

ABSTRACT
The relationship between melanocyte stem cells (MCSCs) and melanoma has been unclear. We recently demonstrated that melanoma-prone MCSCs are able to initiate cutaneous melanoma following stem cell activation through ultraviolet-B (UVB) exposure or natural stem cell cycling. Conversely, MCSC quiescence is sufficient to suppress tumorigenesis. This provides new insight into the role of environmental factors in tumor initiation from adult stem cells.
KEYWORDS: Melanoma origin, melanocyte stem cells, skin, UVB
Defining the cellular origin of cancers (also known as cancer cells of origin, CCOs) and the environmental and/or physiological contexts that can lead to tumor initiation provides insights into the identification of early diagnostic markers, as well as effective preventative strategies. It is particularly important to define which cells can act as CCOs for cutaneous melanoma, the most lethal form of skin cancer.
Adult stem cells are able to act as the cellular origin for many types of cancers.1 However, it has been unclear whether adult stem cells expressing oncogenic combinations known to be sufficient for tumorigenesis can always develop tumors or if their proliferative status governs tumorigenic potential.2 To address this question, we previously determined that hair follicle stem cells, which can act as CCOs for cutaneous squamous cell carcinoma,3 resist tumor initiation when in a quiescent state.2,4 In our recent study, we turned our attention to understanding the nature of tumor initiation and CCOs for cutaneous melanoma.5
Melanocyte stem cells (MCSCs) are a long-lived source for differentiated, pigment producing melanocytes. In the dorsal skin of an adult mouse, MCSCs are located within hair follicles. Hair follicles in the murine dorsal skin cycle from a quiescent state (telogen), to an active hair producing state (anagen), to a destructive phase (catagen), and then finally return to the resting telogen state. Moreover, it is possible to artificially induce stem cell activation and transition to anagen through depilation, and we exploited this methodology to determine the role of MCSC state in melanoma initiation using previously described mouse models of melanoma in addition to methods which activate MCSCs independently of the hair cycle.5,6 Our data demonstrate that, via the oncogenic combinations of mutant proto-oncogene B-Raf and v-Raf murine sarcoma viral oncogene homolog B (BrafV600E) or mutant Kirsten rat sarcoma viral oncogene homolog (KrasG12D) expression combined with conditional loss of the tumor suppressor phosphatase and tensin homolog (Pten), melanoma cell expansion emerges near the bulge, the anatomical residence of MCSCs in anagen, soon after induced telogen to anagen transition. This suggests that MCSCs are able to initiate melanomagenesis upon activation. In contrast, MCSCs containing the same tumorigenic combinations described above were refractory to tumor initiation during inactive MCSC periods including telogen, late anagen and catagen (Fig. 1a). Intriguingly, our results show that induction of oncogenic expression in post-mitotic, fully differentiated melanocytes (late anagen ∼ catagen) did not result in melanoma, and rather these cells underwent programed apoptosis during the hair follicle regression period of catagen. One thing to consider for future work, however, is that a transit-amplifying or intermediate cell state between activated stem cell and post-mitotic terminally differentiated melanocyte has not been well determined in the MCSC hierarchy, and it is possible that melanomas may also be capable of initiating from this undefined state. Taken together, our work revealed that MCSCs can directly act as CCOs for cutaneous melanoma, following stem cell activation.
Figure 1.
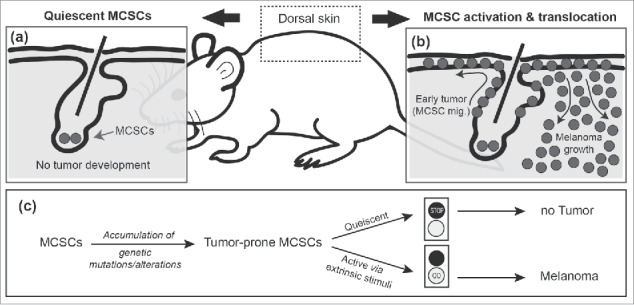
Cutaneous melanoma initiation from melanocyte stem cells is related to adult stem cell status. (a) Quiescent melanocyte stem cells (MCSCs) are refractory to oncogenic expression. However, (b) exposure to ultraviolet-B (UVB) in the environment can induce MCSC activation and migration (MCSC mig.), which in turn causes early melanoma formation throughout the epidermis. Following expansion within the epidermis, the tumor then grows invasively into the dermis (Melanoma growth). (c) Schematic of the role of extrinsic stimuli in melanoma initiation from tumor-prone, quiescent MCSCs.
