

ABSTRACT
Autophagy is required for benign hepatic tumors to progress into malignant hepatocellular carcinoma. In our recent studies, we found that autophagy, or more specifically mitophagy, was required to suppress TP53 and induce the expression of the transcription factor NANOG to maintain hepatic cancer stem cells and promote hepatocarcinogenesis.
KEYWORDS: Autophagy, mitophagy, p53, PINK1, NANOG, hepatocarcinogenesis, cancer stem cells
A specific form of autophagy (i.e., macroautophagy; hereafter autophagy) removes protein aggregates and damaged organelles and is important for maintaining cellular homeostasis. Previous studies using mice with hepatocyte-specific knockout of Atg5 or Atg7, two genes essential for autophagy, indicated that autophagy was required to suppress the initiation of hepatocarcinogenesis.1,2 In these mice, dysfunctional mitochondria accumulated in hepatocytes, resulting in the increase of reactive oxygen species and oxidative DNA damage. The causative role of oxidative stress in the development of liver tumors in these mice was confirmed by the treatment of these mice with the antioxidant N-acetylcysteine, which reduced the incidence of liver tumors.2 Interestingly, these knockout mice developed only benign hepatic tumors with no malignant hepatocellular carcinoma (HCC), not even after they were treated with the carcinogen diethylnitrosamine (DEN), which induced HCC in control mice.2 These results indicated that, although autophagy was required to suppress the initiation of hepatocarcinogenesis, it was also required to promote hepatocarcinogenesis once this process had been initiated. How autophagy might be involved in promoting hepatocarcinogenesis was no clear.
To understand the role of autophagy in the promotion of hepatocarcinogenesis, we isolated liver tumors from control mice and Atg5-knockout mice that had been treated with DEN and examined the possible effect of autophagy on CD133+ and CD49f+ cells. CD133 and CD49f are markers of cancer stem cells (CSCs),3 which are a rare type of cancer cells that are capable of self-renewal and the production of a heterogeneous population of progeny cells. CSCs can also induce vasculogenic mimicry, followed by the formation of blood vessels and increased blood supply, which is critical for the survival of cancer cells in solid tumors.4 We found that the loss of ATG5 would greatly reduce the number of CD133+CD49f+ cells.5 This observation suggested an important role of autophagy in the maintenance of hepatic CSCs. This possibility was confirmed by analyzing the effect of autophagy in vitro using human hepatoma cell lines. We found that the inhibition of autophagy reduced CD133+ cells and the induction of autophagy increased it. The CSC properties of CD133+ cells were confirmed by their ability to self-renew and proliferate in low-attachment plates and to grow tumors when grafted into immunodeficient nude mice.
Mitophagy is the selective removal of mitochondria by autophagy. It plays a critical role in the removal of damaged mitochondria and the control of mitochondrial quality.6 Interestingly, we found that, the same as autophagy, the inhibition and the induction of mitophagy also increased and decreased, respectively, CD133+ cells, raising the possibility that the effect of autophagy on CSCs might be solely mediated by mitophagy. Indeed, our further studies indicated that autophagy and mitophagy had the same effect on the tumor suppressor TP53, best known as p53. We found that the inhibition of either autophagy or mitophagy could increase the p53 level and its phosphorylation at serine-392 (S392), and the induction of either one of them had the opposite effect. Since p53 phosphorylated at S392 could bind to the NANOG promoter and transcriptionally suppress the expression of NANOG,5 a transcription factor critical for the self-renewal and stemness of CSCs, these results provided an explanation as to how autophagy or mitophagy regulate hepatic CSCs.
To further understand the relationship between mitophagy and p53, we analyzed the subcellular localization of p53 that was phosphorylated at S392. This phosphorylated form was found to localize primarily to the nucleus in control cells. It was also found to localize to the nucleus when mitophagy was inhibited. Surprisingly, this S392-phosphorylated form of p53 was found to colocalize with mitochondria when mitophagy was induced. As the induction of mitophagy reduced the level of p53 and its S392-phosphorylated form, this observation indicated that p53 was likely removed with mitochondria by mitophagy.
PINK1 is PTEN-induced kinase. It plays an important role in the induction of mitophagy.7 To further investigate how p53 was depleted when mitophagy was induced, we analyzed its possible relationship with PINK1. To our surprise, we found that the suppression of PINK1 expression reduced the phosphorylation of p53 at S392 and the over-expression of PINK increased it. By conducting in vitro protein phosphorylation assays using p53 as the substrate, we concluded that PINK1 was the elusive kinase that phosphorylates p53 at S392. Interestingly, in a subcellular fractionation experiment, we found that PINK1 physically interacted with p53 only in the mitochondrial fraction. Based on these results, we proposed that, when the basal mitophagy is not perturbed, a low level of PINK1 that is associated with mitochondria recruits and phosphorylates p53 at S392, which may dislodge from mitochondria and localize to the nucleus to partially control the expression of NANOG and the population of CSCs. When mitophagy is induced, p53 recruited by PINK1 to mitochondria is entrapped by autophagosomes and subsequently removed by mitophagy, resulting in the increased expression of NANOG and the CSC population (Fig. 1). However, when mitophagy is impaired, p53 phosphorylated by PINK1 cannot be removed by mitophagy and is transported into the nucleus where it suppresses the expression of NANOG, resulting in the reduction of CSCs and the suppression of hepatocarcinogenesis (Fig. 1).
