best fit of the mean values to an exponential reaction course (considering only values in which less than half of the substrate had reacted), which generated the rate constants (k, shown ± 95% confidence intervals).
See also Figure S2 and S3.
To test the effect of these two substitutions, we made mRNAs that encoded drAgo2 proteins with the D and Y reverted back to their ancestral identities, confirmed comparable expression of these proteins in injected embryos (Figure S3A), and examined the ability of these proteins to slice a miR-430 target in embryos. Each of the single reversions (drAgo2D–E and drAgo2Y–F) conferred detectable slicing activity to the zebrafish protein, and the double reversion (drAgo2DY–EF) imparted activity approaching that of the human protein (Figure 2C). Moreover, the reciprocal human-to-zebrafish substitutions within hsAGO2 eliminated detectable miR-430–guided slicing activity (Figure S3B). Together, these results showed that the E-to-D and F-to-Y substitutions both contributed to the loss of efficient slicing in zebrafish embryos.
To provide more quantitative measurements of slicing activities, we turned to the in vitro slicing assay, monitoring single-turnover reactions with purified miR-430–programmed Ago2 variants. As in the embryo, each of the single reversions (drAgo2D–E and drAgo2Y–F) had improved slicing activity, and drAgo2 slicing activity approached that of hsAGO2 only when both key residues were reverted to their ancestral identities (drAgo2DY–EF) (Figure 2D). Confirming that activity depended on the drAgo2 active site, slicing was abolished when the first aspartate of the DEDH catalytic tetrad was changed to alanine (drAgo2D–A) (Figure 2D).
The E-to-D and F-to-Y substitutions, which together imparted this substantial diminution in slicing activity, are broadly distributed among teleost fish, which comprise most of the extant fish species. With the exception of a presumed D-to-E reversion in the Cichlidae family of Euteleosteomorpha, both the E-to-D and F-to-Y substitutions were present in all 29 teleost species examined (Figure S3C). These 29 species included all teleosts with sequenced genomes and fell within the three teleost subgroups that encompass the vast majority of the extant teleost species (Broughton et al., 2013). Because these substitutions did not extend to more basal jawed fish, represented by gar (Figure 2A), they presumably occurred approximately 300,000,000 years ago, in a common ancestor of most extant teleosts.
An Ancestral G–G Mismatch Within Pre-miR-451 Enhances Cleavage
The inefficiency of drAgo2-catalyzed slicing helps explain why endogenous slicing products have not been reported in zebrafish and why RNAi is described as an ineffective tool for knocking down gene expression in zebrafish (Oates et al., 2000; Mangos et al., 2001; Zhao et al., 2001; Gruber et al., 2005; Kelly and Hurlstone, 2011). The inefficiency of drAgo2-catalyzed slicing also suggested that the activity observed for shRNA expressed from a miR-30e backbone in zebrafish (Dong et al., 2009; De Rienzo et al., 2012) occurs primarily through the mRNA deadenlyation pathway typical of miRNA-mediated repression (Jonas and Izaurralde, 2015) rather than through Ago2-catalyzed slicing. This inefficient slicing was nonetheless unexpected because genetic analyses indicate that a reaction analogous to slicing is required for miR-451 biogenesis in zebrafish (Cifuentes et al., 2010), which prompted us to examine the ability of drAgo2 to process pre-miR-451. Accordingly, we developed an assay for pre-miR-451 binding and cleavage, in which mRNA for FLAG-tagged Ago2 was co-injected with 5′ end-labeled pre-miR-451 into single-cell embryos, and RNA co-purifying with Ago2 was then isolated and analyzed on a denaturing gel (Figure 3A). Consistent with the genetic results (Cifuentes et al., 2010), wild-type drAgo2 was able to bind and cleave pre-miR-451, although cleavage was not as efficient as that observed for hsAGO2 (Figure 3B). Interestingly, some mature miR-451 was also detected with Ago2 active-site mutants (Ago2D–A), perhaps the result of cleavage within the loop of the injected pre-miRNA by another endonuclease. However, the amount of cleaved pre-miR-451 associated with wild-type drAgo2 was substantially greater, indicating that drAgo2 was indeed able to cleave the pre-miRNA, albeit at lower efficiency than that observed for hsAGO2 (Figure 3B). As observed for target slicing, this lower efficiency of drAgo2-mediated pre-miR-451 cleavage was attributed to the E-to-D and F-to-Y substitutions found in zebrafish and other teleosts, in that activity for the constructs with single and double reversions of these substitutions approached that of hsAGO2 (Figure 3C, Figure S2C).
Figure 3. A G–G Mismatch that Promotes Maturation of Fish Pre-miR-451.
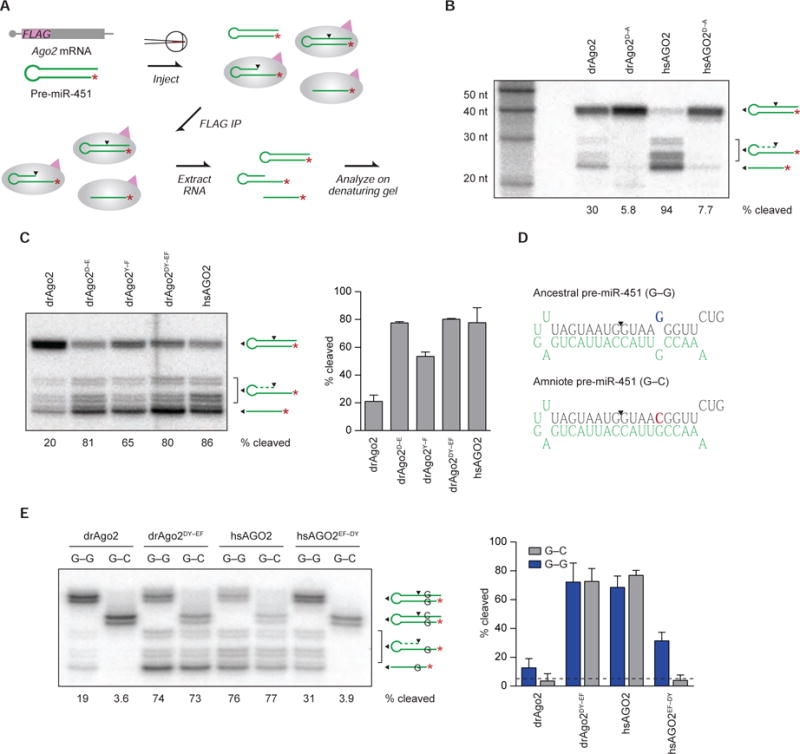
(A) Schematic of the in vivo assay for pre-miR-451 binding and cleavage. End-labeled (red asterisk) pre-miR-451 and mRNA encoding a FLAG-tagged Ago2 protein are co-injected into zebrafish embryos at the one-cell stage. As the embryo develops, some pre-miR-451 is loaded into tagged Ago2 and cleaved at the site indicated (arrowhead), and some cleaved RNA is further resected to generate mature miR-451. At 4 hpf, embryos are lysed, and Ago protein translated from the injected mRNA is immunoprecipitated (IP) based on affinity to the 3X-FLAG tag (pink triangle). Co-immunoprecipitating RNAs, including loaded pre-miR-451, mature miR-451, and processing intermediates, are isolated and analyzed on a denaturing gel.
(B) The ability of the zebrafish and human proteins to cleave pre-miR-451 in zebrafish embryos. Shown are results of binding-and-cleavage assays for the indicated proteins and their respective catalytically dead variants (D-to-A mutants). Each lane contained RNA co-immunoprecipitated from approximately 300 embryos. Below each lane is the percentage of the loaded pre-miR-451 that was cleaved (calculated using all cleaved species, including fully and incompletely resected cleavage products).
