

Abstract
A clonal cytogenetic abnormality was observed in Philadelphia chromosome negative bone marrow cells of 6/27 chronic myeloid leukemia patients (+8 in 4, −7 in 1, and 20q- in 1) with dasatinib-induced remissions. The X-linked human androgen receptor gene assay demonstrated clonality in one additional patient. Single nucleotide polymorphism array analysis revealed somatic uniparental disomy involving chromosome 17(p12-pter) in another patient. The TP53 gene had a 5′ splice site deletion of exon 6 that caused alternative splicing, frame shifting and introduction of a premature stop codon. After three years, no patient developed myelodysplastic syndrome or acute myeloid leukemia.
Keywords: Chronic myeloid leukemia, dasatinib, cytogenetics, single nucleotide polymorphism array, myelodysplastic syndrome, TP53
Introduction
The BCR-ABL kinase inhibitors have revolutionized the treatment of chronic myeloid leukemia (CML).1 Patients with CML have experienced dramatically improved outcomes as a result of imatinib therapy.2 The second generation ABL kinase inhibitor dasatinib has proven to be effective for the treatment of CML patients who do not respond to, or tolerate, imatinib.3,4 Some CML patients with prolonged remissions on tyrosine kinase inhibitors (TKIs) harbor clonal cytogenetic abnormalities (CCA) in the Philadelphia chromosome negative (Ph−) bone marrow cells.5–12 The most commonly reported CCA have included +8, −7, 20q-, −5, and -Y, which are among the most frequent chromosomal changes in myelodysplastic syndrome (MDS).13 In spite of the chromosomal abnormalities observed, the frequency of developing a manifest second hematologic malignancy, such as MDS or acute myeloid leukemia (AML), appears to be low.11,14 Neither progression-free survival nor overall survival appear to be negatively impacted by the presence of a Ph-CCA in CML patients who achieve a major cytogenetic response to imatinib therapy.11
Although Ph-CCA are identified in a small percentage of CML patients who achieve a cytogenetic remission on kinase inhibitor therapy, many more have peripheral blood abnormalities, especially cytopenias, involving one or more cell lineage. These cytopenias could be due to suppression of normal hematopoeisis by the kinase inhibitors, inadequate normal stem cell reserve, or a second clonal bone marrow disorder. Routine metaphase cytogenetics (MC) are a relatively insensitive method to identify clonal hematopoiesis, as chromosomal abnormalities are found in only about 50–60% of patients with MDS. Another clonality assay that can be used in females relies on a CAG trinucleotide repeat polymorphism adjacent to methylated restriction endonuclease sites in the human androgen receptor gene (HUMARA) on the X chromosome.15 This assay has demonstrated the presence of skewed X chromosome inactivation (XCI) in approximately half of patients with newly diagnosed MDS.16 High-resolution single nucleotide polymorphisms arrays (SNP-A) can discover chromosomal lesions that cannot be detected using MC, including microdeletions, microduplications, and gene copy-number neutral loss of heterozygosity (LOH) or uni-parental disomy (UPD).17–20 The HUMARA and SNP-A assays could complement routine MC in the detection of clonal hematopoietic processes. Therefore, bone marrow and peripheral blood samples from CML patients who achieved a major cytogenetic remission (MCyR) on dasatinib therapy were evaluated for evidence of clonal hematopoiesis using MC, HUMARA and SNP-A analysis.
Materials and Methods
Experimental Subjects
A total of 79 consecutive patients with imatinib-resistant or intolerant, Ph positive CML were treated with dasatinib (Sprycel) at UCLA Medical Center. There were 49 patients in chronic phase, 11 in accelerated phase, 11 in myeloid blast crisis, and 8 in lymphoid blast crisis. Of these, 27 patients experienced a sustained MCyR and had ≤15% Ph+ cells on bone marrow MC testing (Table 1). These patients had a median disease duration of 7 years (range 1–15 years) and had received a median of 3 prior therapies (range 1–4). One patient (#9) had relapsed after an allogeneic bone marrow transplant and another (#21) had received an autologous peripheral blood stem cell transplant. Six patients had advanced disease prior to therapy and 21 were in chronic phase. At least one peripheral blood lineage was abnormal in 16 patients, of whom 4 had pancytopenia. Peripheral blood cytopenias were defined as hemoglobin <10 mg/dL, absolute neutrophil count <1,000/mcL, and platelet count <100,000/mcL. All patients signed an IRB-approved informed consent prior to donating samples for this study. The characteristics of the study subjects at the time of sampling for SNP-A analysis are provided in Table I.
Table 1.
