

Short stem-loop RNAs activate RIG-I in mice, activating the innate immune system via specific patterns of gene expression.
Abstract
We have developed highly potent synthetic activators of the vertebrate immune system that specifically target the RIG-I receptor. When introduced into mice, a family of short, triphosphorylated stem-loop RNAs (SLRs) induces a potent interferon response and the activation of specific genes essential for antiviral defense. Using RNA sequencing, we provide the first in vivo genome-wide view of the expression networks that are initiated upon RIG-I activation. We observe that SLRs specifically induce type I interferons, subsets of interferon-stimulated genes (ISGs), and cellular remodeling factors. By contrast, polyinosinic:polycytidylic acid [poly(I:C)], which binds and activates multiple RNA sensors, induces type III interferons and several unique ISGs. The short length (10 to 14 base pairs) and robust function of SLRs in mice demonstrate that RIG-I forms active signaling complexes without oligomerizing on RNA. These findings demonstrate that SLRs are potent therapeutic and investigative tools for targeted modulation of the innate immune system.
INTRODUCTION
Retinoic acid-inducible gene I (RIG-I) is an innate immune sensor that plays a key role in recognizing and responding to infection by RNA viruses (1, 2). RIG-I is activated under conditions that introduce a terminal, double-stranded RNA (dsRNA) molecule into the cell. These molecules can include viral genomes, replication intermediates, and any other species containing a stable RNA duplex that is terminated with a 5′-triphosphate or diphosphate group (3, 4). RIG-I activation by RNA leads to the induction of type I interferon (IFN) genes through the activation of its adaptor molecule, mitochondrial antiviral signaling protein (MAVS) (5). Another member of the RIG-I–like receptors (RLRs), MDA5, binds to long stretches of dsRNA and similarly triggers type I IFN production through MAVS (6). Type I IFNs induce hundreds of genes, collectively known as IFN-stimulated genes (ISGs), which have a variety of antiviral effector functions (7, 8). Both the RNA duplex and 5′-triphosphate moieties are important for specific, high-affinity binding and signaling by RIG-I (9–11), and through a series of recent crystal structures and functional studies of RNA recognition by the RIG-I receptor, the molecular basis for these effects has been elucidated (3, 12).
When appropriately delivered and modulated, RIG-I agonists would be promising tools for application in immuno-oncology (13–16), antiviral prophylaxis (17–20), and vaccine adjuvant development (21). However, all these applications require a specific and potent RIG-I ligand that is functional in vivo. Previous work has suggested that RIG-I function can be controlled and exploited pharmacologically through stimulation with small, well-defined RNA ligands that are no larger than other therapeutically administered oligonucleotides (9, 10, 16). Structural studies, quantitative biochemical work, cell-based assays, and imaging studies have all established that RIG-I is an “RNA end-capper” that encircles a 10–base pair (bp) RNA duplex as a monomer and forms a network of specific interactions with the terminal base pair and the 5′-triphosphate (3, 10–12, 22–27). RIG-I binding to short dsRNA is sufficient to trigger MAVS activation, and this process is enhanced by K63-ubiquitin, which promotes multimerization of the RIG-I CARD domains (28–30). On longer dsRNA (>40 bp), RIG-I forms aggregated filaments in vitro, and signaling in cell culture is less dependent upon K63-ubiquitin (29, 31, 32). However, we do not know whether RIG-I oligomerization on RNA is necessary for signaling in vivo. We sought to determine the ability of a minimal RIG-I ligand to signal in vivo and to monitor the effects on downstream gene expression in a living animal, providing the foundation for the development of an RNA therapeutic.
To this end, we designed a set of stem-loop RNA (SLR) molecules that present a single duplex terminus and therefore bind only one RIG-I molecule. The opposite end of the duplex is blocked with a stable RNA tetraloop, thereby ensuring that RIG-I binds the triphosphorylated duplex terminus in a single, structurally defined orientation. SLRs can be visualized as a short cord with a knot at one end that blocks protein binding. When RIG-I is constrained in this way, a robust IFN response is observed in mammalian cells. Here, we compare selective RIG-I activation by SLRs to that of longer RNA molecules that have been used in previous studies of RIG-I function. We then introduce SLRs into mice and observe potent, MAVS-dependent IFN induction, thereby establishing the physical mechanism for RNA-stimulated RIG-I activation in animals and providing a powerful set of synthetic RIG-I agonists that can be applied as probes, mechanistic tools, and pharmacological agents.