(C) The effects of restoring the ancestral residues to drAgo2 on pre-miR-451 binding and cleavage. Assays were as in (B), injecting mRNA for the indicated proteins. The graph plots mean values from two experiments (error bars, range).
(D) The ancestral and amniote pre-miR-451 sequences, indicating residues of the mature miR-451 guide RNA (green), the site of Ago2-catalyzed cleavage (arrowhead), and the G-to-C change (blue and red, respectively) that creates a G–C match with miRNA position 6.
(E) The effects of the position-6 G–C match on pre-miR-451 binding and cleavage. Assays were as in (B), co-injecting mRNA for the indicated Ago2 proteins with either end-labeled ancestral (G–G) or amniote (G–C) pre-miR-451. The graph plots mean values from two experiments (error bars, range), with the background level of activity indicated (dashed line), as determined from the cleavage observed for the active-site mutants in (B).
See also Figure S4 and S5.
The >30-fold difference between the miR-430–guided slicing activities of hsAGO2 and drAgo2 (Figures 1–2) seemed much greater than the difference between their respective pre-miR-451 cleavage activities (Figure 3B–C). Although we could not rule out the possibility that the smaller apparent difference for pre-miR-451 cleavage might be attributable to differences in the assays (as would be the case, if for instance the results for the more efficient constructs were beyond the dynamic range of the pre-miR-451 cleavage assay), we explored the more interesting possibility that the smaller apparent difference for pre-miR-451 cleavage might be attributable to differences between the substrates of the two types of reactions. Apart from the loop in pre-miR-451, the most prominent structural difference between the two substrates was at miRNA position 6, which was perfectly paired in the slicing substrate but formed a G–G mismatch in the zebrafish pre-miR-451 hairpin (Figure 3D).
Examination of the whole-genome alignments (Tyner et al., 2017) revealed that the G–G mismatch within pre-miR-451 has been conserved among the fish and amphibian species. However, G35 mutated to a C in an amniote ancestor that gave rise to reptiles, birds, and mammals (Figure S4), thereby changing the ancestral G–G mismatch to a G–C Watson–Crick match (Figure 3D), as occurs in our slicing substrate. Although C35 has been retained within most amniote lineages, it mutated again in some linages, most often to the U transition (at least nine times) but sometimes transverting back to a G (at least 3 times) (Figure S4). For example, all three possibilities were observed within primates, with humans and most other apes acquiring the U, gibbons retaining the C, and old-world monkeys reverting back to the G. The variability of this position among mammalian pre-miR-451 sequences has been previously noted, and structure-function studies of human pre-miR-451 show that changing U35 to either a G or an A slightly enhances miR-451 activity in HeLa cells (Yang et al., 2012). This increased activity corresponds to increased miR-451 accumulation, which is attributable to more efficient resection of cleaved pre-miR-451 (Yang et al., 2012). These experiments comparing the human G–U wobble to the G–G and G–A mismatches at positions 6 and 35, respectively, establish an interesting tolerance for mismatches at this position, raising the question of how the G–C Watson–Crick match, which is found in most amniotes but untested in previous studies, might compare with the ancestral G–G mismatch.
To answer this question, we tested ancestral and amniote pre-miR-451 structures in our assay for pre-miR-451 binding and cleavage and found that drAgo2 had a surprising preference for the ancestral G–G mismatch structure (Figure 3E). Indeed, although drAgo2 could bind the G–C structure, cleavage did not exceed the background level observed for the D-to-A active-site mutants (Figures 3B, E). Similar results were observed for the hsAGO2 with the zebrafish substitutions, hsAGO2EF–DY (Figure 3E), and when comparing to the pre-miR-451 hairpin with a C–G rather than a G–C pair at this position (Figure S5). Thus, for these slicing-impaired enzymes possessing the teleost E-to-D and F-to-Y substitutions, the benefit of the G–G mismatch appeared binary—either activity with the G–G mismatch or merely background activity with the G–C match. In contrast, for both repaired drAgo2 (drAgo2DY–EF) and wild-type hsAGO2, no advantage of the mismatch was observed, perhaps reflecting a limited dynamic range of this assay (Figure 3E).
The adequate cleavage of fish pre-miR-451 despite inefficient slicing of perfectly matched targets leads us to speculate that in most teleost fish the only biological role for Ago2 catalytic activity is to produce miR-451. In this scenario, the reason that the catalytically impaired Ago2 of most teleosts has been able to play this role is the presence of an ancestral G–G mismatch within pre-miR-451, which helps compensate for the impaired cleavage activity, allowing these fish to produce enough miR-451 to avoid erythropoiesis defects.
Specific Mismatches at miRNA Position 6 Enhance Slicing of Bound Target
Intrigued by the strong benefit of the G–G mismatch for drAgo2-mediated pre-miR-451 cleavage, we tested whether an analogous mismatch might also enhance target slicing. To isolate the slicing step from substrate binding and product release, we started with single-turnover reactions, in which miR-430–programed Ago2 was in 10-fold excess over the slicing substrate and in large excess over the expected KD’s (Figure 4A–B). hsAGO2 sliced the bound G–G substrate 3.1-fold more rapidly than it sliced the G–C substrate, whereas drAgo2 sliced the G–G substrate 4.8-fold more rapidly. When testing the other two possibilities across from a G at position 6 of the miR-430 guide, the G–G mismatch was also preferred over the G–A mismatch (3.3 and 7.8 fold for hsAGO2 and drAgo2, respectively) and the G–U wobble (3.0 and 3.7 fold for hsAGO2 and drAgo2, respectively), indicating that for this guide RNA slicing enhancement is specific to a G–G mismatch in both species (Figure 4A–B).
Figure 4. Effects of Mismatches on Slicing of Bound Target.

(A) The effects of position-6 mismatches on slicing by miR-430–programmed hsAGO2. In vitro slicing assays were as in Figure 1D, plotted as in Figure 2D.
(B) The effects of position-6 mismatches on slicing by miR-430–programmed drAgo2. Otherwise, this panel is as in (A).
(C) The effects of position-6 mismatches on slicing by miR-451–programmed hsAGO2. Otherwise, this panel is as in (A).
See also Figure S6.
To examine whether the preference for G–G mismatch was specific to position 6 of the guide, which is known to tolerate an abasic guide residue (Lee et al., 2015), we tested the same matched and mismatched possibilities to position 4 of the miR-430 guide, which is also a G. At this position, bound substrate with the G–C match was sliced most rapidly—at least 2-fold more rapidly than substrates with either a wobble or mismatch (Figure S6). Thus, the benefit of a G–G mismatch does not apply throughout the seed region.
To test whether the benefit of a G–G mismatch at position 6 occurs in other miRNA contexts, we examined substrates of miR-451–programmed hsAGO2. miR-451 and miR-430 have similar seed regions, with one difference being that the nucleotides immediately flanking position 6 are swapped (positions 5–7 of miR-451 are CGU, whereas those of miR-430 are UGC). As observed for miR-430–guided slicing, bound substrate with the G–G mismatch at position 6 was sliced more rapidly than those with the G–C match or G–U wobble (Figure 4C). The difference was that the bound substrate with the G–A mismatch was sliced as rapidly was that with the G–G mismatch. Thus, in some nearest-neighbor contexts, a G–A mismatch at position 6 can impart the same benefit as a G–G mismatch.
