

Abstract
Mutations in isocitrate dehydrogenase 1/2 (IDH1/2MT) are drivers of a variety of myeloid neoplasms. As they yield the same oncometabolite, D-2-hydroxyglutarate, they are often treated as equivalent, and pooled. We studied the validity of this approach and found IDH1/2 mutations in 179 of 2119 myeloid neoplasms (8%). Cross-sectionally, the frequencies of these mutations increased from lower- to higher-risk disease, thus suggesting a role in clinical progression. Variant allelic frequencies indicated that IDH1MT and IDH2MT are ancestral in up to 14/74 (19%) vs. 34/99 (34%; P=0.027) of cases, respectively, illustrating the pathogenic role of these lesions in myeloid neoplasms. IDH1/2MT was associated with poor overall survival, particularly in lower-risk myelodysplastic syndromes. Ancestral IDH1MT cases were associated with a worse prognosis than subclonal IDH1MT cases, whereas the position of IDH2MT within clonal hierarchy did not impact survival. This may relate to distinct mutational spectra with more DNMT3A and NPM1 mutations associated with IDH1MT cases, and more ASXL1, SRSF2, RUNX1, STAG2 mutations associated with IDH2MT cases. Our data demonstrate important clinical and biological differences between IDH1MT and IDH2MT myeloid neoplasms. These mutations should be considered separately as their differences could have implications for diagnosis, prognosis, and treatment with IDH1/2MT inhibitors of IDH1/2MT patients.
Keywords: isocitrate dehydrogenase, acute myeloid leukemia, myelodysplastic syndromes, 2-hydroxyglutarate, clonal hierarchy
INTRODUCTION
Mutations in isocitrate dehydrogenases 1 and 2 (IDH1MT and IDH2MT) are implicated in the development of numerous types of cancer, including glioma, chondrosarcoma, cholangiocarcinoma, angioimmunoblastic T-cell lymphoma and certain myeloid neoplasms1. Within myeloid malignancies, IDH1/2MT are present in a significant proportion of acute myeloid leukemia (AML) and, while less common, in myelodysplastic syndromes (MDS) and myeloproliferative neoplasms (MPN)2, 3.
IDH1/2MT impart a gain of function by causing single amino acid changes in the active sites of the enzymes. Whereas (wild-type) IDH1/2WT convert isocitrate and NADP+ to α-ketoglutarate (αKG) and NADPH, mutant IDH1/2 convert NADPH and αKG to NADP+ and D-2-hydroxyglutarate (D-2HG)4. D-2HG and αKG have similar structures; resultant D-2HG accumulation in IDH1/2MT cells inhibits many αKG-dependent dioxygenases, such as TET2 DNA hydroxyl demethylases5, 6 and Jumanji domain-containing histone demethylases (e.g., KDM6A/UTX and KDM3B/JMJD1B). Dysfunction of these enzymes is considered to be responsible for global DNA hypermethylation, inhibition of differentiation, and preservation of stemness7, 8. In addition, D-2HG activates, rather than inhibits Egg Laying Defective Nine (EGLN), which induces HIF1α degradation and growth factor-independent proliferation9, 10.
As direct evidence that IDH1/2MT are oncogenic, introduction of IDH2R140Q or IDH2R172K into 10T1/2 mesenchymal progenitor cells yielded an AML-like disease in mice11–13. IDH2R140Q was also necessary for AML maintenance in mice12, indicating that IDH1/2MT may be useful as therapeutic targets. These findings motivated the development of specific inhibitors of IDH1/2-mutant enzymes14–18. The currently available inhibitors, AG-221 that targets IDH2R140Q and AG-120 that targets IDH1R132H, restrict the production of D-2HG and thereby induce differentiation of AML cells ex vivo15. Both agents have shown promising preliminary results in phase I clinical trials19.
Genomic studies of molecular landscapes in human cancer have frequently combined IDH1MT and IDH2MT as a single functional group despite physiological differences: IDH1 is localized in the cytosol and IDH2 in the mitochondrial matrix. Remarkably, the spectrum of cancers and their subtypes differ in the distribution of IDH1/2MT. IDH1MT predominates in glioma (95%), chondrosarcoma (95%) and cholangiocarcinoma (80%), whereas the IDH1MT:IDH2MT ratio is more balanced or even skewed towards a higher frequency of IDH2MT in AML1. In addition, only IDH2MT have been reported in angioimmunoblastic T-cell lymphoma, osteosarcoma and gastric cancer, again suggesting that there are pathophysiologic differences between IDH1MT and IDH2MT. Biochemical investigations have shown that the specific amino acid substitutions IDH1R132H, IDH1R132C, IDH2R140Q or IDH2R172K differ in D-2HG production potency. IDH2R172K is the most potent, followed by IDH1R132H, IDH1R132C and IDH2R140Q. There are more than 60 different αKG-dependent dioxygenases that can theoretically be inhibited by D-2HG with likely distinct IC50 values for D-2HG. Thus, different IDH1/2MT inhibit different sets of αKG-dependent enzymes and this may partially explain differing distributions of IDH1/2MT between cancers1.
To investigate the clinical impact of IDH1/2MT in myeloid neoplasms, we studied a cohort of 2119 patients with myeloid neoplasms. We performed whole-exome/targeted multi-amplicon sequencing on the samples obtained from these patients from different institutions. We compared the mutational landscapes of IDH1MT and IDH2MT samples, their clinical associations, overall patient survival, and clonal hierarchies. Our aim was to provide insights into IDH1/2MT myeloid malignancy pathogenesis, especially with respect to the clonal architecture of IDH1/2MT cases, and to determine whether IDH1 and IDH2 mutations should be grouped or considered separately, particularly with respect to the potential benefits of IDH1/2-mutant inhibitors in various clinical contexts.