Patient Characteristics
| Patient | Age/Gender | Duration (yrs) | Prior Therapy | Stage | PB Abnormalities | BM Cytogenetics | HUMARA | SNP Array |
|---|---|---|---|---|---|---|---|---|
| 1 | 68/F | 12 | IFN, Hydrea, IM | CP | Anemia | 46,XX[19]/46,XX,t(9;22)[1] | negative | negative |
| 2 | 43/M | 2 | HU, IM | CP | None | 46,XY[20] | negative | |
| 3 | 66/M | 7 | HU, IFN, IM | AP | Thrombocytopenia | 46,XY[20] | negative | |
| 4 | 50/F | 2 | IM | CP | Pancytopenia | 46,XX[12]/47,XX,+8[5]/46,XX,t(9;22)[3] | negative | negative |
| 5 | 48/M | 7 | HU, IFN, IM | CP | None | 46,XX[18]/46,XX,t(9;22)[2] | negative | |
| 6 | 77/M | 9 | HU, IFN, AraC, IM | CP | None | 46,XY[20] | negative | |
| 7 | 57/F | 13 | HU, IFN, IM | CP | None | 46,XX[20] | negative | negative |
| 8 | 76/M | 8 | IFN, HU, AG, IM | CP | Thrombocytosis | 46,XY[20] | UPD 17p | |
| 9 | 62/M | 4 | IM, BMT | BC | Neutropenia | 46,XY[20] | negative | |
| 10 | 53/F | 12 | HU, IFN, IM | CP | Anemia | 46,XX[19]/46,XX,t(9;22)[1] | negative | negative |
| 11 | 80/F | 1 | IM | CP | None | 46,XX[20] | negative | negative |
| 12 | 55/F | 4 | HU, IM | CP | Anemia | 46,XX[18]/46,XX,del(20q)[2] | negative | negative |
| 13 | 56/F | 15 | HU, IFN, IM | AP | Pancytopenia | 46,XX[18]/46,XX,t(9;22)[2] | negative | negative |
| 14 | 44/M | 10 | HU, IFN, IM | CP | None | 46,XY[20] | negative | |
| 15 | 69/F | 9 | IFN, HU, IM, 17AAG | CP | None | 46,XX[20] | negative | negative |
| 16 | 59/F | 3 | IM | CP | None | 46,XX[20] | negative | negative |
| 17 | 41/M | 3 | IM | CP | Pancytopenia | 46,XY[1]/45,XY,−7[18]/47,idem,+21,+22[2] | −7 | |
| 18 | 59/M | 5 | IFN, IM, HU | CP | Thrombocytopenia | 46,XX[18]/46,XX,t(9;22)[2] | negative | |
| 19 | 42/M | 6 | HU, IFN | CP | Thrombocytopenia | 46,XY[14]/47,XY,+8[6] | +8 | |
| 20 | 77/F | 11 | IFN, HU, IM | AP | Anemia | 46,XX[20] | negative | negative |
| 21 | 52/F | 11 | ICE,IFN, BMT, IM | CP | Anemia | 46,XY[17]/47,XY,+8[4] | negative | negative |
| 22 | 52/F | 7 | IFN, HU, AG, IM | BC | Anemia | 46,XX[18]/47,XX,+8[2] | negative | negative |
| 23 | 78/F | 7 | IFN, HU, AG, IM | CP | Anemia | 46,XX[18]/46,XX,t(9;22)[2] | positive | negative |
| 24 | 68/M | 13 | HU, IFN, IM | CP | None | 46,XY[20] | negative | |
| 25 | 70/F | 12 | HU, IFN, AraC, IM | AP | Pancytopenia | 46,XX[17]/46,XX,t(9;22)[1]/46,idem,complex[2] | negative | negative |
| 26 | 78/M | 8 | HU, IFN, IM | CP | None | 46,XX[18]/46,XX,t(9;22)[2] | negative | |
| 27 | 80/F | 6 | HU, IFN, IM | CP | None | 46,XX[30] | negative | negative |
Abbreviations: HU = hydroxyurea, ICE = ifosfamide/carboplatin/etoposide, IFN = interferon, IM = imatinib, 17AAG = 17-allylaminodemethoxy geldanamycin, AG = anagrelide, AraC = cytarabine, BMT = autologous bone marrow transplant, CP = chronic phase, AP = accelerated phase, BC = blast crisis, PB = peripheral blood, BM = bone marrow, complex = inv(3)(p21p25),t(10;18;4)(q22;q21;p16),inv(13)(q12q22), HUMARA = human androgen receptor polymerase chain reaction assay, SNP = single nucleotide polymorphism, UPD = uniparental disomy
Cell Processing and DNA Isolation
Mononuclear cell fractions of peripheral blood and bone marrow were isolated using Ficoll-Hypaque and were cryopreserved until they were used. The CD3 positive (CD3+) T lymphocytes were separated from these samples using anti-CD3 MicroBeads and the MiniMACS separator. This yielded CD3+ cells with purity in excess of 90%. DNA was prepared from CD3+ and CD3 negative (CD3−) fractions of the peripheral blood and bone marrow mononuclear cells using a Puregene DNA Purification System (Gentra Systems).
