

ABSTRACT
Defects in mitosis can lead to aneuploidy, which is a common feature of human cancers. Spindle Assembly Checkpoint (SAC) controls fidelity of chromosome segregation in mitosis to prevent aneuploidy. The ubiquitin receptor protein Ubiquitin Associated and SH3 Domain Containing B (UBASH3B) was recently found to control SAC silencing and faithful chromosome segregation by relocalizing Aurora B kinase to the mitotic microtubules. Accordingly, loss and gain of function of UBASH3B have strong effects on mitotic progression. Downregulation of UBASH3B prevents SAC satisfaction leading to inhibition of chromosome segregation, mitotic arrest, and cell death. In contrast, increased cellular levels of UBASH3B trigger premature and uncontrolled chromosome segregation. Interestingly, elevated levels of UBASH3B were found in aggressive tumors. Therefore, we raised the question whether the oncogenic potential of UBASH3B is linked to its role in chromosome segregation. Here we show that in cancer cells expressing high levels of UBASH3B and SAC proteins, downregulation of UBASH3B, can further potentiate SAC response inducing mitotic arrest and cell death. Moreover, data mining approaches identified a correlation between mRNA levels of UBASH3B and SAC components in a set of primary patient tumors including kidney and liver carcinomas. Thus, inhibition of UBASH3B may offer an attractive therapeutic perspective for cancers.
KEYWORDS: Aneuploidy, Aurora B, kidney carcinoma, liver carcinoma, SAC, UBASH3B
Introduction
Cell division requires duplication of genetic material during S-phase, which is incorporated into chromosomes and equally partitioned between 2 daughter cells during mitosis. The assembly of the mitotic spindle and its attachment to the sister kinetochores allows for proper chromosome segregation. The kinetochore attachment is a stochastic process and is strictly controlled by the Spindle Assembly Checkpoint (SAC) (also known as the mitotic checkpoint), which ensures that chromosome segregation in anaphase does not take place before all chromosomes are properly aligned at the metaphase plate.1 The kinetochore attachment defects prevent SAC satisfaction and inhibit chromosome segregation, often leading to prolonged mitotic arrest and ultimately cell death. The SAC activity implies assembly of the Mitotic Checkpoint Complex (MCC), which includes mitotic arrest deficient 2 (MAD2) and Bub1-related kinase (BubR1) proteins that localize to the kinetochores of misaligned chromosomes and inhibit onset of the anaphase.1 Defects in SAC response or MCC complex are associated with premature and unequal chromosome segregation. Aberrant segregation of chromosomes could result in chromosomal instability (CIN), a persistently high rate of gain and loss of whole chromosomes, a phenomenon, which may in turn lead to a state of aneuploidy, or abnormal number of chromosomes in a cell. CIN cells are very often characterized by increased misorientation of chromosomes, which could be a result of defects in the sister chromatid cohesion, spindle assembly, and inability to resolve errors of microtubule-kinetochore attachments. Indeed, kinetochore microtubule attachments are abnormally stabilized in CIN positive cell lines.2 Overall, impaired expression of MCC components and altered SAC signaling could be associated with an increased risk of aneuploidy and tumor formation. For example, mutations in SAC protein BubR1 lead to mosaic variegated aneuploidy, an extremely rare syndrome associated with microcephaly, growth deficiency, and childhood cancer.3 Possible connections between aneuploidy and tumorigenesis have been demonstrated already over 100 years ago by Theodor Boveri. In the 1960s the karyogram of cancer tissues was shown to differ from that of normal tissues.4 Through systematic genetic analysis of many cancer tissues over the last decades, the presence of severe chromosome abnormalities was demonstrated in thousands of cancer samples.5,6 Consistent with these observations, aneuploidy and CIN were suggested to drive tumorigenesis.7–9 However, increased rates of aneuploidy and CIN were rather implicated in tumor suppression,10 suggesting that the level of damage might determine the oncogenic or oncosuppressor role of aneuploidy and CIN. Consequently, deregulated expression of chromosome segregation factors observed in various cancers stimulated development of therapeutic antimitotic drugs. However, inhibitors of microtubule function, the first generation of antimitotics currently used in therapies, are very nonspecific, which apparently leads to targeting of noncancer cells. Furthermore, none of the specific small molecule inhibitors of mitotic kinases entered into clinic routine so far.11 These rather disappointing clinical outcomes and low therapeutic potential of antimitotics might be due to a general cell toxicity.12 Thus, the current challenge is to identify the factors that control mitosis of cancer cells but at the same time have no effect on the normal noncancer cells. Since aneuploidy is the feature that distinguishes cancer from normal cells, the future drugs may need to exploit this tumor-specific vulnerability. The idea is that such drugs can overwhelm cancer cells with intolerable levels of chromosome instability, which are not compatible with cell survival. Defects in cell cycle regulation, chromosome cohesion, dynamics of kinetochore–microtubule attachment can all lead to chromosome instability in cancer cells.13 Interestingly, in many cancer cells the SAC is weaker but not absent, which is related to chromosome instability-driving malignancy and at the same time secures the cancer cells from acquiring too many errors in chromosome segregation which impacts cell viability.14 It was therefore suggested that more severe inactivation of SAC components may cause a higher level of aneuploidy that exceeds the adaptation capacity of cancer cells.12
In accordance with this, prolonged mitotic progression that often leads to mitotic catastrophe and cell death is a desired outcome for targeting cancer cells.15 Indeed, as compared with normal cells, cancer cells appear to be more sensitive to mitotic catastrophe.16 This suggests that upregulation of SAC response resulting in a prolonged mitosis and mitotic catastrophe should also be exploited as a potential anticancer therapeutic strategy. In particular, the regulators but not the core components of SAC might represent the attractive therapeutic targets, as such regulators are preferentially expressed in cancer cells and promote tolerable levels of aneuploidy and survival.