METHODS
Patient population
Blood and bone marrow samples were obtained from 2119 patients diagnosed with lower-risk MDS (868) and higher-risk MDS (536), defined per World Health Organization classification; secondary AML (sAML; 153); myeloproliferative neoplasms (MPN; 63); MDS/MPN (165); or primary AML (pAML; 334). From this cohort, 418 samples from 409 patients were subjected to whole-exome sequencing (WES). Furthermore, 1815 samples from 1761 patients were tested for a subset of genes (including IDH1 and IDH2; Supplementary Table S1) using targeted sequencing (TS). The total of 2179 samples listed above is greater than our total of 2119 samples because serial samples of 60 patients were included in the analysis. The sum of 409 patients subjected to WES and 1761 patients subjected TS is larger than 2119 because the WES and TS cohorts are partially overlapping.
Informed consent was obtained from patients according to protocols approved by the institutional review boards and in accordance with the Declaration of Helsinki. These patients were seen and treated at the Cleveland Clinic, the University of Tokyo and the Munich Leukemia Laboratory, diagnosis was confirmed on the basis of World Health Organization classification criteria. Patients with refractory anemia (RA), RA with ringed sideroblasts (RARS), refractory cytopenias with multilineage dysplasia (RCMD), RCMD with ringed sideroblasts (RCMD-RS) and MDS-unclassifiable (MDS-u) were classified as lower-risk MDS. Those with refractory anemia with excess blasts (RAEB-1 and RAEB-2) were classified as higher-risk MDS. Details of MDS/MPN and MPN subcategories are provided in Supplementary Table S2. Clinical details of the patients, including blood counts, demographics, and survival times, were obtained from medical records.
Sequencing technology
For WES, DNA was obtained from tumor and germline paired samples. The 50 Mb of protein-coding sequences was enriched from total genomic DNA by liquid-phase hybridization using SureSelect (version 4) (Agilent Technology, Santa Clara, CA), followed by massively parallel sequencing with HiSequation 2000 (Illumina, San Diego, CA). Somatic mutations were called as previously described20, 21, using the GATK algorithm (Broad Institute). To minimize false positives and focus on the most prevalent or relevant somatic events, we implemented a rational bioanalytic filtering approach and applied heuristic bioanalytic pipelines. For confirmation of somatic mutations, we analyzed paired germline DNA from CD3+ lymphocytes. For TS of a specific panel of genes we applied multi-amplicon deep sequencing (TrueSeq; Illumina) to frequently-affected exons of 60 selected genes. The sequencing libraries were generated according to an Illumina paired-end library protocol and subjected to deep sequencing on MiSeq (Illumina) instrumentation according to standard protocol. Copy number information at the locus of each mutation was assessed as previously reported22. For samples sequenced in Munich, 104 known or putative mutational gene targets in MDS were examined for mutations from the cohort using massively parallel sequencing (Illumina) or SureSelect (Agilent) captured target sequences23. High-probability oncogenic mutations were called by eliminating sequencing/mapping errors and known/possible SNPs based on available databases and frequencies of variant reads. Genomic copy number status was calculated by directly enumerating corresponding sequencing reads in each exon.
Analysis of clonal architecture
Variant allelic frequencies (VAF) were calculated as the fraction of mutated reads of the total number of reads of a certain gene. VAFs were adjusted using copy number information at the locus of each mutation. We recapitulated the clonal architecture of a patient using these copy number-adjusted VAFs. Ancestral vs subclonal events were determined using a copy number-adjusted VAF difference between two events, with a higher VAF indicating ancestral origin. We used an VAF threshold of 5% (absolute) to reliably discriminate ancestral from subclonal events. Events below this threshold were considered to be of undeterminable ancestry. This threshold was chosen based on previous studies24 and statistical calculations of our own data, based on the average depth of sequencing in our samples.
Statistical analysis
Comparisons of proportions were performed using the χ2 and Fisher’s exact tests and differences in values and in ranks were assessed by Student t tests and Mann-Whitney U tests, respectively. Cox models were used to identify correlates with overall survival. Kaplan-Meier curves were generated to graphically depict survival differences. Throughout, 2-sided tests were used with significance defined as α<.05. These analyses were performed using SPSS, GraphPad Prism 6 and the R statistical programming language.
RESULTS
Clinical characterization of the IDH1/2 mutation
Among the 2119 patient samples tested (Table 1), we identified IDH1 and IDH2 mutations in 78 (4%) and 101 (5%) cases, respectively, in a mutually exclusive fashion. Of the IDH1MT cases, IDH1R132C (49%, n=36) and IDH1R132H (38%, n=28) were the most common; 12% and 1% harbored IDH1R132X and IDH1T98I, respectively. Amongst IDH2MT cases, IDH2R140Q (85%, n=84) was the most frequent mutation, followed by IDH2R140X (5%, n=5), IDH2R172K (9%, n=9) and IDH2R172W (1%, n=1) (Figure 1A). IDH1MT patients were younger than IDH1/2WT patients (P=.01), an observation similar to that reported for IDH1MT glioblastoma patients25. Across the spectrum of MDS, the frequency of IDH1/2MT increased with progression, as IDH1MT occurred in 2% of lower-risk MDS cases, in 3% of higher-risk MDS cases and in 7% of sAML cases. The average number of mutations in each of these categories as determined by WES did not increase as much with 21, 30, and 27, respectively (Supplementary Figure S1). IDH2MT frequency increased as MDS progressed (in 2% of lower-risk MDS, in 7% of higher-risk MDS and in 6% of sAML cases). These findings suggest that in IDH1/2MT MDS cases, a high proportion of IDH1/2MT are associated with progression to advanced disease. In addition, IDH1 and IDH2 mutation frequencies were relatively lower in MDS/MPN and MPN patients, and showed the highest frequency in pAML patients (Figure 1B). While IDH2MT were enriched for a normal karyotype, IDH1MT patients more often had a deletion of chromosome Y in comparison with IDH1/2WT patients. Other clinical characteristics were similar between IDH1/2MT and IDH1/2WT patients (Table 1; Supplementary Table S3). More detailed clinical information on IDH1MT and IDH2MT patients with myeloid malignancies is shown in Supplementary Tables S4 and S5.