HUMARA PCR Assay
DNA from bone marrow mononuclear cells was used to screen for clonality using the HUMARA PCR assay, as described.21 Clonal hematopoiesis was further assessed using CD3− peripheral blood mononuclear cells (experimental samples) and germ-line DNA from CD3+ T cells (control samples). Briefly, DNA (250 ng) was digested overnight with RsaI, or RsaI and HpaII. The restriction enzymes were heat-inactivated, then a portion of the digested DNA (50 mg) was PCR amplified using fluorescently-labeled primers to the HUMARA gene. The PCR products were diluted and loaded on a capillary electrophoresis apparatus (ABI 3730, Applied Biosystems). The data were analyzed using Genemapper 4.0 software. An allele ratio was calculated for the two peaks in each sample by dividing the area under the RsaI digested peaks by the area under the RsaI/HpaII digested peaks to yield an XCI ratio. The allele ratios of the CD3− samples were divided by the ratios of the CD3+ samples to generate a corrected XCI ratio. By convention, a corrected XCI ratio of >3 or <0.33 was considered to be non-random and indicative of a clonal cell population.22–24
SNP-A Analysis
The Gene Chip Mapping 250K Assay Kit (Affymetrix, Santa Clara, CA) was used according to manufacturer’s instructions, as previously described.19,20 Lesions identified by SNP-A were compared with the Cancer Genome Anatomy Project database (http://cgap.nci.nih.gov) and our own internal control series (N=713) to exclude known copy number variants (CNVs). In these analyses, CNVs and non-clonal areas of pseudo-UPD or germ-line encoded UPD were excluded using a stringent algorithm. Lesions that were seen by MC and confirmed by SNP-A were not further validated. For other lesions, paired germ-line DNA derived from corresponding CD3+ lymphocytes were analyzed to confirm the somatic nature of the observed defects.
Analysis of TP53 mutations
Screening for TP53 mutations in patient #8 was performed using genomic DNA and cDNA. Exons 5, 6, 7, 8 and 9 of the TP53 gene were amplified from genomic DNA by PCR as three separate amplicons using the following primer sets:
exons 5–6 E5-6F 5′-GTTTCTTTGCTGCCGTCTTCCAGT-3′
E5-6R 5′-AGGGAGGTCAAATAAGCAGCAGG-3′
exon 7 E7F 5′-TGGGCCTGTGTTATCTCCTAGGTT-3′
E7R 5′-TGTGATGAGAGGTGGATGGGTAGT-3′
exons 8–9 E8-9F 5′-GCCTCTTGCTTCTCTTTTCC-3′
E8-9R 5′-CGGCATTTTGAGTGTTAGACTG-3′
TP53 cDNA was amplified with the following primers:
P53cF 5′-GCCGTCCCAAGCAATGGATGATTT-3′
P53cR 5′-AGTTCCAAGGCCTCATTCAGCTCT-3′.
Amplicons were electrophoresed on a 1.5 % agarose gel to confirm specificity. TP53 mutation analysis was done by direct sequencing in both orientations. PCR products were prepared for direct sequencing using standard conditions and analyzed using a 3730×1 DNA analyzer (Applied Biosystems). In addition, PCR products of exons 5–6 and cDNA were cloned into pCR 2.1 vector (Invitrogen), and the products were analyzed by sequencing.
Results
Clonal Hematopoiesis Detected by Cytogenetic Analysis
Routine MC demonstrated Ph-CCA in 6 of 27 patients with a sustained MCyR to dasatinib therapy (Table I). The patients had a mean disease duration of 4.5 years (range 1–11) and had received a median of 2 therapies prior to dasatinib (range 1–4). Three patients developed the Ph-CCA previously, while receiving imatinib. The pre-dasatinib cytogenetics for these patients were: (patient #4) 47,XX,+8[10]/46,XX,t(9;22)[10], (patient #17) 46,XY[3]/45,XY,−7[9]/46,XX,t(9;22)[8], and (patient #21) 46,XX[16]/47,XX,+8[6]/46,XX,t(9;22)[8]. In the remaining 3 patients, the Ph-CCA were +8, +8 and 20q-, and were first detected 6, 8 and 12 months after starting dasatinib, respectively. All 6 patients had one or more peripheral blood cytopenia when the Ph-CCA were detected. All CCA persisted for at least 6 months (range 6 to >27 months). Changes in the size of Ph− clones over time are illustrated for the patients with the longest follow up (Figure 1). Only the −7 clone, observed in patient #17, comprised >50% of the bone marrow metaphases at any time. In 2 patients (#4, #19), the +8 clones spontaneously disappeared after 27 and 21 months, respectively. Morphological evidence of MDS did not develop in any of the patients.
Figure 1. Time course of Ph-CCA in patients with the longest follow up.
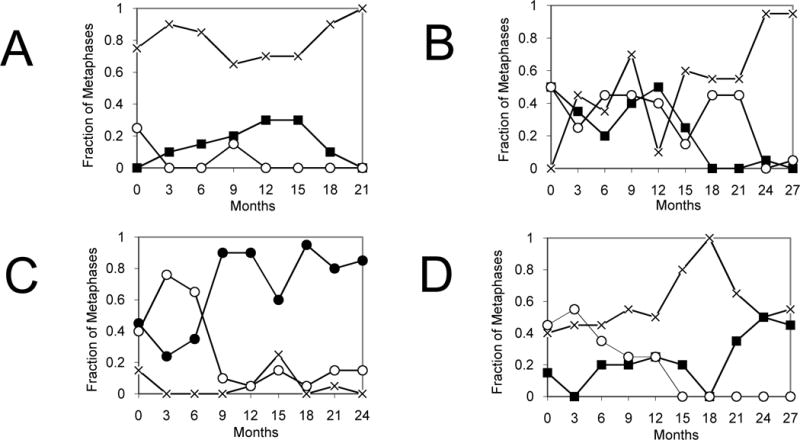
The abundance of the Ph+ and Ph− clonal populations are shown over time for (A) patient #19, (B) patient #4, (C) patient #17, and (D) patient #21. Karyotypes: normal -x- ; 46, t(9;22) -○- ; 47,+8 -■- ; 45,−7 -●- .