Our recent findings show that UBASH3B is the ubiquitin receptor protein for mitotic kinase Aurora B, which drives Aurora B recruitment to the microtubules in cancer cells.17 Spatiotemporal regulation of Aurora B localization plays a crucial role in the control of mitotic progression and SAC function.18 Thus, UBASH3B is a negative SAC regulator that might be specifically used by cancer cells to promote their propagation and survival. Levels of UBASH3B protein were shown to be elevated in highly aggressive breast and prostate cancers, promoting malignant growth, invasion, and metastasis.19 We hypothesize that downregulation of UBASH3B in cancer cells will prevent SAC satisfaction, induce mitotic arrest, and promote cancer cell death.
Results
During mitosis all chromosomes have to be attached to the mitotic spindle before they are segregated to the opposite poles during anaphase. Our recent findings in cervical cancer-derived HeLa cells show that ubiquitin receptor protein UBASH3B plays a critical role in this process.17 Upon microtubule attachment UBASH3B forms a complex with the microtubule motor protein Mitotic kinesin-like protein 2 (MKlp2) and drives relocalization of Aurora B kinase from chromosomes to microtubules. This event satisfies SAC signaling and triggers chromosome segregation (Fig. 1A). Downregulation of UBASH3B leads to the accumulation of Aurora B on chromosomes, thus preventing SAC satisfaction and inducing mitotic arrest and apoptosis (Fig. 1B). In contrast, overexpression of UBASH3B drives premature recruitment of Aurora B to microtubules, SAC silencing, erroneous chromosome segregation, and aneuploidy17 (Fig. 1C).
Figure 1.

A model of the regulation of Aurora B localization by UBASH3B and thereby SAC signaling in cancer cells. (A) Ubiquitylated Aurora B (green) interacts with microtubule-associated UBASH3B (red) on aligned metaphase chromosomes before chromosome segregation. UBASH3B is required and sufficient to transfer Aurora B from the centromeric regions of chromosomes (blue) to the spindle microtubules (black lines) thereby controlling its centromeric and kinetochore (gray) as well as spindle functions. UBASH3B acts in concert with MKlp2 motor protein (yellow) allowing for dynamic Aurora B localization and microtubules targeting, which controls the timing and fidelity of chromosome segregation and Spindle Assembly Checkpoint (SAC). (B) Downregulation of UBASH3B levels leads to the loss of the centromeric localization of Aurora B and its accumulation on the chromosome arms, imparing kinetochore functions of Aurora B. SAC response is high and cells arrest in mitosis without chromosome segregation, causing cell death. (C) High levels of UBASH3B are sufficient to induce relocalization of centromeric Aurora B to microtubules, a signal that silences SAC and induces premature and uncontrolled chromosome segregation, which may lead to aneuploidy.