Previous kinetic analyses of Ago-catalyzed slicing reactions show that mismatches to the guide RNA enhance the rate of product release, thereby enhancing the rate of multiple-turnover slicing of substrates for which release of the fully matched product is rate limiting (Wee et al., 2012). To investigate whether the G–G mismatch to miR-430 might confer this additional, post-slicing rate enhancement, we examined its effect on hsAGO2-catalzyed multiple-turnover slicing. Indeed, as expected for rate-limiting product release, accumulation of product was biphasic, with an initial burst of rapid slicing corresponding to slicing of a stoichiometric amount of enzyme-bound substrate followed by a second phase corresponding to rate-limiting product release for subsequent enzyme turnover (Figure 5A, Figure S7). For the G–G substrate, the rate constant for the second, slower phase (k2) was 2.1-fold faster than that for the G–C substrate (k2 = 0.074 min−1 and 0.035 min−1, respectively), consistent with the idea that following slicing, dissociation of the 3′ cleavage product (which differed for the two substrates) was at least partially rate limiting, and the G–G mismatch enhanced this dissociation rate constant. As expected from the single-turnover results, the initial burst for the G–G substrate was also faster than that for the G–C substrate (Figure 5A). We observed a 3.6-fold enhancement for the G–G mismatch substrate during the initial burst (k1 = 0.94 min−1 and 0.26 min−1 for the G–G and G–C substrates, respectively), which resembled the 3.1-fold enhancement observed in the single-turnover reactions (Figure 4A and Figure 5A). To the extent that the effect of the G–G mismatch was somewhat greater in this burst phase than in the single-turnover regime, the difference can be attributed to the fundamental rate constants of the reaction and the relative concentrations of enzyme and substrate used in the single- versus multiple-turnover assays (Figure S7). These considerations imply that the 3.6-fold enhancement observed in the first phase of the multiple-turnover reaction with hsAGO2 and the 4.8-fold enhancement observed in the single-turnover reaction with drAgo2 best represent the degree to which the G–G mismatch confers enhanced slicing of bound substrate.
Figure 5. Effects of the G–G Mismatch on Target Association, Slicing, and Product Release.

(A) The effect of a position-6 G–G mismatch on multiple-turnover slicing by hsAGO2. These in vitro assays were as in Figure 4A, except substrate (5 nM) was in excess over AGO2 (0.5 nM). Results for the G–C matched (grey) and G–G mismatched (blue) substrates are shown, distinguishing the three replicates (circles, squares, triangles). The graph on the right shows results for the earliest time points (boxed in the graph on the left). The line for each substrate represents the best fit of the mean values to a biphasic reaction course (Wee et al., 2012), which generated the initial and steady-state rate constants (k1 and k2, respectively, shown ± 95% confidence intervals).
(B) Schematic of the in vitro competitive binding and cleavage assay with long (168-nt) and short (80-nt) cap-labeled miR-430 targets.
(C) The effect of the G–G mismatch on competitive binding and cleavage. These in vitro assays were as in Figure 5A, with substrates (2.5 nM each) in excess over hsAGO2 (0.5 nM). The percent of long and short substrates cleaved is shown below the gel. For each substrate pair, the fold difference observed between long and short substrates was normalized using the data from the left half of the gel to account for the differences observed for the same site in the long and short contexts, and these normalized fold differences are shown at the bottom.
(D) Minimal kinetic scheme for Ago2-catalyzed slicing, annotating for each step the effect of the position-6 G–G mismatch compared to the G–C match.
See also Figure S7.
Having found a surprising benefit of a G–G mismatch at miRNA position 6 when slicing of bound substrate (Figures 4 and 5A) and having confirmed that this mismatch also enhances release of bound product (Figure 5A), we turned to substrate association, a step in the slicing-reaction pathway for which a mismatch within the seed region is expected to be detrimental (Salomon et al., 2015). To confirm and quantify the presumed detrimental effect of the G–G mismatch on substrate association, we developed a competitive-cleavage assay, in which excess long (168-nt) and short (80-nt) cap-labeled slicing substrates—one with a perfectly paired site, the other with a G–G-mismatched site—were incubated with limiting miR-430–programmed Ago2 (Figure 5B). In this assay, the two substrates compete for Ago2 binding and complex formation, and these differences in association rates lead to differences in sliced product. As controls, short and long substrates with the same sites were tested. These controls revealed a slight preference for the longer version of each substrate and reiterated the observation of faster slicing of G–G substrates in the second phase of miR-430–guided multiple-turnover slicing (Figure 5C, left half of gel). In the experimental lanes, in which the two site types competed with each other for limiting programed hsAGO2, the G–C site was bound and sliced 2.4-fold more efficiently when it resided in the longer substrate and 1.9-fold more efficiently when it resided in the shorter substrate, indicating a 2.2-fold overall preference for the G–C site over the G–G site (Figure 5C, right half of gel). Because slicing is much faster than substrate release (Wee et al., 2012), and because the first time point provided ample time for slicing (30 min for reactions proceeding at ≥ 0.2 min−1), most of the molecules that bound to programed hsAGO2 were also sliced, and thus differences in slicing rates had a negligible effect on the proportions sliced at the time points of this experiment. Moreover, differences in product dissociation rates were also inconsequential because dissociation was not required for the product to be detected on the gel, and under the competitive conditions, any programed hsAGO2 that is freed upon product release will chose the next substrate molecule based on its relative association rate, irrespective of either the identity or dissociation rate of the recently released product. Thus, the observed 2.2-fold preference for slicing of the G–C site in this competitive assay was primarily attributable to correspondingly more rapid association of the G–C substrate compared to the G–G substrate.
In sum, our kinetic analyses of hsAGO2-catalyzed slicing show that particular mismatches to the seed of the miRNA can have opposing effects on binding and slicing. In addition to the anticipated effects on binding, in which a G–G mismatch at position 6 of the guide RNA slows substrate association and enhances produce release, this mismatch has an unanticipated effect on slicing, in which it enhances slicing of bound substrate. In the context of our sequences and conditions, the effects on substrate association and product release were each ~2-fold, and the effect on slicing of bound substrate was 3–5-fold (Figure 5D).
DISCUSSION
Our discovery that zebrafish lacks efficient slicing demonstrates that a vertebrate species can persist in the wild despite lacking effective RNAi, a powerful gene-silencing pathway that many other eukaryotic species deploy to silence viruses and transposons (Tomari and Zamore, 2005; Malone and Hannon, 2009). Indeed, the two point substitutions that confer this loss of effective RNAi appear to have occurred 300,000,000 years ago in a common ancestor of the sequenced teleost fish. This lineage includes most of the extant fish species—and indeed, most of the vertebrate species currently inhabiting the planet, yet this vertebrate lineage that has lost effective RNAi has not only persisted but thrived.
Perhaps the possession of alternative pathways to combat viruses and transposons has allowed the RNAi pathway to be lost without consequence. Alternatively, the cost of losing the pathway in teleosts might have been offset by a benefit. This type of cost–benefit tradeoff explains why the presence of RNAi is so variable among fungi: Losing RNAi imparts a cost of decreased protection against transposons but also imparts a benefit, in that it enables the acquisition and retention of Killer, a dsRNA element that encodes a toxin that kills neighboring cells that lack Killer (Drinnenberg et al., 2009; Drinnenberg et al., 2011). For fish, we can only speculate on the potential benefits of losing efficient slicing. One possibility is that it would confer resistance to polyoma viruses or other DNA viruses that produce miRNAs that direct slicing of complementary mRNAs transcribed from the opposite viral strand (Grundhoff and Sullivan, 2011).
Whether losing RNAi was essentially neutral or conferred a net benefit to the teleost lineage, the lack of efficient RNAi is clearly not a benefit for the use of zebrafish as a model organism to study the molecular basis of vertebrate development and physiology. Our identification of the two point substitutions that conferred the loss of efficient slicing in teleosts suggests how, with the use of modern gene-editing methods, this activity might be restored to zebrafish. The generation of a zebrafish line that possess efficient slicing activity might enable RNAi-based gene-knockdown tools in this model organism and would also reveal the consequences of regaining efficient slicing in a lineage that has not experienced it in 0.3 billion years.