Figure 1.
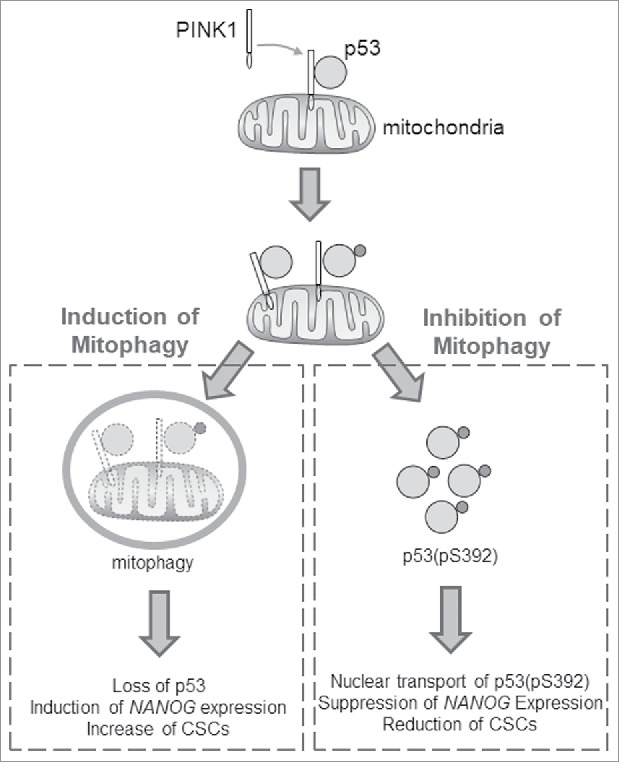
Effects of autophagy on hepatic cancer stem cells. PINK1 (PTEN-induced kinase 1) binds to the tumor suppressor p53 on mitochondria. When mitophagy is induced, p53 is removed with mitochondria by mitophagy. This leads to the induction of expression of the transcription factor NANOG and the increase of cancer stem cells (CSCs). When mitophagy is inhibited, p53 is phosphorylated by PINK1 at serine-392 (S392). This phosphorylated form of p53 (i.e., p53(pS392)) is then transported into the nucleus to suppress the expression of NANOG and the production of CSCs.
In conclusion, our findings demonstrated a critical role of mitophagy in the maintenance of the hepatic CSC population via the control of p53 activities. Although our studies were focused on liver tumors, it is likely that mitophagy may also play similar roles in the control of CSCs of other tumor types.
Funding Statement
This work was funded by the U.S. Public Health Service Grants CA177337, DK094652, DK100257 and AI129540.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800. doi: 10.1101/gad.2016211. PMID:21498569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tian Y, Kuo CF, Sir D, Wang L, Govindarajan S, Petrovic LM, Ou JH. Autophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesis. Cell Death Differ. 2015;22(6):1025–1034. doi: 10.1038/cdd.2014.201. PMID:25526090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uthaya Kumar DB, Chen CL, Liu JC, Feldman DE, Sher LS, French S, DiNorcia J, French SW, Naini BV, Junrungsee S, et al.. TLR4 Signaling via NANOG Cooperates With STAT3 to Activate Twist1 and Promote Formation of Tumor-Initiating Stem-Like Cells in Livers of Mice. Gastroenterology. 2016;150(3):707–719. doi: 10.1053/j.gastro.2015.11.002. PMID:26582088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu TJ, Sun BC, Zhao XL, Zhao XM, Sun T, Gu Q, Yao Z, Dong XY, Zhao N, Liu N. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene. 2013;32(5):544–553. doi: 10.1038/onc.2012.85. PMID:22469978 [DOI] [PubMed] [Google Scholar]
- 5.Liu K, Lee J, Kim JY, Wang L, Tian Y, Chan ST, Cho C, Machida K, Chen D, Ou JH. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol Cell. 2017;68(2):281-292 e285. doi: 10.1016/j.molcel.2017.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Redmann M, Dodson M, Boyer-Guittaut M, Darley-Usmar V, Zhang J. Mitophagy mechanisms and role in human diseases. Int J Biochem Cell Biol. Aug 2014;53:127–133. doi: 10.1016/j.biocel.2014.05.010. PMID:24842106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. doi: 10.1038/nrm3028. PMID:21179058 [DOI] [PMC free article] [PubMed] [Google Scholar]