Since high levels of UBASH3B were detected in highly aggressive breast and prostate cancers,19 we therefore wondered whether different cancer cells can utilize UBASH3B to override SAC. If this is the case, depletion of UBASH3B is expected to cause a mitotic delay and accumulation of cells in preanaphase stages. HeLa cells are characterized by high levels of SAC due to increased expression of BubR1.20 Likewise, adenocarcinoma-derived MDA-MB-231 cells express high MAD2 and BubR1 levels,20 suggesting the presence of a strong SAC response (Fig. 2A). Interestingly, and similar to HeLa cells,17 UBASH3B knockdown in MDA-MB-231 cells markedly increased numbers of prometa- and metaphase-like cells indicating a delay in mitosis (Fig. 2B and C). In contrast, in UBASH3B-negative colorectal adenocarcinoma-derived Colo 320DM (human Dukes' type C, colorectal adenocarcinoma with double minutes) cell line the numbers of preanaphase cells were not altered upon downregulation of UBASH3B. Moreover, epidermoid carcinoma-derived A431 cells, characterized by low levels of SAC components did not increase preanaphase cell numbers upon UBASH3B knockdown (Fig. 2C), in line with SAC-dependent mechanism of UBASH3B-mediated mitotic progression. Importantly, protein levels of SAC components were not affected by depletion of UBASH3B, which however efficiently reduced the protein levels of UBASH3B by at least 70% in all analyzed cell lines (Ref.17, Fig. 2B and data not shown). These observations suggest that mitotic progression in UBASH3B-expressing cancers characterized by strong SAC is sensitive to alterations of UBASH3B levels. To test this hypothesis, we have analyzed the level of cell death in the four cancer cell lines (HeLa, MDA-MB-231, Colo 320DM, A431). Interestingly, downregulation of UBASH3B induced approximately twofold increase in the death rate in HeLa and MDA-MB-231 cell lines but not in Colo 320DM and A431 cells (Fig. 2D) as compared with the corresponding control siRNA treated cells. To further corroborate these findings, we have analyzed the strength of the SAC response in the four cancer cell lines and in the primary fibroblasts (F31, R5). Interestingly, strong SAC response and high numbers of mitotic figures following treatment with the microtubule stabilizing agent paclitaxel were identified in HeLa and MDA-MB-231 in contrast to Colo 320DM, A431, and F31, R5 cells (Fig. 2E). Accordingly, the levels of SAC protein BubR1 were found low in the 2 primary cell lines (Fig. 2E). Importantly, despite high levels of UBASH3B found in the primary fibroblasts, their treatment with control or UBASH3B siRNAs did not lead to any differences in the cell death rate (Fig. 2F). These preliminary correlative findings suggest that cancer cells with a strong SAC response and expressing high levels of SAC as well as UBASH3B are specifically vulnerable to reduction of UBASH3B levels. Thus, downregulation or inhibition of UBASH3B in these cancer cells could be a mechanism to interrupt mitosis in tumors and serve as a novel therapeutic avenue in cancer treatment.
Figure 2.
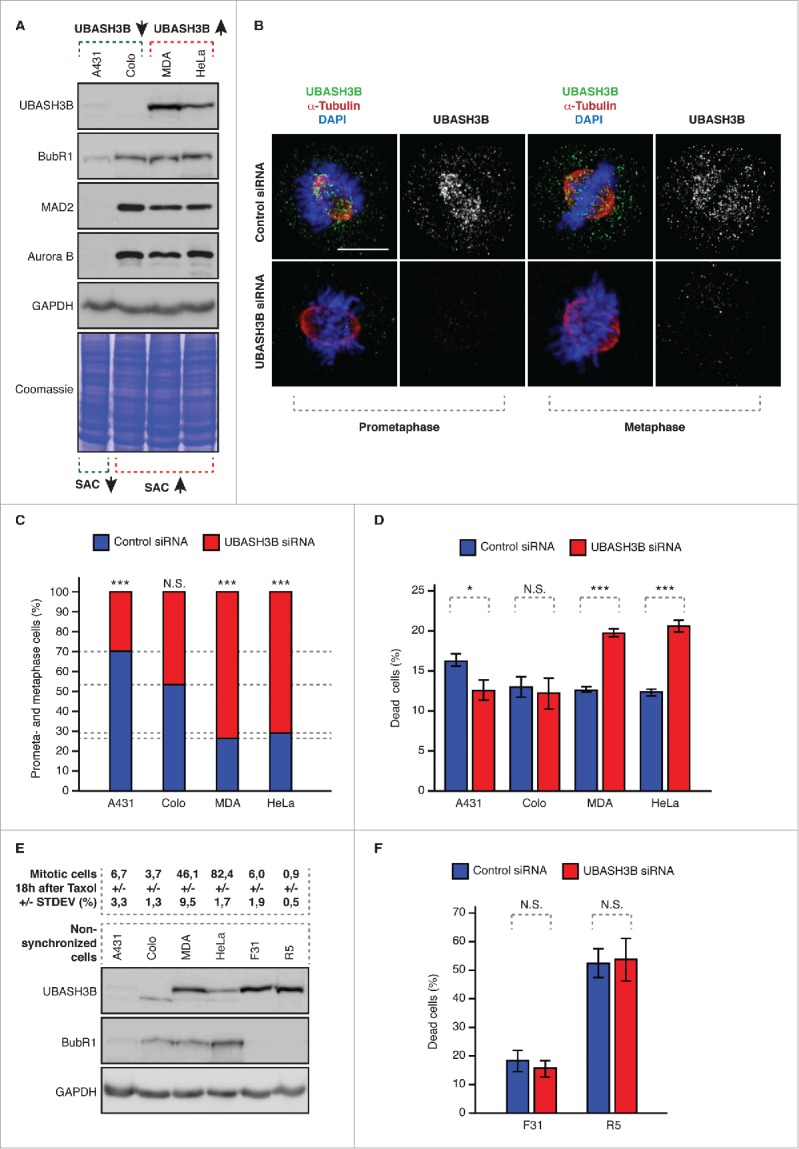