RESULTS
SLRs are an optimal design for RIG-I recognition
Unlike other RNA molecules that have been developed as RIG-I activators, complexes between SLRs and RIG-I molecules have been characterized crystallographically (3, 10, 23), making it possible to visualize and optimize the molecular interaction networks that stabilize active RIG-I–RNA complexes. Relative to two-piece duplexes, the stem-loop design provides simplicity, structural stability, and resistance to nucleases while presenting a single duplex terminus that fits precisely into the RNA binding pocket of RIG-I (figs. S1 and S2).
To examine the relative potency of SLRs and compare them with larger, more complex ligands, we used a well-established cell-based reporter assay in which IFN induction is monitored upon transfection with RNA (22). We tested the potency of two SLRs that vary in duplex length: SLR10 (containing 10 bp) and SLR14 (containing 14 bp). Recent studies indicate that RNAs bearing a diphosphate at the 5′ terminus are bona fide ligands for RIG-I (33). Thus, we compared the SLRs with a corresponding diphosphorylated stem-loop (pp-SLR14), as well as stem-loops lacking 5′-phosphate moieties (OH-SLR10 and OH-SLR14). We also included another control RNA, which is a 5′-triphosphorylated RNA that is the same length as SLR10, but is single-stranded and nonstructural (ppp-NS). In parallel, we examined the IFN response of a 19-bp two-piece duplex that is commercially marketed as a RIG-I ligand (19-mer dsRNA), and various other two-piece duplexes that have been used for previous structure/function studies (Fig. 1A and fig. S1). In addition, all the duplexes are compared with polyinosinic:polycytidylic acid [poly(I:C)], which is polydisperse and of unknown structure.
Fig. 1. SLRs are optimally recognized by RIG-I.
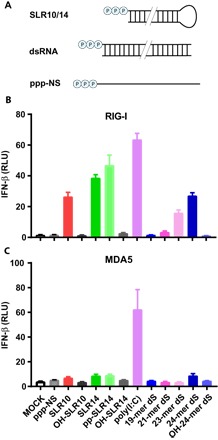
(A) SLRs were designed to fold stably into a minimal RIG-I ligand containing 10 or 14 bp and a triphosphorylated 5′ terminus. SLRs were compared against other reported double-stranded RIG-I ligands (dsRNA) 19 to 24 bp in length and control RNAs lacking structure (ppp-NS) or 5′-triphosphates (OH-). Human embryonic kidney (HEK) 293T cells lacking endogenous RIG-I and MDA5 were transfected with plasmids expressing RIG-I (B) or MDA5 (C) and a luciferase reporter under the control of an IFN-β promoter. Cells were then stimulated by various RNAs, and luciferase production was measured as a proxy for IFN-β response. Poly(I:C) stimulates both receptors, whereas SLRs are specific for RIG-I. SLR response is dependent upon the di- or triphosphate moiety and stimulates RIG-I as well or better than other reported RIG-I ligands. RLU, relative luciferase unit.
We observe that SLR10, SLR14, and pp-SLR14 are potent activators of RIG-I, inducing high levels of type I IFNs in the cell-based assay (Fig. 1B). Relative to the 14-bp duplexes, SLR10 displays somewhat reduced activity, but this trend is not universal (vide infra). As expected, the IFN response requires both the determinants that are required for RIG-I activation: a 5′-diphosphate or 5′-triphosphate and a duplex RNA structure, as OH-SLR10, OH-SLR14, and ppp-NS ligands failed to stimulate a significant level of IFN induction (Fig. 1B).
Notably, we failed to detect a significant IFN response with the RNA duplex (19-mer dsRNA) that is being marketed and used as a specific RIG-I ligand (fig. S1) (32, 34–36). We observed a small response from two RNA duplexes that have been used as RIG-I ligands in previous studies (9) (21- and 23-mer dsRNAs) and a moderate response from a dsRNA that we specifically designed to have a high thermodynamic stability relative to previously published duplex designs (24-mer dsRNA) (Fig. 1B). The IFN responses of the dsRNA ligands track closely with their relative thermodynamic stability (fig. S1), establishing that the efficacy of RIG-I ligands depends on a stable terminal duplex structure, which is enforced by the stem-loop design in the SLRs. In keeping with previous findings, we observe a strong response from poly(I:C) (Fig. 1B). Together, these findings confirm previous work demonstrating the potency of SLRs in cell culture (10, 37, 38) and benchmark SLR potency relative to other putative RIG-I ligands.