Table 1.
Clinical characterization of the IDH1/2 mutation in 2119 patients
|
IDH1MT (N=78) |
p-value |
IDH2MT (N=101) |
p-value |
IDH1/2WT (N=1940) |
Total (N=2119) |
|
|---|---|---|---|---|---|---|
| Age (median) | 60 | 0.01 | 65 | 0.58 | 66 | 66 |
| Sex* | ||||||
| Male | 46 (59%) | 64 (63%) | 1154 (59%) | 1264 | ||
| Female | 30 (38) | 33 (33%) | 761 (39%) | 824 | ||
| Diagnosis | ||||||
| Low-risk MDS | 21 (2%) | 0.0049 | 15 (2%) | 0.0001 | 832 | 868 |
| High-risk MDS | 18 (3%) | 0.5947 | 37 (7%) | 0.0186 | 481 | 536 |
| sAML | 11 (7%) | 0.0239 | 8 (6%) | 0.6869 | 134 | 153 |
| MDS/MPN | 2 (1%) | 0.08 | 7 (4%) | 0.85 | 156 | 165 |
| MPN | 0 | 0.176 | 2 (3%) | 0.76 | 61 | 63 |
| pAML | 26 (8%) | 0.0001 | 32 (10%) | 0.0001 | 276 | 334 |
| Karyotype** | ||||||
| Normal | 46 (59%) | 0.2789 | 65 (64%) | 0.0138 | 1010 (52%) | 1121 (53%) |
| Aberrant | 27 (35%) | 0.2917 | 29 (29%) | 0.0164 | 792 (41%) | 848 (40%) |
| Complex | 5 (6%) | 1 | 7 (7%) | 1 | 136 (7%) | 150 (7%) |
Sex NA: IDH1MT: 2, IDH2MT: 4, IDH1/2WT: 23
Karyotype NA:IDH1MT: 0, IDH2MT: 0, IDH1/2WT: 2
Overview of clinical characteristics of the IDH1MT, IDH2MT and IDH1/2WT patients in the study cohort. Percentages in the diagnosis rows are calculated as percentage of patients with a certain diagnosis with IDH1MT or IDH2MT. Percentages in the karyotype rows are calculated as percentage of IDH1MT/IDH2MT patients with a certain karyotype of their myeloid neoplasm. P values are calculated using the Student’s t test (Age) or Fisher’s Exact test (all other characteristics).
Figure 1. Breakdown of IDH1/2 mutations among disease types and specific amino acid substitutions.
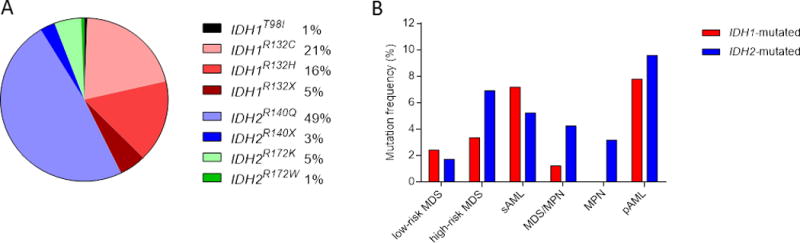
(A) Frequencies of IDH1 and IDH2 mutations in various myeloid neoplasms. (B) Pie chart showing the percentages of the specific IDH1/2 mutational amino acid substitutions in the cohort.
Abbreviations: pAML, primary acute myeloid leukemia; sAML, secondary acute myeloid leukemia.
Molecular characterization of IDH1 and IDH2-mutated myeloid neoplasms
To characterize the molecular features of patients with myeloid neoplasms with IDH1MT or IDH2MT, we analyzed associations between IDH1/2MT and other mutational events (Figure 2A; Supplementary Table S6). Overall, NPM1 mutations were more frequent in both IDH1MT and IDH2MT cases, compared with IDH1/2WT patients (Figure 2B and Supplementary Tables S6-8). Other somatic events were not significantly associated with both IDH1MT and IDH2MT cases compared with IDH1/2WT cases.
Figure 2. Mutational spectrum and most and least frequently co-occurring mutations in IDH1/2-mutated cases.