It has been suggested that extrinsic factors may regulate the initiation of tumor formation from tumor-prone stem cells, potentially by increasing their division rate.7,8 In line with this, our results demonstrated that MCSCs require a transition from a non-proliferative quiescent state to an actively dividing state for melanomagenesis. Excessive ultraviolet radiation exposure, through sunlight, is a well-known risk factor for skin cancers including cutaneous melanoma. It has also been previously reported that ultraviolet-B (UVB) exposure can cause MCSC activation and translocation from the follicular niche to the interfollicular epidermis.9 Thus, we hypothesized that sunlight exposure, especially UVB, could trigger cutaneous melanoma development through aberrant activation of quiescent tumor-prone MCSCs. Strikingly, UVB-induced migration significantly triggered the induction of MCSC-originating melanoma (Fig. 1b).5 These results propose how extrinsic factors can promote melanoma initiation from CCOs by changing their activity status (Fig. 1c). Furthermore, we demonstrated that acute regional inflammation plays an important role in facilitating UVB-mediated MCSC-originating melanoma initiation. This further suggests a potential contribution of other physiological stresses in MCSC-originating melanomagenesis. Regional or systemic stress factors such as chronic inflammation or metabolic imbalance may affect MCSC status directly, or alter the micro-environment, which may lead to the activation of tumor-prone MCSCs. Additional work will be needed to identify the factors that may also induce MCSC-originating melanomas, especially in regions which are not regularly exposed to sunlight.
We also demonstrated that micro-environmental expression of high mobility group AT-hook 2, known as Hmga2, is required for efficient melanoma formation following UVB irradiation from melanoma-prone MCSCs.5 A cell non-autonomous necessity for Hmga2 was inferred through transplantation experiments, in which Hmga2−/− MCSCs expressing the melanomagenic combination were able to generate melanomas on recipient animals in equivalence to tumor-prone Hmga2+/+ MCSCs. While our data show Hmga2 upregulation in both the epidermis and non-epidermal portions of the skin, further studies will be needed to demonstrate in which tissue Hmga2 mediates expression of signaling molecules required for MCSC activation and translocation. Hmga2 is a DNA binding protein that is thought to have the ability to alter transcription factor accessibility, and thus could potentially affect the expression of numerous genes involved in many signaling pathways. Identifying the critical downstream pathways that are required for MCSC activation and translocation in UVB exposed Hmga2−/− skin will enable identification of signaling targets that could be used to suppress MCSC-originating melanoma formation.
While our study demonstrated MCSCs can act as CCOs for melanoma upon stem cell activation, it is still unknown if MCSC heterogeneity or if the immediate MCSC progeny contain inherent differences in tumorigenic potential. In the skin, the hair follicle stem cell hierarchy and heterogeneity within the stem cell compartment have been well defined, and furthermore, it has been suggested that this stem cell heterogeneity can provide the differential tumorigenic potential for cutaneous squamous cell carcinomas.10 In cutaneous melanoma, it is also possible that there are different MCSC populations which can give rise to melanoma more efficiently or are refractory to oncogene-mediated melanomagenesis. Looking forward, identifying additional environmental and physiological factors that promote the critical early steps of cutaneous melanoma formation and understanding the molecular mechanisms of tumor initiation from melanoma CCOs will ultimately provide better molecular therapeutic targets for melanoma prevention and early cancer treatment.
Funding Statement
This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Cancer Research Program (Award No. W81XWH-16-1-0272), a Cornell Center for Vertebrate Genomics Scholar Award and Seed Funding from the Cornell Stem Cell Program.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.White AC, Lowry WE.. Refining the role for adult stem cells as cancer cells of origin. Trends Cell Biol. 2015;25(1):11–20. doi: 10.1016/j.tcb.2014.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moon H, Donahue LR, White AC.. Understanding cancer cells of origin in cutaneous tumors. In: Liu H, Lathia JD, editors. Cancer Stem Cells. London (UK): Elsevier; 2016. p. 263–84. [Google Scholar]
- 3.White AC, Tran K, Khuu J, Dang C, Cui Y, Binder SW, Lowry WE.. Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proc Natl Acad Sci U S A. 2011;108(18):7425–30. doi: 10.1073/pnas.1012670108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White AC, Khuu JK, Dang CY, Hu J, Tran KV, Liu A, Gomez S, Zhang Z, Yi R, Scumpia P, et al.. Stem cell quiescence acts as a tumour suppressor in squamous tumours. Nat Cell Biol. 2014;16(1):99–107. doi: 10.1038/ncb2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moon H, Donahue LR, Choi E, Scumpia PO, Lowry WE, Grenier JK, Zhu J, White AC.. Melanocyte Stem Cell Activation and Translocation Initiate Cutaneous Melanoma in Response to UV Exposure. Cell Stem Cell. 2017;21(5):665–678.e6. doi: 10.1016/j.stem.2017.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr, You MJ, DePinho RA, McMahon M, Bosenberg M.. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41(5):544–52. doi: 10.1038/ng.356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomasetti C, Vogelstein B.. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347(6217):78–81. doi: 10.1126/science.1260825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu S, Powers S, Zhu W, Hannun YA.. Substantial contribution of extrinsic risk factors to cancer development. Nature. 2016;529(7584):43–7. doi: 10.1038/nature16166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chou WC, Takeo M, Rabbani P, Hu H, Lee W, Chung YR, Carucci J, Overbeek P, Ito M.. Direct migration of follicular melanocyte stem cells to the epidermis after wounding or UVB irradiation is dependent on Mc1r signaling. Nat Med. 2013;19(7):924–9. doi: 10.1038/nm.3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowry WE, Flores A, White AC.. Exploiting Mouse Models to Study Ras-Induced Cutaneous Squamous Cell Carcinoma. J Invest Dermatol. 2016;136(8):1543–8. doi: 10.1016/j.jid.2016.03.017 [DOI] [PubMed] [Google Scholar]