Downregulation of UBASH3B leads to potentiation of SAC in cancer cells. (A) Indicated cancer cell lines were analyzed by Western blotting. Colo = Colo 320DM, MDA = MDA-MB-231, HeLa = HeLaWS (B) MDA-MB-231 were treated by control and UBASH3B small interfering RNAs (siRNAs), synchronized by double thymidine block and release in mitosis and analyzed by immunofluorescence microscopy. Examples of prometaphase and metaphase cells are depicted. (C) Indicated cancer cell lines were treated by control (blue bars) and UBASH3B siRNAs (red bars), analyzed as in (B) and the numbers of cells (average of n = 6200 per cell line) in prometa- and metaphase-like stages were quantified. Equal numbers of cells were analyzed per control and UBASH3B siRNA groups for each cell line. Next, numbers of prometaphases and metaphases were quantified per control and UBASH3B siRNAs (equals 100%) and fractions of preanaphase cells in both groups were determined to show the differences between control and UBASH3B siRNA groups. (D) Indicated cancer cell lines were treated by control (blue bars) and UBASH3B siRNAs (red bars) for 48h, and percentage of dead cells was quantified. At least 500 cells were analyzed per each data point. (E) Upper panel: indicated cancer cell lines and primary fibroblasts (F31 and R5) were treated with Taxol (paclitaxel), analyzed by immunofluorescence microscopy and percentage of mitotic cells was quantified (average of n = 3000 per cell line). STDEV indicates standard deviation. Lower panel: indicated cancer cell lines and primary fibroblasts (F31 and R5) were analyzed by Western blotting. (F) Primary fibroblasts (F31 and R5) were analyzed as in (D) and percentage of dead cells was quantified. At least 500 cells were analyzed per each data point. In all experiments “***” indicates the P value of less than 0.001, “*”- less than 0.05, and N.S. indicates nonsignificant difference.
To further explore therapeutic perspectives of UBASH3B-mediated SAC silencing and to identify targetable cancer types, we analyzed the relative mRNA expression levels of UBASH3B as well as SAC components MAD2 (MAD2L1) and BubR1 (BUB1B) in different cancers. To this end we performed expression correlation analyses using data from the The Cancer Genome Atlas (TCGA) database. The positive correlation between MAD2L1 and BUB1B expression was statistically significant in all the tumors analyzed (Fig. 3A). A positive correlation between UBASH3B and BUB1B expression was statistically significant in Luminal B breast cancer, melanoma, pancreatic adenocarcinoma, kidney renal cell carcinoma, and liver hepatocellular carcinoma, while a positive correlation between UBASH3B and MAD2L1 expression was statistically significant in melanoma, pancreatic adenocarcinoma, kidney renal cell carcinoma, and liver hepatocellular carcinoma. In particular, kidney renal cell carcinoma and liver hepatocellular carcinoma tissue samples showed the strongest positive correlation in the mRNA expression levels of the three genes (Fig. 3B and C). This analysis suggests that these two cancer types may possess the molecular profiles suitable for therapeutic targeting of UBASH3B.
Figure 3.

Correlation analysis of mRNA expression of UBASH3B and SAC factors. (A) Spearman's rank correlation coefficient (r) was calculated to analyze the relationship between normalized mRNA expression levels of SAC components (MAD2L1 = MAD2, BUB1B = BubR1), and UBASH3B in several human tumors using expression data from the TCGA database. The results were considered statistically significant (indicated in bold) when P < 0.05. The number of available patients is indicated. Note that kidney renal cell carcinoma and liver hepatocellular carcinoma tissue samples (red) show the strongest positive correlation in the mRNA expression levels of the three genes. (B and C) Scatter plot of UBASH3B, MAD2L1, and BUB1B expression correlation analysis for kidney renal cell carcinoma (B) and liver hepatocellular carcinoma (C).