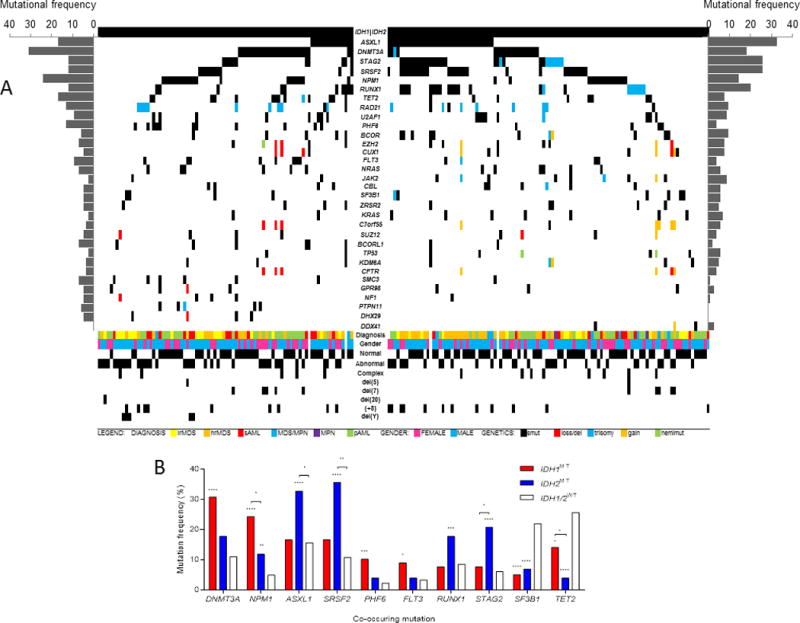
(A) Mutational spectrum and clinical spectrum of IDH1MT cases (left half) and IDH2MT cases (right half) with myeloid neoplasms. (B) Prevalence of co-occurring mutations in IDH1/2MT cases, compared with IDH1/2WT cases.
Abbreviations: lrMDS, low-risk MDS; hrMDS, high-risk MDS; sAML, secondary acute myeloid leukemia; MDS/MPN, myelodysplastic syndromes/myeloproliferative neoplasms; pAML, primary acute myeloid leukemia; smut, somatic mutation; del, deletion; hemimut, hemizygous mutation.
However, when IDH1MT cases were compared with IDH1/2WT cases, they were enriched for DNMT3A (P<.0001), PHF6 (P=.0006) and FLT3 (P=.0195) mutations, whereas IDH2MT cases were enriched for ASXL1 (P<.0001), SRSF2 (P<.0001), RUNX1 (P=.0034) and STAG2 (P<.0001) compared with IDH1/2WT cases. NPM1 mutations occurred more frequently in IDH1MT patients than in IDH2MT patients (P=0.045), whereas IDH2MT patients were enriched for ASXL1 (P=0.016), SRSF2 (P=0.007) and STAG2 (P=0.019) mutations compared with IDH1MT patients. Cohesin complex mutant carriers (STAG2, RAD21, SMC3) were more frequent in IDH1MT (P=.025) and IDH2MT (P<.0001) cases compared with IDH1/2WT cases. Conversely, SF3B1 and TET2 mutations occurred less frequently in IDH1MT (P<.0001 and P=.023) and in IDH2MT (P<.0001 and P<.0001) cases than in IDH1/2WT cases. Although we observed a negative correlation between IDH1 and TET2 mutations, TET2 mutations, if present, were less infrequent in IDH1MT patients than in IDH2MT patients (Figure 2C).
Analysis of ancestry and clonal architecture of IDH1/2-mutated patients
Using copy number-adjusted VAF, we reconstructed the clonal architecture of IDH1/2MT patients to establish whether an IDH1MT or IDH2MT was an ancestral or subclonal mutation. Clonal hierarchy was further confirmed and refined by serial analyses, performed in 60 exemplary cases (Figure 3C). IDH1MT and IDH2MT were ancestral in 19% and 34% of IDH1/2MT patients and subclonal in 55% and 45%, respectively (Figure 3A). As we used a cut-off value of 5% (absolute) in VAF difference, the ancestral vs. subclonal status of some IDH1/2MT remained “undetermined” when there were small VAF differences between IDH1/2 mutations and other mutations (see Methods). A lower proportion of IDH1MT cases were of ancestral origin compared to IDH2MT cases (P=.027). We also observed differences in the clonal succession between specific IDH1/2MT variants (Figure 3B). Whereas IDH1R132H and IDH1R132C mutations were ancestral in equal frequencies (22% and 21%), other IDH1 mutations (IDH1T98I (n=1), IDH1R132L (n=1) and IDH1R132S (n=5)) occurred only as subclonal events. Similarly, IDH2R140Q mutations were ancestral in 39% of IDH2R140Q-mutated cases, whereas other IDH2R140 mutations (IDH2R140L (n=2) and IDH2R140W (n=3)) were always subclonal. IDH2R172 mutations were ancestral in only 10% of IDH2R172-mutated patients. In the different types of myeloid neoplasms we did not observe differences in the distribution between ancestral vs. subclonal mutations in IDH1/2 and there was no difference in the mean VAF of IDH1MT vs IDH2MT (Supplementary Figure S2A-C). We observed higher IDH1/2MT VAFs in patients with ancestral IDH1/2MT than subclonal IDH1/2MT (Figure 3C and Supplementary Figure S2D). IDH1/2MT VAFs were highest in patients with IDH1/2MT of undeterminable ancestry, which may reflect a higher disease burden in patients when IDH1/2MT cooperate with other mutations. WES analyses revealed that IDH1MT and IDH2MT were ancestral in 1/21 (5%) and 2/24 (8%) of IDH1/2MT cases, respectively, whereas TS suggested that IDH1MT and IDH2MT were ancestral in 13/53 (25%) and 32/75 (43%) of cases (P=.0002). This difference probably reflects missed ancestral mutations in the TS samples (Supplementary Table S9). The clonal architecture of representative IDH1/2MT patients (out of 60 studied) that were serially sequenced is shown in Figure 3D and Supplementary Figure S2E.
Figure 3. Analysis of clonal architecture of IDH1/2-mutated cases.