Clonal Hematopoiesis Detected by HUMARA PCR Assay
Samples from 15 female patients were evaluated for clonality using the HUMARA assay (Table I). One patient (#23) without Ph-CCA demonstrated skewing of XCI in the CD3− peripheral blood mononuclear cells that was not present in the CD3+ cells (Figure 2). The mean XCI ratio from two separate experiments performed in duplicate was 4.3, and 3.1 after correction for germline XCI. Using the established criteria that the corrected XCI ratio be >3:1, the HUMARA assay was inadequately sensitive to detect the small clonal populations present in the four female patients with Ph-CCA. These patients had a Ph-clone that comprised 5–20% of the cells analyzed by MC at the time of sampling.
Figure 2. HUMARA assay results of patient #23.
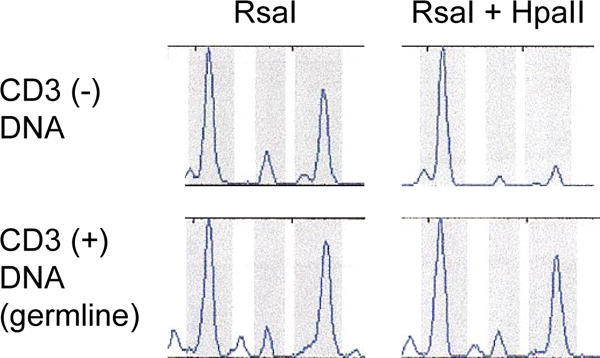
Skewing of the XCI pattern was demonstrated for CD3− peripheral blood mononuclear cells, but not for the CD3+ lymphocytes. The mean XCI ratio determined by two separate experiments performed in duplicate was 4.3, and after correction for germ-line XCI was 3.1.
Clonal Hematopoiesis Detected by SNP-A Analysis
SNP-A analysis confirmed the presence of −7 in patient #17 and +8 in patient #19, whose Ph− CCAs comprised 95% and 30% of the bone marrow metaphases by MC at the time of sampling. Clones present at a frequency lower than 30% by MC were not detectable. In addition, SNP-A analysis demonstrated 6 chromosomal abnormalities not present in a database of copy number SNP variations in normal individuals. When CD3+ T lymphocytes from these patients were analyzed, 5 lesions, including gain 10p15.2-p15.3, del 2q37.3, gain 11q12.5, del 8q24.23 and del 13p14.2 were found to be germline-encoded. However, evidence of UPD 17(p12-pter) was found to be present in CD3− peripheral blood mononuclear cells but not in CD3+ lymphocytes of patient #8 (Figure 3). This patient had persistent thrombocytosis requiring hydroxyurea and anagrelide administration in spite of a sustained complete cytogenetic remission and complete molecular response (PCR negative for BCR-ABL translocation) to dasatinib. Therefore, he had clinical and SNP-A evidence of a second clonal hematologic process.
Figure 3. SNP-A analysis of chromosome 17 using peripheral blood mononuclear cells from patient #8.
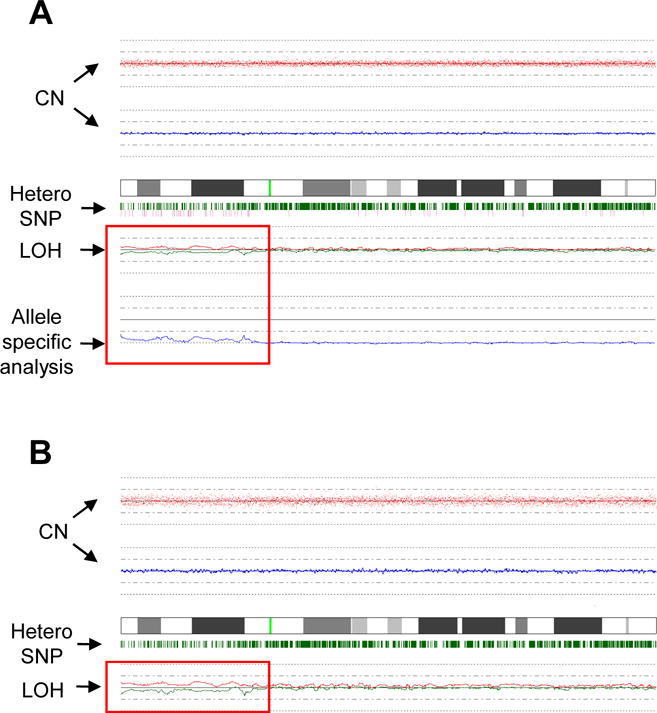
(A) A comparison of DNA from CD3− cells of patient #8 versus DNA from pooled controls demonstrates loss of heterozygosity (LOH) without copy number abnormality involving chromosome 17(p12-pter) is consistent with UPD. (B) SNP-A karyotyping using DNA from CD3− cells and CD3+ cells from patient #8 confirms that the chromosome 17(p12-pter) UPD is an acquired lesion in the Ph− cells.