Discussion
Aneuploidy is one of the hallmarks of tumorigenesis. Aneuploidy observed in human cancers may arise from the defects in the mitotic checkpoint, which normally protects the cells from chromosome missegregation.21 Cancer cells may override the SAC either by downregulation of expression levels of the SAC components or their mutations. Indeed, defects in the mitotic checkpoint signaling are frequently observed in different human cancers,22–24 however mutations in the SAC genes have been rarely reported as the majority of them is very likely to be nonviable.
Here we propose that UBASH3B can indirectly contribute to the mitotic checkpoint defect by triggering the relocalization event of the essential mitotic kinase and SAC regulator Aurora B.17 Interestingly, sustained kinase activity of Aurora B by overexpression of Bub1 kinase induced aneuploidy and formation of various tumors in mice.25 Likewise, high expression of Aurora B promoted aneuploidy and tumor development in a xenograph model.26
We believe that UBASH3B-mediated mechanism can be adopted by cancer cells, which due to high rates of proliferation accumulate chromosomal abnormalities that under normal circumstances prevent SAC satisfaction and lead to apoptosis. These cancers may induce expression of UBASH3B to promote SAC silencing and induce aneuploidy and CIN in a manner that is compatible with their survival. It is particularly intriguing that downregulation of UBASH3B in cancer cell lines with high levels of SAC components and strong SAC response leads to Aurora B mislocalization,17 further potentiation of SAC and accumulation of cells in preanaphase stages (Fig. 2B and C) and finally to cell death (Fig. 2D). On the other hand, overexpression of UBASH3B leads to SAC silencing in these cells (Ref.17 and data not shown). High levels of UBASH3B were observed in highly aggressive forms of breast and pancreatic cancers in humans and in mouse models, in which UBASH3B promoted metastasis.19 Our correlative studies with SAC components presented here suggest that high levels of UBASH3B can be observed in other cancer types, in particular hepatocellular carcinoma (HCC) and kidney renal cell carcinoma (RCC). Hepatocellular carcinoma (HCC) is associated with the chronic exposure to toxins, alcohol, or viral infection and is one of the most common causes of cancer-related death in the world. Moreover, aneuploidy is frequently observed in HCC and is implicated in carcinogenesis process.27–29 Some HCC cell lines also showed impaired SAC response and failed to arrest in mitosis following exposure to microtubule drugs.29 Likewise, the incidence of HCC recurrence after surgical resection was higher in patients with lost expression of SAC MAD1 protein.30 A high risk of kidney RCC is often associated with the von Hippel-Lindau (VHL) disease and alteration in the VHL gene is a frequent event in sporadic clear cell RCC. Interestingly, recent studies in the VHL-deficient renal carcinoma showed impaired mitotic checkpoint and aneuploidy due to reduced expression of MAD2 both in cells31 and in mice.32 Lowering levels of MAD2 can indeed weaken the SAC response in a manner that is compatible with the cellular survival but leads to tumor development.9 Future studies are needed to confirm if these two cancer types are also dependent on the high levels of UBASH3B and sensitive to its downregulation. However, we strongly believe that the oncogenic potential of UBASH3B can be, at least partially, correlated with its role in chromosome segregation in these cancer cells.
High therapeutic potential of UBASH3B inactivation in cancer is further supported by other recent findings, claiming that UBASH3B may promote cancer cell growth and invasion by inactivating E3 Ubiquitin Ligase Casitas B-lineage Lymphoma (CBL), which subsequently leads to upregulation of epidermal growth factor receptor.19,33 Therefore inactivation of UBASH3B might interfere with both cancer cells growth and proliferation.
Irrespectively of the precise mechanisms, UBASH3B function might be restricted to aggressive, aneuploid cancer cells offering an interesting therapeutic perspective exploiting this tumor-specific vulnerability. This is confirmed by our results in the primary fibroblasts where weak SAC response and low levels of SAC protein BubR1 are found (Fig. 2E) and where downregulation of UBASH3B does not affect cellular survival (Fig. 2F). Interestingly, other nonpathogenic cells, in particular embryonic cells are characterized by lack of persistent SAC.34–37 Thus, regulation of faithful chromosome segregation in the primary fibroblasts and other normal cells might not be dependent on UBASH3B. This is also supported by the fact that UBASH3B knockout animals are viable and do not exhibit any developmental phenotypes.38 In future, it will be important to uncover the precise molecular mechanisms of control of UBASH3B function during normal mitosis as well as in tumorigenesis.