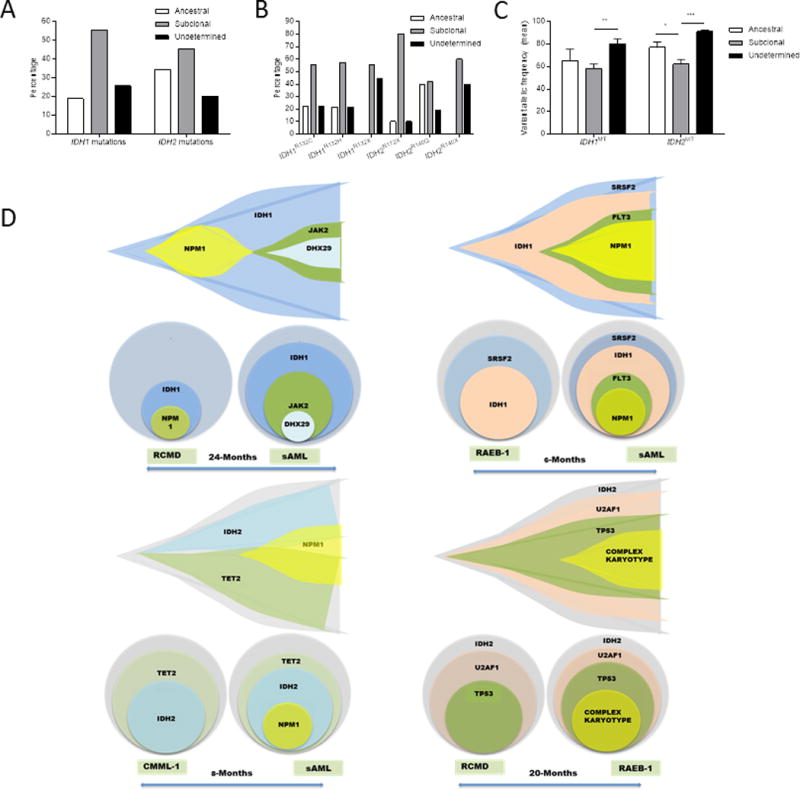
(A) Percentages of IDH1/2MT cases in which the IDH1/2 mutation was an ancestral event, a subclonal event or an event of undeterminable ancestry, based on the variant allelic frequency of the IDH1/2 mutations and other co-occurring mutations. (B) As in (A), but with specific IDH1/2 amino acid substitutions. (C) Mean variant allelic frequencies of IDH1 mutations and IDH2 mutations in IDH1/2MT cases in which the IDH1/2 mutation is an ancestral or subclonal event, or of undeterminable ancestry. (D) Fish plots of serially sequenced IDH1/2MT patients.
In subclonal IDH1/2MT cases, we investigated the corresponding ancestral events. These analyses showed heterogeneity, with 19 and 20 different ancestral mutations in 35 and 38 cases with subclonal IDH1MT or IDH2MT, respectively. Subclonal IDH1MT were most often preceded by an ancestral DNMT3A mutation (30%; Supplemental Figure S2F), whereas ancestral RUNX1 mutations most frequently preceded a subclonal IDH2MT (17%; Supplemental Figure S2G). We observed a RUNX1 mutation preceding a subclonal IDH1MT only once. Hierarchical clonal analyses of neoplasms with indeterminable IDH1/2MT clonal architecture (small VAF differences between IDH1/2 mutations and other mutations) may provide insight into which mutations co-operate (in the case of enrichment of co-occurring mutations; Figure 2) or compete (in the case of mutual exclusivity with co-occurring mutations) with IDH1/2MT. DNMT3A mutations were most frequently the “main competitor” of IDH1MT and IDH2MT of undetermined ancestry (Supplemental Figure S2H-I). We did not observe a single RUNX1 mutation that competed with IDH2MT, indicating that VAF differences between ancestral RUNX1 mutations and subclonal IDH2MT, and vice versa, are rather large.
Prognostic effect of IDH1/2 mutations in myeloid neoplasms
In our cohort, IDH1/2MT were associated with worse overall survival (Figure 4A). In subset analyses, we observed that IDH1/2MT were significantly associated with reduced median overall survival in lower-risk MDS patients (41 vs. 66 months, P=.03), but not in higher-risk MDS (34 vs 30 months, P=.91), sAML (26 vs. 19 months, P=.74) or pAML patients (20 vs. 16 months, P=.79; Figure 4B-E). This finding further illustrates the role of IDH1/2MT in progression to a more malignant disease. Patients with ancestral IDH1MT tended to have a worse survival than patients with subclonal IDH1MT (23 vs. 32 months, P=.09; Figure 4F), whereas there was no survival difference between patients with ancestral vs. subclonal IDH2MT (33 vs. 30 months, P=.35; Figure 4G).
Figure 4. IDH1/2 mutations and associations with overall patient survival in myeloid neoplasms.