Alternative Splicing of the TP53 Gene in Patient #8
Sequencing of the TP53 gene using genomic DNA isolated from CD3− mononuclear cells of patient #8 revealed a deletion of 5 nucleotides from the 5′ splice site of exon 6 (Figure 4A). This deletion was not observed in DNA obtained from CD3+ cells of the patient. Deletion of TP53 exon 6 was observed upon sequencing of cDNA from CD3−, but not CD3+, mononuclear cells (Figure 4B). Therefore, deletion of the 5′ splice site of TP53 exon 6 resulted in skipping of that exon. This was predicted to result in a frame shift and premature introduction of a stop codon, producing a non-functional p53 protein (Figure 4C).
Figure 4. Alternative splicing of the TP53 gene in patient #8.
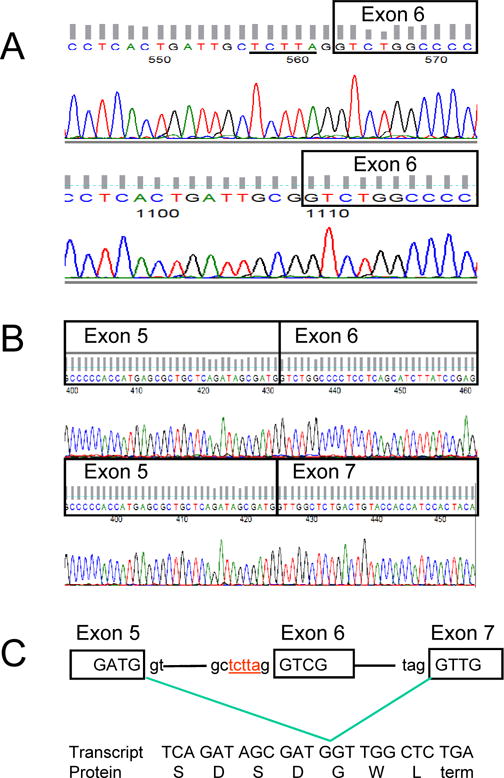
(A) Exon 6 of the TP53 gene was sequenced using genomic DNA from CD3+ cells of patient #8 as germline control (upper sequence). The underlined nucleotides were absent when genomic DNA from CD3− peripheral blood mononuclear cells was sequenced (lower sequence). (B) Sequencing of TP53 using cDNA prepared from CD3− cells demonstrated loss of exon 6 (lower sequence) while cDNA from CD3+ cells revealed normal sequence (upper sequence). (C) Deletion of the 5 nucleotides (shown in red) in the 5′ splice region of exon 6 lead to alternative splicing and a frame shift with introduction of a premature stop codon.
Patient Outcomes
All 6 of the patients with Ph-CCA had peripheral blood cytopenias involving one or more lineage. Two patients received chronic filgrastim administration, one received an erythropoiesis stimulating agent (ESA), and two received both agents. The +8 clone observed repeatedly in two patients (#4, #19) disappeared after 21 and 27 months, respectively (Figure 1). In spite of this, patient #4 continued to require ESA and filgrastim support. Patient #17 with a −7 clone had his Ph− clone expand to comprise >50% of the bone marrow metaphases (Figure 1). However, morphologic evidence of MDS was absent on repeated bone marrow examinations over more than 24 months of follow up. One patient (#22) with +8 Ph-CCA experienced disease progression to blast crisis, but achieved a third chronic phase on the Aurora kinase inhibitor PHA-739358.25 After achieving another CCyR, the +8 clone recurred transiently. No patient developed MDS or AML after a follow up of 6–36 months. The patient who was found to have marked skewing of X chromosome inactivation (#23) had anemia requiring ESA support. She did not experience any morphologic evidence of MDS after 21 months of follow up. Patient #8 with UPD of chromosome 17(p12-pter) by SNP-A analysis had clinical features of a second myeloproliferative disorder resembling essential thrombocytosis. He had a normal karyotype by routine MC and did not have the V617F mutation of JAK2. Anagrelide and hydroxyurea were administered to control the platelet count. Nevertheless, he experienced recurrent strokes and died with progressive cognitive decline.
Discussion
Patients who achieved a sustained MCyR on dasatinib had a high rate of clonal hematopoiesis in the Ph− cells identified through complementary methodologies including MC, HUMARA assay and SNP-A based karyotyping. The HUMARA assay has been used to study clonality in MDS, with approximately half of patients demonstrating a clonal XCI pattern using stringent criteria.24 We identified one patient with clonal XCI who lacked Ph-CCA. Standard criteria for clonality with the HUMARA assay require that the clonal population comprises approximately 75% of the sample.21–23 As a result, the oligoclonal populations in the female patients with Ph-CCA could not be detected using HUMARA. SNP-A analysis confirmed the CCA only in the 2 patients who had a Ph− clone comprising ≥30% of the bone marrow metaphases. This level of sensitivity is consistent with previously reported experience using SNP-A in MDS.19 In addition, one patient without Ph-CCA was found to harbor a UPD of chromosome 17(p12-pter). The overall frequency of clonality in the Ph− cells or our patient population was 8/27 (30%) using MC, HUMARA and SNP-A analyses. Because the clonal populations identified by MC were predominantly small and below the level of detection by HUMARA and SNP-A assays, the true frequency of oligoclonal Ph− cells is probably higher than we could detect.