The cost of losing efficient Ago2 catalytic activity was attenuated in teleosts because they retained the G–G mismatch within pre-miR-451, the precursor of a miRNA required for proper erythrocyte development. We found that this mismatch to position 6 of the miRNA enabled drAgo2-mediated pre-miR-451 cleavage; without the mismatch, pre-miR-451 cleavage was essentially abolished. Although maturation of the G–G mismatched pre-miR-451 within drAgo2 was not as rapid as that observed within hsAGO2, it did appear to be sufficient for adequate miR-451 to be produced within the timeframe of erythropoiesis. The unanticipated advantage of this mismatch to a seed nucleotide was also observed during Ago2-catalyzed slicing of bound target transcripts, which occurred 3–5-fold more rapidly for bound substrates containing the mismatch compared to those that were perfectly matched.
Many lines of evidence point to the strict preference for perfect Watson–Crick pairing to the miRNA seed during target binding (Bartel, 2009), and with no evidence to the contrary, this seed pairing, together with pairing to the midsection of the guide RNA, has been assumed to be also preferred for slicing of bound target. Our results reveal that in fact there is a tradeoff between the preferences for binding and those of the subsequent conformational and chemical steps required for slicing. Moreover, for miR-430–directed slicing, the post-chemistry advantage of the G–G mismatch, with its 2-fold more favorable product release, essentially negates the 2-fold disadvantage that this mismatch imparts on target association. Similar results are anticipated for this mismatch in the context of other guide RNAs, although more needs to be learned about the influence of neighboring base-pair identity, as illustrated by the effects of the position-6 G–A mismatch, which enhances slicing of the bound miR-451 substrate but not that of miR-430.
siRNAs and artificial miRNAs are important research tools for gene-knockdown studies, and siRNAs are showing promise in the clinic (Bobbin and Rossi, 2016). In the current design of these gene-knockdown tools, pairing to the last few nucleotides of the guide is considered unimportant (Elbashir et al., 2001b), as is pairing to the first nucleotide of the guide, which is bound to Ago2 in a configuration that prevents pairing to the mRNA (Ma et al., 2005; Parker et al., 2005). However, the remainder of the guide is typically designed to pair perfectly to the target mRNA. Knowledge that mismatches between the guide and target can enhance product dissociation rates and thereby potentially increase the multiple-turnover rate of slicing (Wee et al., 2012) is not typically exploited to improve these reagents, presumably out of concern that the benefits to multiple turnover would be offset by less efficient target binding and slicing. Our results revealing the tradeoff between pairing preferences for binding and slicing suggest that, depending on the relative importance of target association, target slicing, and product release, a suitable mismatch at position 6 might impart an overall benefit. This strategy for enhancing slicing and product release might be particularly useful for improving siRNAs with nucleotide modifications that protect them from nucleases, as these modifications often also enhance pairing stability, which could shift the balance with respect to the relative importance of target association, predicted to become less of a concern, and product release, predicted to become more of a concern.
The discovery that a G–G mismatch at position 6 of the guide enhances both pre-miR-451 cleavage and target slicing raises the question of whether other mismatches or wobbles at other seed positions might also enhance these activities. Thus far, we have not found a mismatch that confers a benefit at position 4, but within the context of miR-451, a G–A mismatch at position 6 also imparts a benefit. Another key question is how the G–G (or G–A) mismatch confers its benefit. The perturbed geometry or increased flexibility imparted by this mismatch presumably favors either the transition-state geometry of the active site or an on-pathway pre-chemistry conformational change. Potentially related to this question is that of how the identity of the guide RNA can have such a large influence on the slicing rate of bound substrate, which differed by >100 fold, depending upon whether the substrate was paired to miR-1 or to miR-451 (hsAGO-catalyzed slicing rates of 2.2 min−1 and 0.019 min−1, respectively; Figure S1C and Figure 4C). Now that these differences and the unanticipated tradeoff between the pairing preferences for binding and slicing are known, systematic biochemical and biophysical studies can be designed to take aim at these questions.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Bartel (dbartel@wi.mit.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293T cells were cultured in DMEM (VWR) with 10% FBS (Clonetech). All cells were cultured at 37°C with 5% CO2. Cell lines were of female origin.
METHOD DETAILS
Plasmids
All plasmids generated for this study are listed in the Key Resources Table and available at Addgene with maps and sequences. To construct Ago2 plasmids, the coding sequence of human or zebrafish Ago2 was inserted into the pCS2+ vector (RZPD). The sequence encoding the 3X-FLAG tag was then added downstream of the start codon by PCR-mediated insertion to generate pCS2+-FLAG3-hsAgo2 and pCS2+-FLAG3-drAgo2. To construct domain-swap plasmids, both the coding sequence of the N domain as well as the remaining domains of human or zebrafish Ago2 were separately amplified by PCR and then spliced together by overlap extension PCR. Point substitutions were introduced by PCR-based mutagenesis using QuikChange Lightning Multi Site-Directed Mutagenesis (Agilent) to generate plasmids encoding FLAG-tagged mutant Ago2 proteins. To construct plasmids used to generate the injected miR-430 target RNAs, the coding sequence of Zeocin was inserted into the pCS2+ vector. A single miR-430 site (perfect, mismatch, or G–G mismatch) was inserted downstream of the Zeocin sequence using QuikChange Lightning Multi Site-Directed Mutagenesis. To construct plasmids used to generate purified programed Ago2 complexes, the coding sequence of human or zebrafish Ago2 was inserted into the pcDNA3 vector (Invitrogen) and then the 3X-FLAG tag and point substitutions were introduced as described above.
In vivo slicing assay
Ago2 mRNAs and target RNAs were transcribed in vitro using mMESSAGE mMACHINE SP6 according to the manufacturer’s instructions (Life Technologies). The transcribed RNA was purified with RNeasy Mini (QIAGEN) according to the manufacturer’s instructions, ethanol precipitated, and stored in water at −80°C. One-cell embryos were injected with target RNA containing one miR-430 site (10 pg/embryo) with or without additional Ago2 mRNA (100 pg/embryo) in a volume of 1 nL (PLI-100 Plus Pico-Injector, Harvard Apparatus). For each condition, 50 embryos were injected. Embryos that developed to the sphere stage (which was approximately 4 hpf and when endogenous miR-430 peaks) were manually de-chorionated, pooled by condition, and placed in 1 mL TRI Reagent (Life Technologies) for RNA isolation. Isolated RNA was chloroform extracted, ethanol precipitated, and stored at −80°C prior to analysis on small-RNA blots. The in vivo assays for miR-1–guided slicing were performed as described (Giraldez et al., 2005; Cifuentes et al., 2010).
Small-RNA blot
Total RNA (2 μg) from each condition was denatured and resolved on a 5% urea-polyacrylamide gel. RNA was then electroblotted onto an Amersham Hybon-NX nylon membrane (VWR) and UV cross-linked at 254 nm. Membranes were pre-incubated with ULTRAhyb Ultrasensitive hybridization buffer (Life Technologies) at 68°C under rotation for 1 hour and then hybridized under the same conditions overnight with a body-labeled RNA probe complementary to the 5′ cleavage product. This RNA probe was transcribed in vitro using MAXIscript T7 RNA polymerase according to the manufacturer’s instructions (Life Technologies), replacing the UTP with [α-32P]UTP (PerkinElmer), and desalted with Micro Bio-Spin P-30 gel columns (BioRad). Radiolabeled RNA was purified on a 4% urea-polyacrylamide gel, eluted from gel slices in 0.3 M NaCl overnight at 4°C, and ethanol precipitated prior to incubation with the membrane. Membranes were then washed twice with low-stringency buffer (2X SSC and 0.1% SDS) for 5 min under rotation at 68°C, and once with high-stringency buffer (0.1X SSC and 0.1 % SDS) for 30 min under rotation at 68°C. The blots were then exposed to a phosphorimaging screen for 1–14 days. Signal was detected using the Typhoon FLA 7000 phosphorimager (GE Healthcare Life Sciences) and analyzed using the MultiGauge software (FujiFilm). RNA blots used to monitor miR-1–guided slicing were prepared following the NorthernMax-Gly Kit (Life Technologies) according to the manufacturer’s instructions. Total RNA (1.5 μg) from each condition was incubated with Glyoxal Loading Dye for 30 min at 50°C and resolved on a 1.5% agarose gel. RNA was then transferred onto a nylon membrane using a Whatman Nytran SuPerCharge TurboBlotter (Sigma) for 2 hours and UV cross-linked at 254 nm. Membranes were pre-incubated with ULTRAhyb-Oligo hybridization buffer (Life Technologies) at 42°C under rotation for 1 hour and then hybridized under the same conditions overnight with an end-labeled DNA probe complementary to the 5′ cleavage product. This DNA probe (IDT) (Table S1) was purified on a urea-polyacrylamide gel, phosphorylated with [γ-32P]ATP using T4 polynucleotide kinase (New England BioLabs), desalted with Micro Bio-Spin P-30 Gel Columns (BioRad), and gel purified again. All subsequent steps were as above.