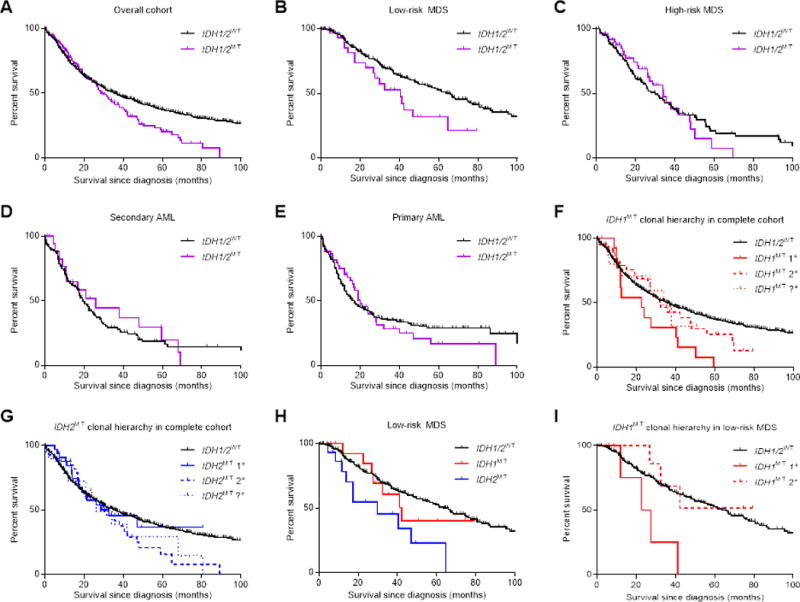
(A) Survival data of IDH1/2MT patients with myeloid neoplasms. (B-E) Subset survival analyses in low-risk MDS (B), high-risk MDS (C), secondary AML (D) and primary AML (E). (F) Survival data of IDH1MT patients with myeloid neoplasms, stratified on the ancestral importance of the IDH1 mutation. (G) as in (F), but with IDH2 mutations. (H) Subset survival analysis in low-risk MDS patients, with IDH1 and IDH2 mutations shown separately. (I) As in (F), but with IDH1 mutations in low-risk MDS patients.
Abbreviations: 1°, ancestral genetic event; 2°, subclonal genetic event; ?°, genetic event of undeterminable ancestry.
The association between IDH1MT or IDH2MT and overall survival was investigated separately in low-risk MDS to assess the impact on subsequent outcomes in early disease. Compared with IDH1/2WT patients, we found that IDH2MT patients (30 vs. 66 months, P=.003), but not IDH1MT patients (42 vs. 66 months, P=.64), had a worse prognosis (Figure 4H). Because IDH1MT were less frequently ancestral than IDH2MT in lower-risk MDS patients (22% vs. 40%), we analyzed whether ancestry determines to what extent IDH1/2MT impacts overall survival. Indeed, patients with ancestral IDH1MT had a worse survival than patients with myeloid neoplasms that had a subclonal IDH1MT (23 vs. 42 months, P=.05) or IDH1/2WT patients (23 vs. 66 months, P=.006; Figure 4I). Ancestral IDH2MT may also be associated with worse prognosis, compared with subclonal IDH2MT. However, definitive conclusions from this subset analysis were hindered by the limited number of cases (Supplementary Figure S3).
DISCUSSION
In this report, we describe the clinical and molecular characterization of IDH1MT and IDH2MT patients with myeloid neoplasms. The size of the cohort allowed for the most comprehensive analysis to date of molecular, morphologic and clinical features associated with IDH1/2 mutations, separately and combined. For the first time, a comprehensive analysis of clonal architecture distinguished ancestral from subclonal somatic lesions and determined differences in their clinical and biological impact. We demonstrate that IDH1MT and IDH2MT can occur as ancestral or subclonal defects. In a substantial proportion of cases, IDH1MT (19%) and IDH2MT (34%) represent ancestral lesions, but more often IDH1/2MT follow other ancestral mutations as subclonal events, likely explaining higher percentages of subclonal events in advanced myeloid disease. When present in lower-risk/early MDS, IDH1/2MT are associated with a poor prognosis, while in higher-risk myeloid neoplasms, prognosis could not be further stratified. Patients with ancestral IDH1MT exhibit worse survival than those with a subclonal IDH1MT mutation, particularly in lower-risk MDS. Such a difference was not found when ancestral vs. subclonal IDH2MT were compared, indicating that IDH2MT more rapidly dominate the clonal hierarchy.
Analyses of mutations that positively or negatively correlate with IDH1/2MT in myeloid neoplasms have heretofore treated IDH1/2MT cases as a single functional entity. In our cross-sectional analysis, this approach of pooling IDH1/2MT was only adequate for their association with NPM1 mutations, and their mutual exclusivity with TET2 mutations, as these are the only lesions that were significant for both IDH1MT and IDH2MT separately. In addition, IDH1MT cases were significantly enriched for DNTM3A and PHF6 mutations, and IDH2MT cases for ASXL1, SRSF2, RUNX1 and STAG2 mutations. When IDH1/2MT cases would be pooled, all these six mutations are significantly more frequently occurring in IDH1/2MT vs. IDH1/2WT cases. This would falsely suggest that they occur more frequently in both IDH1MT and IDH2MT cases, whereas the significant correlation is only true for either IDH1MT or IDH2MT cases. “pAML-associated” DNMT3A and NPM1 mutations occurred significantly more frequently in IDH1MT cases than in IDH2MT cases, whereas the reverse was true for “MDS/sAML/(MPN)-associated” ASXL1, SRSF2, STAG2 and RUNX1 mutations. Of note, there were no significant proportional differences with respect to diagnosis between IDH1MT and IDH2MT cases, with only a trend (P=0.051) towards higher-risk MDS patients among IDH2MT cases than among IDH1MT cases. Apparently, the functional cooperation between certain mutations (e.g. DNTM3A mutations and IDH1MT or RUNX1 mutations and IDH2MT) in the clonal hierarchy is driving the aforementioned positive correlations without resulting in disease phenotypes that are typical for these lesions.