Persistent Ph-CCA were observed in 18% of our patients who had achieved at least a major cytogenetic response to dasatinib (10% of all patients treated in the chronic phase). Our findings are similar to previously reported findings of Ph-CCA in 5 of 33 (15%) patients who experienced a MCyR on dasatinib.26 Somewhat lower frequencies of Ph-CCA have been reported in imatinib-treated patients. Several large series of patients receiving imatinib after interferon failure reported frequencies of Ph-CCA ranging from 3.4% to 6.1% of treated patients.5,9,11 Assuming that 60% of patients treated with imatinib in the second-line achieve a MCyR, the frequency of Ph-CCA in these patients approaches that observed in patients responding to dasatinib. The Ph-CCA were first observed during prior imatinib therapy in 3 of our 6 patients, and 2 of 5 patients in the previous dasatinib series.26 The higher rate of Ph-CCA in dasatinib-treated patients may be due to a longer median disease duration or more prolonged exposure to tyrosine kinase inhibition.
The etiology of Ph-CCA remains unclear. Because a few CML patients were reported to develop Ph-CCA during sustained cytogenetic responses to interferon therapy27, TKIs are not required for the evolution of this process. It is possible that the Ph-CCA resulted from the DNA damage that induced the development of the CML clone. The deep remissions and prolonged survival conferred by TKI therapy may permit the subsequent expansion of the Ph− clones. However, TKIs have been shown to impair DNA repair in Ph− cells through inhibition of c-Abl activity. Following DNA damage, c-Abl expression increases and it translocates from the cytoplasm to the nucleus where it induces cell cycle arrest or apoptosis.28,29 Inhibition of c-Abl kinase activity by imatinib or dasatinib in Ph− cells exposed to radiation results in a higher frequency of DNA mutations and delayed DNA repair.30 Therefore, prolonged exposure to the c-Abl TKIs may contribute to the development of Ph-CCA in normal hematopoietic progenitor cells.
The clinical significance of the second clonal hematopoietic populations remains to be determined. Only 3 patients developed MDS or acute myeloid leukemia (AML) out of 1701 CML patients treated with imatinib at MD Anderson.14 In all, approximately 17 cases of MDS or AML have been reported in CML patients receiving kinase inhibitor therapy, most of which have been associated with −7, 7q-, or complex cytogenetic abnormalities.11,14,31
Although peripheral blood cytopenias were common in our patient population, one patient had clinical features of a second myeloproliferative disorder resembling essential thrombocythemia without V617F mutation of JAK2, and with a normal karyotype by routine MC. SNP-A analysis demonstrated UPD17p in this patient. Recently, UPD17p or del17p have been observed in advanced MDS and secondary AML, especially in association with monosomy 7/deletion 7q and/or monosomy 5/deletion 5q.32 These patients also had multiple deletions and gains of other chromosomal material by SNP-A analysis. Biallelic missense mutations in the DNA-binding domain of p53 were identified in most patients and were associated with poor prognosis. However, in our patient, UPD17 occurred as a sole defect. Because of the association between UPD of chromosome 17p and p53 mutations, genomic sequencing of this gene was performed. A 5 bp deletion that destroyed the 5′ splice site of exon 6 was identified in bone marrow cells from this patient. Alternative splicing resulted in loss of exon 6, which deleted part of the DNA-binding domain and introduced a frame shift resulting in the introduction of a premature stop codon. This same splicing variant was reported to occur frequently in chronic lymphocytic leukemia.33 Mutation of the 5′ splicing consensus region of exon 6 was not observed in these patients however. The truncated p53 protein encoded by the frame-shifted mRNA lacked transcriptional activity by FASAY analysis.33 Deletions of 17p or inactivating mutations of p53 have been described in patients with essential thrombocythemia, especially in the setting of disease progression to MDS or AML, and following prolonged exposure to hydroxyurea.34,35
In summary, patients with longstanding CML frequently have peripheral blood abnormalities in association with a second hematological clone. The combined use of bone marrow MC, HUMARA assay and SNP analysis are complementary in identifying and characterizing these clonal processes, but likely underestimate their frequency due to insensitivity for detecting oligoclonal populations. Supportive care measures for peripheral blood cytopenias are appropriate for most patients with Ph− clones, given their low risk of progression.
Acknowledgments
This research was supported by the NIH National Center for Research Resources. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Additional support came from the Leukemia and Lymphoma Society, and the Greg Wojahn Memorial Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
Ronald Paquette has received speaking honoraria from Bristol Myers Squibb and Novartis. Neil Shah has received speaking honoraria from Bristol Myers Squibb.
Contributions
Ronald Paquette designed the study, acquired and analyzed data, wrote the manuscript and approved the final version. John Nicoll and Meenal Chalukya acquired and analyzed data, revised the manuscript and approved the final version. Lucas Gondek and Monika Jasek acquired and analyzed data, wrote and revised the manuscript, and approved the final version. Charles Sawyers and Neil Shah acquired data, revised the manuscript and approved the final version. Jaroslaw Maciejewski designed the study, analyzed data, revised the manuscript and approved the final version.