Materials and methods
Cell culture, siRNA transfections, and western blotting were performed as described previously.17 Primary fibroblasts (R5 and F31) were obtained from Institut de Genetique et de Biologie Moleculaire et Cellulaire (IGBCM) Cell Culture Facility. Analysis of the percentage of preanaphase cells was performed in cells synchronized in mitosis by double thymidine block and release and by immunofluorescence microscopy as described previously.17 The strength of the SAC response was analyzed by treatment of cells with Taxol (paclitaxel) (at 1 µM) for 18 hours and immunofluorescence microscopy using rabbit polyclonal antibodies to the mitotic marker phospho histone H3 Ser10 (Merck Millipore 06–570, 1:500).
Cellular death was analyzed by LIVE/DEAD® Fixable Dead Cell Stain kit (Thermo Fischer Scientific, catalog number L34969). Cell were grown on the coverslips, treated with indicated siRNAs for 48h as described previously17 and stained with the fluorescent reactive dye according to the manufacturer's protocol. Following treatment, cells were fixed with 3% ParaFormAldehyde (PFA) in Phosphate-buffered saline (PBS) and analyzed by immunofluorescence microscopy as described previously.17
Gene expression data from The Cancer Genome Atlas database (http://cancergenome.nih.gov/) were used to analyze UBASH3B, BUB1B, and MAD2L1 expression in 17 types of cancer. Normalized mRNA expression data were downloaded from cBioportal and analyzed as described previously.39,40 Spearman correlation was performed using GraphPad Prism (GraphPad Software, San Diego, CA, www.graphpad.com). All statistical tests were two-sided, and results were considered statistically significant when P < 0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank the members of the Sumara and Sanz-Moreno groups for helpful discussions on the manuscript. They are grateful to the Imaging Center of the IGBMC (ICI) and the IGBMC Cell Culture Facility for their support on this research.
Funding
K.K. was supported by a PhD contract from Région Alsace and INSERM and a Labex fellowship from IGBMC and Conectus Alsace. S.A. is supported by a Labex fellowship from IGBMC. This study was supported by the grant ANR-10-LABX-0030-INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame program Investissements d'Avenir ANR-10-IDEX-0002–02. Research in I.S. laboratory is supported by the IGBMC, CNRS, Fondation ARC pour la recherche sur le cancer, Institut National du Cancer (INCa), La Ligue Contre le Cancer and University of Strasbourg Institute of Advanced Studies. I.R-H is supported by Fundacion Alfonso Martin Escudero. Research in V.S-M laboratory is supported by Cancer Research UK (CRUK) C33043/A12065 and Royal Society RG110591.
Author contributions
K.K., I.R-H., V.S-M. and I.S. designed the study. K.K., C.K., S.A. I.R-H. conducted the experiments. I.S. and K.K. wrote the manuscript.
References
- 1.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 2007; 8:379-93; PMID:17426725; https://doi.org/ 10.1038/nrm2163 [DOI] [PubMed] [Google Scholar]
- 2.Tanno Y, Susumu H, Kawamura M, Sugimura H, Honda T, Watanabe Y. The inner centromere-shugoshin network prevents chromosomal instability. Science 2015; 349:1237-40; PMID:26359403; https://doi.org/ 10.1126/science.aaa2655 [DOI] [PubMed] [Google Scholar]
- 3.Hanks S, Coleman K, Reid S, Plaja A, Firth H, Fitzpatrick D, Kidd A, Méhes K, Nash R, Robin N, et al.. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet 2004; 36:1159-61; PMID:15475955; https://doi.org/ 10.1038/ng1449 [DOI] [PubMed] [Google Scholar]
- 4.Nowell PC. The minute chromosome (Phl) in chronic granulocytic leukemia. Blut 1962; 8:65-6; PMID:14480647; https://doi.org/ 10.1007/BF01630378 [DOI] [PubMed] [Google Scholar]
- 5.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature 1998; 396:643-9; PMID:9872311; https://doi.org/ 10.1038/25292 [DOI] [PubMed] [Google Scholar]
- 6.Mitelman F. Recurrent chromosome aberrations in cancer. Mutat Res 2000; 462:247-53; PMID:10767636; https://doi.org/ 10.1016/S1383-5742(00)00006-5 [DOI] [PubMed] [Google Scholar]