The differences in at least some of the biologic/clinical features between IDH1MT and IDH2MT cases may stem from the differences in the biochemical consequences of these lesions. For instance, the various IDH1/2MT differ in D-2HG levels they produce, which is likely due to the impact of the specific amino acid substitutions on the catalytic site and thereby the conversion rate of αKG to D-2HG. Whereas IDH1 is cytoplasmic, IDH2 is mitochondrial and αKG levels may not be completely interchangeable between these subcellular compartments, generating different conditions for IDH1MT and IDH2MT to synthetize D-2HG. Furthermore, nuclear DNA and histone demethylases (e.g. TET2, Jumonji) and cytoplasmic prolyl hydroxylases (EGLN) are considered to be drivers of IDH1/2MT-induced oncogenesis1, suggesting that D-2HG mainly functions oncogenically outside mitochondria. It is unknown to what extent D-2HG passes the mitochondrial membrane. Therefore, the impact of the compartmentalization of αKG and D-2HG on the downstream effects of IDH2MT vs. IDH1MT is unclear.
D-2HG functions as an oncometabolite that inhibits various αKG-dependent dioxygenases. Whereas the effects of D-2HG on DNA demethylase TET2, histone demethylase JumonjiC and the HIF1α degrader EGLN have been described thoroughly9, 26, 27, there are in fact over 60 different αKG-dependent human dioxygenases involved in a plethora of cellular functions that may be inhibited, or possibly activated, as in the case of EGLN, by D-2HG. All these enzymes have specific IC50 values of D-2HG for inhibition of αKG-dependent enzymes. Thus, each D-2HG concentration (i.e. each IDH1/2MT variant) is expected to inhibit a specific subset of αKG-dependent dioxygenases and alter cellular functions in a variety of ways.
It has been proposed that the αKG-dependent DNA demethylase TET2 is one of the most important downstream targets of IDH1/2MT 26, 28. D-2HG inhibits TET2 and results in a genome-wide DNA hypermethylation in IDH1/2MT cancers that induces stemness and inhibits differentiation7. In our cohort, and that of others29, IDH1/2 and TET2 mutations are mostly mutually exclusive, supporting the notion of similar cellular downstream effects. TET2 mutations, albeit rare, were more frequent in IDH1MT cases (14%) than in IDH2MT cases (4%). It is possible that IDH1MT result in less TET2 inhibition than IDH2MT and that a synergistic/additive effect of co-occuring IDH1 and TET2 mutations augments TET2 inhibition. Of note, TET2 is widely held to be a downstream element of the pathogenic cascade induced by IDH1/2MT. Despite this, the profoundly distinct nosologic and morphologic spectra associated with TET2 and IDH1/2 mutations speak against this notion. Another putative important downstream target of IDH1/2MT is UTX/KDM6A7, 27. However, we did not observe mutual exclusivity between IDH1/2 and UTX mutations, suggesting that D-2HG-mediated UTX inhibition has a different effect than the UTX mutations that we observed. Notably, we did not observe a negative correlation between IDH1/2 and WT1 mutations, as previously reported30.
Our study provides the first comprehensive subclassification of IDH1/2MT cases with myeloid neoplasms based on their rank within clonal hierarchy, and thus their timing in clonal ontogeny. While cases initiated by IDH1/2MT are a distinct subset of myeloid neoplasms, in many instances IDH1/2MT are subclonal. The impact of the corresponding ancestral lesions, then, may more profoundly shape the individual neoplastic biology. This observation has clinical implications because, at least theoretically, therapeutic targeting of subclonal lesions is less likely to be curative as the ancestral clone cannot be eliminated. Remissions achieved in such cases thus have a greater likelihood of relapse, and durable clinical benefit may depend on combination therapies that also target the ancestral event(s). In addition, we report that ancestral IDH1/2MT are related to poor clinical outcomes, whereas this association is weaker for subclonal IDH1/2MT. These findings illustrate that patients with myeloid neoplasms carrying ancestral IDH1/2MT are the best candidates for therapy with IDH1/2MT inhibitors. Thus, sequencing that targets mutations frequently co-occurring with IDH1/2MT, in particular those which are often ancestral may aid rational clinical treatment of IDH1/2MT patients with IDH1/2MT inhibitors.
IDH1/2MT occur in a myriad of cancer types. Their biology in glioma and acute myeloid leukemia has been studied most intensively. The distribution of IDH1/2MT differs between gliomas and myeloid neoplasms, and fundamental differences exist in IDH1/2MT biology between these 2 tumor types. Whereas IDH1/2MT are very early events in gliomagenesis and has even been proposed as a canonical ancestral event31, 32, our findings show that this is in stark contrast to myeloid neoplasms, in which IDH1/2MT are ancestral in a minority of cases. In addition, IDH1/2MT associate with a strikingly prolonged overall survival in glioma25, 33, whereas our study, and that of others34, showed that these mutations are associated with worse prognosis in myeloid neoplasms.