References
- 1.Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112:4808–4817. doi: 10.1182/blood-2008-07-077958. [DOI] [PubMed] [Google Scholar]
- 2.Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005:2640–2653. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- 3.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–41. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 4.Hochhaus A, Kantarjian HM, Baccarani M, Lipton JH, Apperley JF, Druker BJ, et al. Dasatinib induces notable hematologic and cytogenetic responses in chronic-phase chronic myeloid leukemia after failure of imatinib therapy. Blood. 2007;109:2303–9. doi: 10.1182/blood-2006-09-047266. [DOI] [PubMed] [Google Scholar]
- 5.Medina J, Kantarjian H, Talpaz M, O’Brien S, Garcia-Manero G, Giles F, et al. Chromosome abnormalities in Philadelphia chromosome-negative metaphases appearing during imatinib mesylate therapy in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase. Cancer. 2003;98:1905–11. doi: 10.1002/cncr.11729. [DOI] [PubMed] [Google Scholar]
- 6.Bumm T, Müller C, Al-Ali HK, Krohn K, Shepherd P, Schmidt E, et al. Emergence of clonal cytogenetic abnormalities in Ph− cells in some CML patients in cytogenetic remission to imatinib but restoration of polyclonal hematopoiesis in the majority. Blood. 2003;101:1941–1949. doi: 10.1182/blood-2002-07-2053. [DOI] [PubMed] [Google Scholar]
- 7.O’Dwyer ME, Gatter KM, Loriaux M, Druker BJ, Olson SB, Magenis RE, et al. Demonstration of Philadelphia chromosome negative abnormal clones in patients with chronic myelogenous leukemia during major cytogenetic responses induced by imatinib mesylate. Leukemia. 2003;17:481–487. doi: 10.1038/sj.leu.2402848. [DOI] [PubMed] [Google Scholar]
- 8.McMullin MF, Humphreys M, Byrne J, Russell NH, Cuthbert RJ, O’Dwyer ME. Chromosomal abnormalities in Ph− cells of patients on imatinib. Blood. 2003;102:2700–2701. doi: 10.1182/blood-2003-06-1943. [DOI] [PubMed] [Google Scholar]
- 9.Terre C, Eclache V, Rousselot P, Imbert M, Charrin C, Gervais C, et al. Report of 34 patients with clonal chromosomal abnormalities in Philadelphia negative cells during imatinib treatment of Philadelphia-positive chronic myeloid leukemia. Leukemia. 2004;18:1340–1346. doi: 10.1038/sj.leu.2403399. [DOI] [PubMed] [Google Scholar]
- 10.Loriaux M, Deininger M. Clonal cytogenetic abnormalities in Philadelphia chromosome negative cells in chronic myeloid leukemia patients treated with imatinib. Leuk Lymphoma. 2004;45:2197–2203. doi: 10.1080/10428190410001723278. [DOI] [PubMed] [Google Scholar]
- 11.Deininger MWN, Cortes J, Paquette R, Park B, Hochhaus A, Baccarani M, et al. The prognosis for patients with chronic myeloid leukemia who have clonal cytogenetic abnormalities in Philadelphia chromosome-negative cells. Cancer. 2007;110:1509–1519. doi: 10.1002/cncr.22936. [DOI] [PubMed] [Google Scholar]
- 12.Jabbour E, Kantarjian HM, Abruzzo LV, O’Brien S, Garcia-Manero G, Vertovstek S, et al. Chromosomal abnormalities in Philadelphia chromosome-negative metaphases appearing during imatinib mesylate therapy in patients with newly diagnosed chronic myeloid leukemia in chronic phase. Blood. 2007;110:2991–2995. doi: 10.1182/blood-2007-01-070045. [DOI] [PubMed] [Google Scholar]
- 13.Fenaux P, Morel P, Lai JL. Cytogenetics of myelodysplastic syndromes. Sem Hematol. 1996;33:127–138. [PubMed] [Google Scholar]
- 14.Kovitz C, Kantarjian H, Garcia-Manero G, Abruzzo LV, Cortes J. Myelodysplastic syndromes and acute leukemia developing after imatinib mesylate therapy for chronic myeloid leukemia. Blood. 2006;108:2811–2813. doi: 10.1182/blood-2006-04-017400. [DOI] [PubMed] [Google Scholar]
- 15.Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the androgen-receptor gene correlates with X chromosome inactivation. Am J Human Genetics. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 16.Delabesse E, Aral S, Kamoun P, Varet B, Turhan AG. Quantitative non-radioactive clonality analysis of human leukemic cells and progenitors using the human androgen receptor (AR) gene. Leukemia. 1995;9:1578–1582. [PubMed] [Google Scholar]
- 17.Maciejewski JP, Mufti GJ. Whole genome scanning as a cytogenetic tool in hematologic malignancies. Blood. 2008;112:965–974. doi: 10.1182/blood-2008-02-130435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohamedali A, Gaken J, Twine NA, Ingram W, Westwood N, Lea NC, et al. Prevalence and prognostic significance of allelic imbalance by single-nucleotide polymorphism analysis in low-risk myelodysplastic syndrome. Blood. 2007;110:3365–3373. doi: 10.1182/blood-2007-03-079673. [DOI] [PubMed] [Google Scholar]