- 7.Babu JR, Jeganathan KB, Baker DJ, Wu X, Kang-Decker N, van Deursen JM. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J Cell Biol 2003; 160:341-53; PMID:12551952; https://doi.org/ 10.1083/jcb.200211048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, et al.. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet 2004; 36:744-9; PMID:15208629; https://doi.org/ 10.1038/ng1382 [DOI] [PubMed] [Google Scholar]
- 9.Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty VV, Benezra R. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 2001; 409:355-9; PMID:11201745; https://doi.org/ 10.1038/35053094 [DOI] [PubMed] [Google Scholar]
- 10.Weaver BAA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 2007; 11:25-36; PMID:17189716; https://doi.org/ 10.1016/j.ccr.2006.12.003 [DOI] [PubMed] [Google Scholar]
- 11.Marzo I, Naval J. Antimitotic drugs in cancer chemotherapy: promises and pitfalls. Biochem Pharmacol 2013; 86:703-10; PMID:23886991; https://doi.org/ 10.1016/j.bcp.2013.07.010 [DOI] [PubMed] [Google Scholar]
- 12.Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, Mak TW. Targeting Mitosis in Cancer: Emerging Strategies. Mol Cell 2015; 60:524-36; PMID:26590712; https://doi.org/ 10.1016/j.molcel.2015.11.006 [DOI] [PubMed] [Google Scholar]
- 13.Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol CB 2010; 20:R285-95; PMID:20334839; https://doi.org/ 10.1016/j.cub.2010.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weaver BAA, Cleveland DW. Does aneuploidy cause cancer? Curr Opin Cell Biol 2006; 18:658-67; PMID:17046232; https://doi.org/ 10.1016/j.ceb.2006.10.002 [DOI] [PubMed] [Google Scholar]
- 15.Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol 2011; 12:385-92; PMID:21527953; https://doi.org/ 10.1038/nrm3115 [DOI] [PubMed] [Google Scholar]
- 16.Janssen A, Kops GJPL, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci U S A 2009; 106:19108-13; PMID:19855003; https://doi.org/ 10.1073/pnas.0904343106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krupina K, Kleiss C, Metzger T, Fournane S, Schmucker S, Hofmann K, Fischer B, Paul N, Porter IM, Raffelsberger W, et al.. Ubiquitin Receptor Protein UBASH3B Drives Aurora B Recruitment to Mitotic Microtubules. Dev Cell 2016; 36:63-78; PMID:26766443; https://doi.org/ 10.1016/j.devcel.2015.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krenn V, Musacchio A. The Aurora B Kinase in Chromosome Bi-Orientation and Spindle Checkpoint Signaling. Front Oncol 2015; 5:225; PMID:26528436; https://doi.org/ 10.3389/fonc.2015.00225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee ST, Feng M, Wei Y, Li Z, Qiao Y, Guan P, Jiang X, Wong CH, Huynh K, Wang J, et al.. Protein tyrosine phosphatase UBASH3B is overexpressed in triple-negative breast cancer and promotes invasion and metastasis. Proc Natl Acad Sci U S A 2013; 110:11121-6; https://doi.org/ 10.1073/pnas.1300873110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greene LM, Campiani G, Lawler M, Williams DC, Zisterer DM. BubR1 is required for a sustained mitotic spindle checkpoint arrest in human cancer cells treated with tubulin-targeting pyrrolo-1,5-benzoxazepines. Mol Pharmacol 2008; 73:419-30; PMID:17991869; https://doi.org/ 10.1124/mol.107.039024 [DOI] [PubMed] [Google Scholar]
- 21.Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet 2012; 13:189-203; PMID:22269907 [DOI] [PubMed] [Google Scholar]
- 22.Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature 1998; 392:300-3; PMID:9521327; https://doi.org/ 10.1038/32688 [DOI] [PubMed] [Google Scholar]
- 23.Masuda A, Takahashi T. Chromosome instability in human lung cancers: possible underlying mechanisms and potential consequences in the pathogenesis. Oncogene 2002; 21:6884-97; PMID:12362271; https://doi.org/ 10.1038/sj.onc.1205566 [DOI] [PubMed] [Google Scholar]
- 24.Takahashi T, Haruki N, Nomoto S, Masuda A, Saji S, Osada H, Takahashi T. Identification of frequent impairment of the mitotic checkpoint and molecular analysis of the mitotic checkpoint genes, hsMAD2 and p55CDC, in human lung cancers. Oncogene 1999; 18:4295-300; PMID:10439037; https://doi.org/ 10.1038/sj.onc.1202807 [DOI] [PubMed] [Google Scholar]