In summary, we present results that demonstrate distinct differences between IDH1MT and IDH2MT. The position in the clonal hierarchy may be important for understanding the impact on the biology and clinical consequences of these mutations, and may refine future treatment of IDH1/2MT myeloid neoplasms with IDH1/2MT inhibitors. We conclude that the minority of patients with myeloid neoplasms carrying ancestral IDH1/2MT are the best candidates for therapy with IDH1/2MT inhibitors.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (Bethesda, MD; NIH) grants RO1HL-082983 (J.P.M.), U54 RR019391 (J.P.M.), K24 HL-077522 (J.P.M.), a grant from the AA & MDS International Foundation (Rockville, MD; J.P.M.), the Robert Duggan Charitable Fund (Cleveland, OH; J.P.M.), and Scott Hamilton CARES grant (Cleveland, OH; H.M.), a grant from the AA & MDS International Foundation (H.M.) and Grant-in-Aids from the Ministry of Health, Labor and Welfare of Japan and KAKENHI (23249052, 22134006, and 21790907) (Tokyo; S.O.), project for development of innovative research on cancer therapies (p-direct) (Tokyo; S.O.), the Japan Society for the Promotion of Science (JSPS) through the ‘Funding Program for World-Leading Innovative R&D on Science and Technology’, initiated by the Council for Science and Technology Policy (CSTP) (Tokyo; S.O.), the Dutch Cancer Society (KWF; UVA 2014-6839) (C.J.F.v.N, R.J.M and F.E.B.), an AMC PhD Scholarship (R.J.M), the American Society of Hematology Research Training award for fellows and a Damon Runyon Postdoctoral Fellowship (DRG 117-15, A.D.V.), Grant-in-Aids from the Ministry of Health, Labor and Welfare of Japan and KAKENHI (23249052, 22134006, 24221011 and 21790907; T.H.), by the Industrial Technology Research Grant Program from NEDO (08C46598a; T.H.), by Project for Development of Innovative Research on Cancer Therapeutics (P-Direct; T.H.) and by the Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST, T.H.) and Edward P Evans AA/MDS foundation (M.A.S.). The results presented here are partly based upon the data generated by The Cancer Genome Atlas pilot project established by the NCI and NHGRI.
Footnotes
Conflict of interest:
The authors declare no conflict of interest.
SUPPLEMENTARY INFORMATION
‘Supplementary information is available at Leukemia’s website’
References
- 1.Molenaar RJ, Radivoyevitch T, Maciejewski JP, van Noorden CJ, Bleeker FE. The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim Biophys Acta Rev Cancer. 2014;1846:326–341. doi: 10.1016/j.bbcan.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 2.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ponnaluri VK, Maciejewski JP, Mukherji M. A mechanistic overview of TET-mediated 5-methylcytosine oxidation. Biochem Biophys Res Commun. 2013;436:115–120. doi: 10.1016/j.bbrc.2013.05.077. [DOI] [PubMed] [Google Scholar]
- 6.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen C, Liu Y, Lu C, Cross JR, Morris JPt, Shroff AS, et al. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes Dev. 2013;27:1974–1985. doi: 10.1101/gad.226613.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kats LM, Reschke M, Taulli R, Pozdnyakova O, Burgess K, Bhargava P, et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell. 2014;14:329–341. doi: 10.1016/j.stem.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mylonas E, Janin M, Bawa O, Opolon P, David M, Quivoron C, et al. Isocitrate dehydrogenase (IDH)2 R140Q mutation induces myeloid and lymphoid neoplasms in mice. Leukemia. 2014 doi: 10.1038/leu.2014.18. [DOI] [PubMed] [Google Scholar]
- 14.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 16.Davis MI, Gross S, Shen M, Straley KS, Pragani R, Lea WA, et al. Biochemical, cellular, and biophysical characterization of a potent inhibitor of mutant isocitrate dehydrogenase IDH1. J Biol Chem. 2014;289:13717–13725. doi: 10.1074/jbc.M113.511030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Gorlich K, et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood. 2013;122:2877–2887. doi: 10.1182/blood-2013-03-491571. [DOI] [PubMed] [Google Scholar]
- 18.Deng G, Shen J, Yin M, McManus J, Mathieu M, Gee P, et al. Selective Inhibition of Mutant Isocitrate Dehydrogenase 1 (IDH1) via Disruption of a Metal Binding Network by an Allosteric Small Molecule. J Biol Chem. 2014 doi: 10.1074/jbc.M114.608497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ledford H. Metabolic quirks yield tumour hope. Nature. 2014;508:158–159. doi: 10.1038/508158a. [DOI] [PubMed] [Google Scholar]
- 20.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 21.Makishima H, Yoshida K, Nguyen N, Przychodzen B, Sanada M, Okuno Y, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45:942–946. doi: 10.1038/ng.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J, et al. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J Clin Oncol. 2009;27:6109–6116. doi: 10.1200/JCO.2009.23.7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–247. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stead LF, Sutton KM, Taylor GR, Quirke P, Rabbitts P. Accurately identifying low-allelic fraction variants in single samples with next-generation sequencing: applications in tumor subclone resolution. Hum Mutat. 2013;34:1432–1438. doi: 10.1002/humu.22365. [DOI] [PubMed] [Google Scholar]
- 25.Molenaar RJ, Verbaan D, Lamba S, Zanon C, Jeuken JW, Boots-Sprenger SH, et al. The combination of IDH1 mutations and MGMT methylation status predicts survival in glioblastoma better than either IDH1 or MGMT alone. Neuro Oncol. 2014;16:1263–1273. doi: 10.1093/neuonc/nou005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Losman JA, Kaelin WG., Jr What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27:836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jankowska AM, Makishima H, Tiu RV, Szpurka H, Huang Y, Traina F, et al. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood. 2011;118:3932–3941. doi: 10.1182/blood-2010-10-311019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rampal R, Alkalin A, Madzo J, Vasanthakumar A, Pronier E, Patel J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014;9:1841–1855. doi: 10.1016/j.celrep.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watanabe T, Nobusawa S, Kleihues P, Ohgaki HC. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. 2009;174:1149–1153. doi: 10.2353/ajpath.2009.080958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational Analysis Reveals the Origin and Therapy-Driven Evolution of Recurrent Glioma. Science. 2013 doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 Mutations in Gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou KG, Jiang LJ, Shang Z, Wang J, Huang L, Zhou JF. Potential application of IDH1 and IDH2 mutations as prognostic indicators in non-promyelocytic acute myeloid leukemia: a meta-analysis. Leuk Lymph. 2012;53:2423–2429. doi: 10.3109/10428194.2012.695359. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.