miRNA Duplexes
Synthetic RNA oligonucleotides (IDT) representing the miR-430b and miR-1 duplex (Table S1) were purified on a 15% urea-polyacrylamide gel and incubated in 2X Annealing buffer (60 mM Tris-HCl pH 7.5, 200 mM NaCl, 2 mM EDTA) at 90°C for 2 min and then slow cooled to room temperature over >3 hours. Annealed RNA was separated from ssRNAs on a native 15% polyacrylamide gel, and duplex was eluted from gel slices in 0.3 M NaCl overnight at 4°C, ethanol precipitated, and stored in 1X Annealing buffer at −80°C.
miR-430–, miR-1–, and miR-451–programed Ago2
HEK293T cells were cultured in DMEM (VWR) supplemented with 10% heat-inactivated fetal bovine serum at 37°C with 5% CO2 and split every second or third day at approximately 90% confluency. Cells grown in 150 mm plates were co-transfected with pcDNA3-FLAG3-Ago2 and pMAX-GFP (control) using Lipofectamine 2000 (Life Technologies) at approximately 50% confluency, according to the manufacturer’s instructions. At 24 hours post-transfection, cells were transferred to 254 mm square plates and allowed to grow for another 48 hours. Cells were harvested, and S100 extracts were prepared as described (Wee et al., 2012), except that the cells were lysed with a 23G needle and syringe. miR-430 or miR-1 duplex or pre-miR-451was incubated in 1 ml extract at a final concentration of 50 nM for 2 hours at 25°C, and then programmed Ago2 was affinity purified using a protocol modified from that of Flores-Jasso et al. (2013). Assembled Ago2-RISC was first captured with a 3′ biotinylated 2′–O–methyl-modified oligonucleotide that paired with nucleotides 2–8 of the miRNA (Table S1) and displaced with a competitor DNA oligonucleotide (Table S1) as described (Wee et al., 2012; Flores-Jasso et al., 2013), except the complex was displaced from the capture oligonucleotide in two successive rounds of elution, each with 100 μL of elution solution (10 μM competitor oligo in 18 mM HEPES pH 7.4, 1 mM potassium acetate, 3 mM magnesium acetate, 0.01% NP-40, 0.2 mg/mL BSA, 0.01 mg/mL yeast tRNA) for 2 hours. To remove complex that had formed with endogenous Agos from the extract, the complex with the ectopically expressed Ago2 was immunopurified based on affinity to the FLAG tag. Anti-FLAG M2 Magnetic Beads (Sigma) were equilibrated with binding buffer (18 mM HEPES pH 7.4, 100 mM potassium acetate, 1 mM magnesium acetate, 0.01 mg/mL yeast tRNA, 0.01% NP-40, 0.2 mg/mL BSA) and incubated with the pooled elution fractions for 2 hours, shaking at 1100 rpm on a ThermoMixer (Eppendorf) at 25°C. The beads were washed with binding buffer three times and eluted with FLAG peptide (Sigma) in binding buffer. The eluted protein in storage buffer [binding buffer supplemented with glycerol and DTT (13% and 1 mM final concentrations, respectively), which diluted the binding buffer by 25%] was flash frozen in liquid nitrogen and stored at −80°C. To measure the binding capacity of the complex and thereby determine its concentration, complex was incubated with excess radiolabeled target RNA that contained a phosphorothioate linkage flanked by 2′–O–methyl-ribose at positions 10 and 11 to block cleavage (Table S1), and layered nitrocellulose-nylon filter-binding assays were performed to quantitate bound and unbound RNA.
RNA targets for in vitro slicing assays
Targets for in vitro slicing assays were transcribed in vitro with T7 RNA polymerase, treated with TURBO DNase (Life Technologies) and purified on a urea-polyacrylamide gel. Purified RNA was capped in two batches to generate RNA with high and low specific activity, using the Vaccinia capping system (New England BioLabs) according to the manufacturer’s directions. RNA of high specific activity was prepared by incubating 10 picomol RNA with only [α-32P]GTP for 2 hours in a 10 μl reaction, before adding 0.5 nanomol GTP for another hour, and RNA of low specific activity was prepared by using a 140:1 molar ratio of GTP:[α-32P]GTP. Capped RNA was gel purified, phenol-cholorform and chloroform extracted, ethanol precipitated, resuspended in water, quantified by UV absorbance (NanoDrop) and stored at −80°C. RNA of high-specific activity was used for the single-turnover assays, and mixtures of high- and low-specificity were used for the multiple-turnover assays, using the high-specificity RNA to optimize the amount of radioactivity but including mostly low-specificity RNA to maintain accurate concentrations.
In vitro slicing assay
Slicing assays were performed in the 37°C warm room. Pre-mixed, cap-labeled target RNA was incubated in reaction buffer (18 mM HEPES pH 7.4, 100 mM potassium acetate, 1 mM magnesium acetate, 0.01 mg/ mL yeast tRNA, 0.01% NP-40, 5 mM DTT) for 15 min at 37°C, and then miR-430–, miR-1–, or miR-451–programed Ago2 was added to initiate the slicing reaction. Reactions were incubated at 37°C, and 2 μL aliquots were removed at each indicated time point and quenched with Gel Loading Buffer II (95% formamide, 18 mM EDTA, 0.025% SDS, 0.025% xylene cyanol, and 0.025% bromophenol blue; Life Technologies). To monitor slicing, RNAs were resolved on a urea-polyacrylamide gel, and radiolabeled target and product were visualized on the Typhoon FLA 7000 phosphorimager.