- 25.Ricke RM, Jeganathan KB, van Deursen JM. Bub1 overexpression induces aneuploidy and tumor formation through Aurora B kinase hyperactivation. J Cell Biol 2011; 193:1049-64; PMID:21646403; https://doi.org/ 10.1083/jcb.201012035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen HG, Makitalo M, Yang D, Chinnappan D, St Hilaire C, Ravid K. Deregulated Aurora-B induced tetraploidy promotes tumorigenesis. FASEB J Off Publ Fed Am Soc Exp Biol 2009; 23:2741-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attallah AM, Tabll AA, Salem SF, El-Sadany M, Ibrahim TA, Osman S, El-Dosoky IM. DNA ploidy of liver biopsies from patients with liver cirrhosis and hepatocellular carcinoma: a flow cytometric analysis. Cancer Lett 1999; 142:65-9; PMID:10424782; https://doi.org/ 10.1016/S0304-3835(99)00165-2 [DOI] [PubMed] [Google Scholar]
- 28.Huang SF, Hsu HC, Fletcher JA. Investigation of chromosomal aberrations in hepatocellular carcinoma by fluorescence in situ hybridization. Cancer Genet Cytogenet 1999; 111:21-7; PMID:10326586; https://doi.org/ 10.1016/S0165-4608(98)00215-5 [DOI] [PubMed] [Google Scholar]
- 29.Saeki A, Tamura S, Ito N, Kiso S, Matsuda Y, Yabuuchi I, Kawata S, Matsuzawa Y. Frequent impairment of the spindle assembly checkpoint in hepatocellular carcinoma. Cancer 2002; 94:2047-54; PMID:11932908; https://doi.org/ 10.1002/cncr.10448 [DOI] [PubMed] [Google Scholar]
- 30.Nam CW, Park NH, Park BR, Shin JW, Jung SW, Na YW, Seo JH. Mitotic checkpoint gene MAD1 in hepatocellular carcinoma is associated with tumor recurrence after surgical resection. J Surg Oncol 2008; 97:567-71; PMID:18491369; https://doi.org/ 10.1002/jso.20999 [DOI] [PubMed] [Google Scholar]
- 31.Thoma CR, Toso A, Gutbrodt KL, Reggi SP, Frew IJ, Schraml P, Hergovich A, Moch H, Meraldi P, Krek W. VHL loss causes spindle misorientation and chromosome instability. Nat Cell Biol 2009; 11:994-1001; PMID:19620968; https://doi.org/ 10.1038/ncb1912 [DOI] [PubMed] [Google Scholar]
- 32.Hell MP, Duda M, Weber TC, Moch H, Krek W. Tumor suppressor VHL functions in the control of mitotic fidelity. Cancer Res 2014; 74:2422-31; PMID:24362914; https://doi.org/ 10.1158/0008-5472.CAN-13-2040 [DOI] [PubMed] [Google Scholar]
- 33.Goyama S, Schibler J, Gasilina A, Shrestha M, Lin S, Link KA, Chen J, Whitman SP, Bloomfield CD, Nicolet D, et al.. UBASH3B/Sts-1-CBL axis regulates myeloid proliferation in human preleukemia induced by AML1-ETO. Leukemia 2016; 30:728-39; PMID:26449661; https://doi.org/ 10.1038/leu.2015.275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hara K, Tydeman P, Kirschner M. A cytoplasmic clock with the same period as the division cycle in Xenopus eggs. Proc Natl Acad Sci U S A 1980; 77:462-6; PMID:6928638; https://doi.org/ 10.1073/pnas.77.1.462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Encalada SE, Willis J, Lyczak R, Bowerman B. A spindle checkpoint functions during mitosis in the early Caenorhabditis elegans embryo. Mol Biol Cell 2005; 16:1056-70; PMID:15616189; https://doi.org/ 10.1091/mbc.E04-08-0712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sluder G. Role of spindle microtubules in the control of cell cycle timing. J Cell Biol 1979; 80:674-91; PMID:572367; https://doi.org/ 10.1083/jcb.80.3.674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang M, Kothari P, Lampson MA. Spindle assembly checkpoint acquisition at the mid-blastula transition. PloS One 2015; 10:e0119285; PMID:25741707; https://doi.org/ 10.1371/journal.pone.0119285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carpino N, Turner S, Mekala D, Takahashi Y, Zang H, Geiger TL, Doherty P, Ihle JN. Regulation of ZAP-70 activation and TCR signaling by 2 related proteins, Sts-1 and Sts-2. Immunity 2004; 20:37-46; PMID:14738763; https://doi.org/ 10.1016/S1074-7613(03)00351-0 [DOI] [PubMed] [Google Scholar]
- 39.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al.. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2:401-4; PMID:22588877; https://doi.org/ 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6:pl1; PMID:23550210; https://doi.org/ 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]