- 19.Gondek LP, Tiu R, O’Keefe CL, Sekeres MA, Theil KS, Maciejewski JP. Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood. 2008;111:1534–1542. doi: 10.1182/blood-2007-05-092304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunbar AJ, Gondek LP, O’Keefe CL, Makishima H, Rataul MS, Szpurka H, et al. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 2008;68:10349–10357. doi: 10.1158/0008-5472.CAN-08-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kopp P, Jaggi R, Tobler A, Borisch B, Oestreicher M, Sabacan L, et al. Clonal X-inactivation analysis of human tumors using the human androgen receptor gene (HUMARA) polymorphism: a non-radioactive and semiquantitative strategy applicable to fresh and archival tissue. Mol Cell Probes. 1997;11:217–228. doi: 10.1006/mcpr.1997.0099. [DOI] [PubMed] [Google Scholar]
- 22.Vogelstein B, Fearon ER, Hamilton SR, Preisinger AC, Willard HF, Michelson AM, et al. Clonal analysis using recombinant DNA probes from the X-chromosome. Cancer Res. 1987;47:4806–4813. [PubMed] [Google Scholar]
- 23.Willman CL, Busque L, Griffith BB, Favara BE, McClain KL, Duncan MH, et al. Langerhans’-cell histiocytosis (Histiocytosis X) – a clonal proliferative disease. N Engl J Med. 1994;331:154–160. doi: 10.1056/NEJM199407213310303. [DOI] [PubMed] [Google Scholar]
- 24.Delabesse E, Aral S, Kamoun P, Varet B, Turhan AG. Quantitative non-radioactive clonality analysis of human leukemic cells and progenitors using the human androgen receptor (AR) gene. Leukemia. 1995;9:1578–1582. [PubMed] [Google Scholar]
- 25.Paquette RL, Shah NP, Sawyers CL, Martinelli G, Nicoll J, et al. PHA-739358, an Aurora kinase inhibitor, induces clinical responses in chronic myeloid leukemia harboring T315I mutations of BCR-ABL. Blood. 2007;110:312a. [Google Scholar]
- 26.Fabarius A, Haferlach C, Muller MC, Erben P, Lahaye E, Giehl M, et al. Dynamics of cytogenetic aberrations in Philadelphia chromosome positive and negative hematopoiesis during dasatinib therapy of chronic myeloid leukemia patients after imatinib failure. Haematologica. 2007;92:834–837. doi: 10.3324/haematol.11064. [DOI] [PubMed] [Google Scholar]
- 27.Fayad L, Kantarjian H, O’Brien S, Seong D, Albitar M, Keating M, et al. Emergence of new clonal abnormalities following interferon-alpha induced complete cytogenetic response in patients with chronic myeloid leukemia: report of three cases. Leukemia. 1997;11:767–771. doi: 10.1038/sj.leu.2400642. [DOI] [PubMed] [Google Scholar]
- 28.Kharbanda S, Yuan Z-M, Weichselbaum R, Kufe D. Determination of cell fate by c-Abl activation in the response to DNA damage. Oncogene. 1998;17:3309–3318. doi: 10.1038/sj.onc.1202571. [DOI] [PubMed] [Google Scholar]
- 29.Levav-Cohen Y, Goldberg Z, Zuckerman V, Grossman T, Haupt S, Haupt Y. C-Abl as a modulator of p53. Biochem Biophys Res Commun. 2005;331:737–749. doi: 10.1016/j.bbrc.2005.03.152. [DOI] [PubMed] [Google Scholar]
- 30.Fanta S, Sonnenberg M, Skorta I, Duyster J, Miething C, Aulitzky WE, et al. Pharmacologic inhibition of c-Abl compromises genetic stability and DNA repair in Bcr-Abl-negative cells. Oncogene. 2008;27:4380–4384. doi: 10.1038/onc.2008.68. [DOI] [PubMed] [Google Scholar]
- 31.Kovitz C, Kantarjian H, Garcia-Manero G, Abruzzo LV, Cortes J. Myelodysplastic syndromes and acute leukemia developing after imatinib mesylate therapy for chronic myeloid leukemia. Blood. 2006;108:2811–2813. doi: 10.1182/blood-2006-04-017400. [DOI] [PubMed] [Google Scholar]
- 32.Jasek M, Gondek LP, Bejanyan N, Tiu RV, Huh J, Theil KS, et al. SNP array-based analysis of chromosome 17 reveals biallelic TP53 mutations due to uniparental disomy 17p in advanced MDS and AML with cooperating deletions of chromosomes 5 and 7. Blood. 2008;112:875a. [Google Scholar]
- 33.Pekova S, Cmejla R, Smolej L, Kozak T, Spacek M, Prucha M. Identification of a novel, transactivation-defective splicing variant of p53 gene in patients with chronic lymphocytic leukemia. Leukemia Res. 2008;32:395–400. doi: 10.1016/j.leukres.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 34.Sterkers Y, Preudhomme C, Laï JL, Demory JL, Caulier MT, Wattel E, et al. Acute myeloid leukemia and myelodysplastic syndromes following essential thrombocythemia treated with hydroxyurea: high proportion of cases with 17p deletion. Blood. 1998;91:616–22. [PubMed] [Google Scholar]
- 35.Neri A, Fracchiolla NS, Radaelli F, Boletini A, Ribera S, Migliorini C, et al. p53 tumour suppressor gene and RAS oncogenes: molecular analysis in the chronic and leukaemic phases of essential thrombocythaemia. Br J Haematol. 1996;93:670–3. doi: 10.1046/j.1365-2141.1996.d01-1690.x. [DOI] [PubMed] [Google Scholar]