pre-miR-451 binding and cleavage assay
Synthetic pre-miR-451 RNAs (IDT) (Table S1) were purified on a urea-polyacrylamide gel, phosphorylated with [γ-32P]ATP using T4 polynucleotide kinase (New England BioLabs), desalted with Micro Bio-Spin P-30 gel columns (BioRad), and gel purified again. One-cell embryos were co-injected with end-labeled pre-miR-451 (10 pg/embryo) and Ago2 mRNA (100 pg/embryo), injecting 250–300 embryos for each condition (i.e., each lane on the gel). At 4 hpf, injected embryos were manually de-chorionated in the presence of 1 mg/mL pronase (EMD Millipore), washed 3 times with E3 (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, and 0.33 mM MgSO4), and transferred to a 0.6 mL Eppendorf tube. To break the yolk sak, embryos were gently pipetted with 400 μL of de-yolking buffer [55 mM NaCl, 1.8 mM KCl, 1.25 mM NaHCO3, protease inhibitor cocktail tablet (cOmplete, mini, EDTA-free, Sigma; one tablet per 10 mL buffer)]. The embryos were then shaken at 1100 rpm on a ThermoMixer for 5 min and centrifuged at 300 g for 1 min to separate the yolk from the cells. The yolk-containing supernatant was removed, and this de-yolking process was repeated 3 times. Embryo lysis buffer (25 mM Tris pH 7.5, 2 mM EDTA, 150 mM NaCl, 10% glycerol, 1% Triton X-100) was then added and vortexed with 0.5 mm glass beads (BioSpec) for 5 seconds every 30 seconds for 4 min at 4°C. The 0.6 mL Eppendorf tube was punctured at its bottom with a 26G needle and placed inside a 1.5 mL Eppendorf tube. The lysate was separated from the glass beads and clarified by centrifugation at 21,130 g for 10 min at 4°C. Clarified lysates were flash frozen in liquid nitrogen and stored at −80°C prior to FLAG immunoprecipitation. To immunoprecipitate FLAG-tagged Ago2, the lysate was incubated with Anti-FLAG M2 Magnetic Beads (Sigma) under rotation at 4°C overnight in binding buffer (25 mM Tris pH 7.5, 2 mM EDTA, 150 mM NaCl, 10% glycerol, 1% Triton X-100). The lysate was removed, and the beads were washed three times with wash buffer (25 mM Tris pH 7.5, 2 mM EDTA, 1 M NaCl, 10% glycerol, 1% Triton X-100) for 10 min each, under rotation, at 4°C. 1 mL TRI Reagent (Life Technologies) was added directly to the beads to extract total RNA from the immunoprecipitated material. After chloroform extraction and ethanol precipitation, extracted RNA was resolved on urea-polyacrylamide gels, visualized on the Typhoon FLA 7000 phosphorimager, and analyzed using the MultiGauge software.
Immunoblotting
Lysate from ~10 embryos from each condition were boiled and denatured in SDS loading dye (125 mM Tris pH 6.8, 4% SDS, 20% glycerol, bromophenol blue, 5% β-mercaptoethanol) for 10 min at 90°C and resolved on a NuPAGE 4–12% Bis-Tris protein gel (Life Technologies). Protein was then blotted onto a PVDF membrane (Life Technologies) at 110 mA for 2 hours. Blots probed for FLAG were blocked in PBST (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 0.1% Tween-20) containing 5% milk for 1 hour and probed with mouse anti-FLAG antibody (Sigma, diluted 1:5000 in PBST containing 5% milk), rocking for 1 hr. After washing with PBST for 10 min three times, blots were probed for the primary antibody using HRP-conjugated donkey anti-mouse IgG (GE Healthcare Life Sciences, diluted 1:10,000) for 1 hour. Blots probed for GAPDH were blocked with PBST containing 1% BSA, probed with the primary antibody, rabbit anti-GAPDH (Abcam, diluted 1:5000 in PBST containing 1% BSA), and the primary antibody was probed using HRP-conjugated donkey anti-rabbit IgG (GE Healthcare Life Sciences, diluted 1:10,000). After washing with PBST for 10 min three times, HRP detection was through electrogenerated chemiluminescence using Amersham ECL prime western blotting detection reagent (Thermo Fisher Scientific), according to the manufacturer’s instructions, with light detected on Amersham Hyperfilm ECL (Thermo Fisher Scientific).
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of in vitro slicing assays
Single-turnover slicing data were analyzed using the MultiGauge software to calculate percent cleaved. The data were fit in MATLAB to the exponential equation,
where F(t) is target cleaved over time, and k is rate constant according to the following scheme,
Multiple-turnover slicing data were fit in MATLAB to the burst and steady-state equation (Wee et al., 2012),
where F(t) is target cleaved over time, E is the enzyme concentration, and a and b are rate constants (reported as k1 and k2, respectively) according to the following scheme,
DATA AND SOFTWARE AVAILABILITY
Raw experimental data from this study have been deposited to Mendeley Data and can be found at https://data.mendeley.com/datasets/fjg3b788k5/draft?a=8a5b69b5-2580-498e-b03a-cdd8492a183f.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal ANTI-FLAG M2 antibody | Sigma | F9291-1MG |
| Anti-GAPDH antibody | Abcam | ab9485 |
| Amersham ECL Mouse IgG, HRP-linked Ab (from sheep) | GE Healthcare Life Sciences | NXA931-1ml |
| Bacterial and Virus Strains | ||
| One Shot TOP10 chemically competent E. coli | Life Technologies | C404006 |
| Biological Samples | ||
| DMEM | VWR | 45000-304 |
| Opti-MEM | Life Technologies | 31985062 |
| FBS | Clontech | 631367 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| [γ-32P]ATP | PerkinElmer | NEG035C001MC |
| [α-32P]UTP | PerkinElmer | NEG007H001MC |
| [α-32P]GTP | PerkinElmer | NEG006H500UC |
| Pronase | EMD Millipore | 537088-50KU |
| Notl-HF | New England Biolabs | R3189L |
| cOmplete, Mini, EDTA-free protease inhibitor cocktail tablets | Sigma | 11836170001 |
| TRI Reagent solution | Life Technologies | AM9738 |
| Phenol:chloroform:isoamyl alcohol 25:24:1 | Sigma | P2069-100ML |
| Chloroform | J.T.Baker Analyical | 9180-01 |
| SUPERase-In | Life Technologies | AM2696 |
| TURBO DNase | Life Technologies | AM2239 |
| Yeast tRNA | Life Technologies | 15401011 |
| Dynabeads MyOne Streptavidin C1 | Life Technologies | 65002 |
| ANTI-FLAG M2 magnetic beads | Sigma | M8823 |
| 3X-FLAG peptide | Sigma | F4799-4MG |
| GE Healthcare Amersham Hybond-XL membranes | Thermo Fisher Scientific | 45001147 |
| GE Healthcare Amersham Hybond-NX membrane | VWR | 95038-412 |
| Whatman Protran nitrocellulose membrane | Sigma | Z670898 |
| PVDF pre-cut blotting membrane, 0.2 μm pore size | Life Technologies | LC2002 |
| GE Healthcare Amersham Hyperfilm ECL | Thermo Fisher Scientific | 45001504 |
| GE Healthcare Amersham ECL prime western blotting detection reagent | Thermo Fisher Scientific | 45002401 |
| Gel loading buffer II (denaturing PAGE) | Life Technologies | AM8547 |
| Whatman Nytran SuPerCharge (SPC) TurboBlotter | Sigma | WHA10416302 |
| Critical Commercial Assays | ||
| mMESSAGE mMACHINE SP6 | Life Technologies | AM1340 |
| RNeasy Mini Kit | QIAGEN | 74104 |
| QuikChange Lightning Multi Site-Directed Mutagenesis | Agilent | 210515 |
| Phusion High-Fidelity DNA polymerase | New England Biolabs | M0530L |
| T4 PNK | New England Biolabs | 101228-172 |
| MAXIscript T7 RNA polymerase | Life Technologies | AM1312 |
| Vaccinia capping system | New England Biolabs | M2080S |
| Lipofectamine 2000 | Life Technologies | 11668019 |
| NuPAGE Novex 4-12% Bis-Tris Gel 1.0 mm | Life Technologies | NP0321BOX |
| NuPAGE MOPS SDS running buffer | Life Technologies | NP0001 |
| NuPAGE transfer buffer | Life Technologies | NP00061 |
| Micro Bio-Spin P-30 gel columns | Bio-Rad | 7326250 |
| NorthernMax-Gly kit | Life Technologies | AM1946 |
| ULTRAhyb Ultrasensitive hybridization buffer | Life Technologies | AM8670 |
| ULTRAhyb-Oligo hybridization buffer | Life Technologies | AM8663 |
| Deposited Data | ||
| Raw data | This paper, Mendeley Data | https://data.mendeley.com/datasets/fjg3b788k5/draft?a=f466501d-4dac-4b86-8d57-4cecb1fc3376 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Zebrafish | ZIRC | AB |
| Oligonucleotides | ||
| See Table S1 | This study | Table S1 |
| Recombinant DNA | ||
| pCS2+-eGFP | This study | N/A |
| pCS2+-drAgo2 | This study | N/A |
| pCS2+-FLAG3-drAgo2 | This study | N/A |
| pCS2+-FLAG3-drAgo2D–A | This study | N/A |
| pCS2+-FLAG3-drAgo2D–E | This study | N/A |
| pCS2+-FLAG3-drAgo2Y–F | This study | N/A |
| pCS2+-FLAG3-drAgo2DY–EF | This study | N/A |
| pCS2+-FLAG3-hsN-drAgo2 | This study | N/A |
| pCS2+-hsAgo2 | This study | N/A |
| pCS2+-FLAG3-hsAgo2 | This study | N/A |
| pCS2+-FLAG3-hsAgo2D–A | This study | N/A |
| pCS2+-FLAG3-hsAgo2E–D | This study | N/A |
| pCS2+-FLAG3-hsAgo2F–Y | This study | N/A |
| pCS2+-FLAG3-hsAgo2EF–DY | This study | N/A |
| pCS2+-FLAG3-drN-hsAgo2 | This study | N/A |
| pCS2+-Zeocin | This study | N/A |
| pCS2+-Zeocin-miR-430 | This study | N/A |
| pCS2+-Zeocin-miR-430-10–11 mm | This study | N/A |
| pCS2+-Zeocin-miR-430-G–G mm | This study | N/A |
| pMAX GFP | Amaxa | VDF-1012 |
| pcDNA3-drAgo2 | This study | N/A |
| pcDNA3-FLAG3-drAgo2 | This study | N/A |
| pcDNA3-FLAG3-drAgo2D–E | This study | N/A |
| pcDNA3-FLAG3-drAgo2Y–F | This study | N/A |
| pcDNA3-FLAG3-drAgo2DY–EF | This study | N/A |
| pcDNA3-FLAG3-drAgo2D–A | This study | N/A |
| pcDNA3-hsAgo2 | This study | N/A |
| pcDNA3-FLAG3-hsAgo2 | This study | N/A |
| GFP-3xPTmiR-1 | Giraldez lab | N/A |
| Software and Algorithms | ||
| MATLAB | Mathworks | |
| Multi Gauge 2.2 | Fujifilm | |
| GraphPad Prism | GraphPad Software | |
| Other | ||
Supplementary Material
Acknowledgments
We thank B. Kleaveland and N. Bisaria for helpful discussions, and S. McGeary for experimental and technical advice. This research was supported by NIH grants GM061835 and GM118135 (to D.P.B). D.P.B is an investigator of the Howard Hughes Medical Institute. D.P.B. is a member of the scientific advisory board of Alnylam Pharmaceuticals. A patent has been filed on this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
G.R.C. and D.P.B. conceived the project, designed the study, and wrote the manuscript. G.R.C. performed the experiments. H.S. provided guidance and expertise.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and one table.
References
- Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008;22:2773–2785. doi: 10.1101/gad.1705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi G, Wiley EO, Mansour H, Miller MR, Orti G, Haussler D, O’Brien SJ, Ryder OA, Venkatesh B. The fishes of Genome 10K. Mar Genomics. 2012;7:3–6. doi: 10.1016/j.margen.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Betancur RR, Broughton RE, Wiley EO, Carpenter K, Lopez JA, Li C, Holcroft NI, Arcila D, Sanciangco M, Cureton Ii JC, et al. The tree of life and a new classification of bony fishes. PLoS Curr. 2013;5 doi: 10.1371/currents.tol.53ba26640df0ccaee75bb165c8c26288. currents.tol.53ba26640df0ccaee75bb165c8c26288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobbin ML, Rossi JJ. RNA Interference (RNAi)-Based Therapeutics: Delivering on the Promise? Annu Rev Pharmacol Toxicol. 2016;56:103–122. doi: 10.1146/annurev-pharmtox-010715-103633. [DOI] [PubMed] [Google Scholar]
- Broughton RE, Betancur RR, Li C, Arratia G, Orti G. Multi-locus phylogenetic analysis reveals the pattern and tempo of bony fish evolution. PLoS currents. 2013;5 doi: 10.1371/currents.tol.2ca8041495ffafd0c92756e75247483e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature. 2010;465:584–589. doi: 10.1038/nature09092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, et al. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science. 2010;328:1694–1698. doi: 10.1126/science.1190809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis E, Caiment F, Tordoir X, Cavaille J, Ferguson-Smith A, Cockett N, Georges M, Charlier C. RNAi-mediated allelic trans-interaction at the imprinted Rtl1/Pegl1 locus. Curr Biol. 2005;15:743–749. doi: 10.1016/j.cub.2005.02.060. [DOI] [PubMed] [Google Scholar]
- De Rienzo G, Gutzman JH, Sive H. Efficient shRNA-mediated inhibition of gene expression in zebrafish. Zebrafish. 2012;9:97–107. doi: 10.1089/zeb.2012.0770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong M, Fu YF, Du TT, Jing CB, Fu CT, Chen Y, Jin Y, Deng M, Liu TX. Heritable and lineage-specific gene knockdown in zebrafish embryo. PLoS One. 2009;4:e6125. doi: 10.1371/journal.pone.0006125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinnenberg IA, Fink GR, Bartel DP. Compatibility with killer explains the rise of RNAi-deficient fungi. Science. 2011;333:1592. doi: 10.1126/science.1209575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinnenberg IA, Weinberg DE, Xie KT, Mower JP, Wolfe KH, Fink GR, Bartel DP. RNAi in budding yeast. Science. 2009;326:544–550. doi: 10.1126/science.1176945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001a;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001b;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faehnle CR, Elkayam E, Haase AD, Hannon GJ, Joshua-Tor L. The making of a slicer: activation of human Argonaute-1. Cell reports. 2013;3:1901–1909. doi: 10.1016/j.celrep.2013.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemr M, Malik R, Franke V, Nejepinska J, Sedlacek R, Vlahovicek K, Svoboda P. A retrotransposon-driven dicer isoform directs endogenous small interfering RNA production in mouse oocytes. Cell. 2013;155:807–816. doi: 10.1016/j.cell.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Flores-Jasso CF, Salomon WE, Zamore PD. Rapid and specific purification of Argonaute-small RNA complexes from crude cell lysates. Rna. 2013;19:271–279. doi: 10.1261/rna.036921.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome research. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez AJ, Cinalli RM, Glasner ME, Enright AJ, Thomson JM, Baskerville S, Hammond SM, Bartel DP, Schier AF. MicroRNAs regulate brain morphogenesis in zebrafish. Science. 2005;308:833–838. doi: 10.1126/science.1109020. [DOI] [PubMed] [Google Scholar]
- Gruber J, Manninga H, Tuschl T, Osborn M, Weber K. Specific RNAi mediated gene knockdown in zebrafish cell lines. RNA Biol. 2005;2:101–105. doi: 10.4161/rna.2.3.2060. [DOI] [PubMed] [Google Scholar]
- Grundhoff A, Sullivan CS. Virus-encoded microRNAs. Virology. 2011;411:325–343. doi: 10.1016/j.virol.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, Kjems J. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011;30:4414–4422. doi: 10.1038/emboj.2011.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauptmann J, Dueck A, Harlander S, Pfaff J, Merkl R, Meister G. Turning catalytically inactive human Argonaute proteins into active slicer enzymes. Nature structural & molecular biology. 2013;20:814–817. doi: 10.1038/nsmb.2577. [DOI] [PubMed] [Google Scholar]
- Hauptmann J, Kater L, Loffler P, Merkl R, Meister G. Generation of catalytic human Ago4 identifies structural elements important for RNA cleavage. Rna. 2014;20:1532–1538. doi: 10.1261/rna.045203.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki YW, Siomi MC, Siomi H. PIWI-Interacting RNA: Its Biogenesis and Functions. Annu Rev Biochem. 2015;84:405–433. doi: 10.1146/annurev-biochem-060614-034258. [DOI] [PubMed] [Google Scholar]
- Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421–433. doi: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- Kelly A, Hurlstone AF. The use of RNAi technologies for gene knockdown in zebrafish. Briefings in functional genomics. 2011;10:189–196. doi: 10.1093/bfgp/elr014. [DOI] [PubMed] [Google Scholar]
- Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- Lee HS, Seok H, Lee DH, Ham J, Lee W, Youm EM, Yoo JS, Lee YS, Jang ES, Chi SW. Abasic pivot substitution harnesses target specificity of RNA interference. Nat Commun. 2015;6:10154. doi: 10.1038/ncomms10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor L, Hannon GJ. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305:1437–1441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- Ma JB, Yuan YR, Meister G, Pei Y, Tuschl T, Patel DJ. Structural basis for 5′-end-specific recognition of guide RNA by the A. fulgidus Piwi protein. Nature. 2005;434:666–670. doi: 10.1038/nature03514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Hannon GJ. Small RNAs as guardians of the genome. Cell. 2009;136:656–668. doi: 10.1016/j.cell.2009.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangos S, Vanderbeld B, Krawetz R, Sudol K, Kelly GM. Ran binding protein RanBP1 in zebrafish embryonic development. Mol Reprod Dev. 2001;59:235–248. doi: 10.1002/mrd.1028. [DOI] [PubMed] [Google Scholar]
- Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell. 2004;15:185–197. doi: 10.1016/j.molcel.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Ascano M, Gogakos T, Ishibe-Murakami S, Serganov AA, Briskin D, Morozov P, Tuschl T, Patel DJ. Eukaryote-specific insertion elements control human ARGONAUTE slicer activity. Cell reports. 2013;3:1893–1900. doi: 10.1016/j.celrep.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Weinberg DE, Bartel DP, Patel DJ. Structure of yeast Argonaute with guide RNA. Nature. 2012;486:368–374. doi: 10.1038/nature11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oates AC, Bruce AE, Ho RK. Too much interference: injection of double-stranded RNA has nonspecific effects in the zebrafish embryo. Dev Biol. 2000;224:20–28. doi: 10.1006/dbio.2000.9761. [DOI] [PubMed] [Google Scholar]
- Parker JS, Roe SM, Barford D. Structural insights into mRNA recognition from a PIWI domain-siRNA guide complex. Nature. 2005;434:663–666. doi: 10.1038/nature03462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick DM, Zhang CC, Tao Y, Yao H, Qi X, Schwartz RJ, Jun-Shen Huang L, Olson EN. Defective erythroid differentiation in miR-451 mutant mice mediated by 14-3-3zeta. Genes Dev. 2010;24:1614–1619. doi: 10.1101/gad.1942810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen KD, Simmini S, Abreu-Goodger C, Bartonicek N, Di Giacomo M, Bilbao-Cortes D, Horos R, Von Lindern M, Enright AJ, O’Carroll D. The miR-144/451 locus is required for erythroid homeostasis. J Exp Med. 2010;207:1351–1358. doi: 10.1084/jem.20100458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon WE, Jolly SM, Moore MJ, Zamore PD, Serebrov V. Single-Molecule Imaging Reveals that Argonaute Reshapes the Binding Properties of Its Nucleic Acid Guides. Cell. 2015;162:84–95. doi: 10.1016/j.cell.2015.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle NT, MacRae IJ. The crystal structure of human Argonaute2. Science. 2012;336:1037–1040. doi: 10.1126/science.1221551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina SA, Koonin EV. Origins and evolution of eukaryotic RNA interference. Trends in ecology & evolution. 2008;23:578–587. doi: 10.1016/j.tree.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Nam JW, Farh KK, Chiang HR, Shkumatava A, Bartel DP. Expanding the microRNA targeting code: functional sites with centered pairing. Mol Cell. 2010;38:789–802. doi: 10.1016/j.molcel.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JJ, Smith SK, Hannon GJ, Joshua-Tor L. Crystal structure of Argonaute and its implications for RISC slicer activity. Science. 2004;305:1434–1437. doi: 10.1126/science.1102514. [DOI] [PubMed] [Google Scholar]
- Song R, Hennig GW, Wu Q, Jose C, Zheng H, Yan W. Male germ cells express abundant endogenous siRNAs. Proc Natl Acad Sci U S A. 2011;108:13159–13164. doi: 10.1073/pnas.1108567108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein P, Rozhkov NV, Li F, Cardenas FL, Davydenko O, Vandivier LE, Gregory BD, Hannon GJ, Schultz RM. Essential Role for endogenous siRNAs during meiosis in mouse oocytes. PLoS genetics. 2015;11:e1005013. doi: 10.1371/journal.pgen.1005013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtelny AO, Eichhorn SW, Chen GR, Sive H, Bartel DP. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature. 2014;508:66–71. doi: 10.1038/nature13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam OH, Aravin AA, Stein P, Girard A, Murchison EP, Cheloufi S, Hodges E, Anger M, Sachidanandam R, Schultz RM, et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008;453:534–538. doi: 10.1038/nature06904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomari Y, Zamore PD. Perspective: machines for RNAi. Genes Dev. 2005;19:517–529. doi: 10.1101/gad.1284105. [DOI] [PubMed] [Google Scholar]
- Tuschl T. RNA interference and small interfering RNAs. Chembiochem. 2001;2:239–245. doi: 10.1002/1439-7633(20010401)2:4<239::AID-CBIC239>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Tyner C, Barber GP, Casper J, Clawson H, Diekhans M, Eisenhart C, Fischer CM, Gibson D, Gonzalez JN, Guruvadoo L, et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 2017;45:D626–D634. doi: 10.1093/nar/gkw1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Totoki Y, Toyoda A, Kaneda M, Kuramochi-Miyagawa S, Obata Y, Chiba H, Kohara Y, Kono T, Nakano T, et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008;453:539–543. doi: 10.1038/nature06908. [DOI] [PubMed] [Google Scholar]
- Wee LM, Flores-Jasso CF, Salomon WE, Zamore PD. Argonaute divides its RNA guide into domains with distinct functions and RNA-binding properties. Cell. 2012;151:1055–1067. doi: 10.1016/j.cell.2012.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick EM, Miska EA. piRNAs: from biogenesis to function. Development. 2014;141:3458–3471. doi: 10.1242/dev.094037. [DOI] [PubMed] [Google Scholar]
- Yang JS, Maurin T, Lai EC. Functional parameters of Dicer-independent microRNA biogenesis. Rna. 2012;18:945–957. doi: 10.1261/rna.032938.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Maurin T, Robine N, Rasmussen KD, Jeffrey KL, Chandwani R, Papapetrou EP, Sadelain M, O’Carroll D, Lai EC. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc Natl Acad Sci U S A. 2010;107:15163–15168. doi: 10.1073/pnas.1006432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594–596. doi: 10.1126/science.1097434. [DOI] [PubMed] [Google Scholar]
- Yoda M, Cifuentes D, Izumi N, Sakaguchi Y, Suzuki T, Giraldez AJ, Tomari Y. Poly(A)-specific ribonuclease mediates 3′-end trimming of Argonaute2-cleaved precursor microRNAs. Cell reports. 2013;5:715–726. doi: 10.1016/j.celrep.2013.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Cao Y, Li M, Meng A. Double-stranded RNA injection produces nonspecific defects in zebrafish. Dev Biol. 2001;229:215–223. doi: 10.1006/dbio.2000.9982. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.