SLRs stimulate a potent IFN response in animals
To examine the ability of SLRs to induce type I IFN responses in vivo, we used a well-established RNA delivery method to introduce RNA ligands intravenously in living mice (17). Wild-type (WT) C57BL/6 mice were injected intravenously with RNA ligands complexed to polyethylenimine (PEI), and 5 hours later, IFN-α in mouse sera was measured by enzyme-linked immunosorbent assay (ELISA). A dose-response study of SLR10 showed an optimal response at 25 μg of RNA (fig. S3). An RNA stem-loop lacking a 5′-triphosphate (OH-SLR10) failed to induce IFN-α at any concentration (fig. S3).
We then examined the IFN-α response to SLR10 molecules that were either synthesized (synth SLR10) or transcribed in vitro (trans SLR10) at the optimal RNA dose. We observed elevated levels of IFN-α after injection of synthetic SLR10 compared to transcribed SLR10 (Fig. 2A). These results indicated that chemically synthesized SLR10, likely due to the enrichment in complete product, is more potent than transcribed RNA. Therefore, from here on, we exclusively use SLRs and dsRNA that are synthesized in house.
Fig. 2. SLR injection induces robust type I IFN responses in vivo.
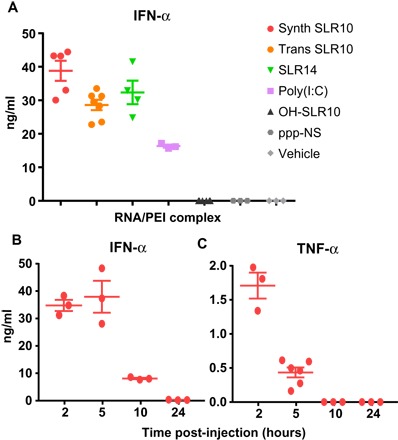
(A) C57BL/6 mice were injected intravenously with 25 μg of SLR10, SLR14, OH-SLR10, poly(I:C), ppp-NS, or vehicle control complexed with in vivo-jetPEI, and sera were collected 5 hours later. (B and C) C57BL/6 mice were injected intravenously with 25 μg of synthetic SLR10, and sera were collected at the indicated time points. Synthetic RNA was used for all experiments except in (A), where in vitro transcribed SLR (trans SLR) was used for comparison. The concentrations of IFN-α and TNF-α were measured by ELISA. Synthetic SLR10 was superior to transcribed SLR10 in inducing serum IFN-α.
We examined the extent of the IFN response induced by the various RNA ligands. Systemic IFN-α response was robustly induced by SLR10 and SLR14 (Fig. 2A). In contrast, no induction of IFN-α was observed after OH-SLR10 or ppp-NS injection in vivo. The widely used ligand poly(I:C) induced IFN-α, but to a considerably lower degree (Fig. 2A), thereby providing a benchmark for the relative in vivo activity of the SLR10 and SLR14 ligands.
Next, we analyzed the time course of IFN-α and tumor necrosis factor–α (TNF-α) secretion in response to in vivo injected SLR10. By 2 hours after intravenous injection, SLR10 elicited robust IFN-α and TNF-α responses (Fig. 2, B and C). By 5 hours after injection, SLR10 sustained higher levels of IFN-α, which dropped off by 10 hours after injection and were undetectable after 24 hours. In contrast, TNF-α levels peaked at 2 hours and were undetectable by 10 hours after injection. These data indicate that SLRs induce rapid, robust, and transient levels of IFN-α and TNF-α response in mice.
Diphosphorylated SLRs are also potent inducers of type I IFN in vivo
To evaluate the ability of diphosphorylated RNA to trigger IFN responses in vivo, we injected 25 μg of pp-SLR10, OH-SLR10, or SLR10 into mice as described above, collected the spleen after 5 hours of induction, and measured the IFN-α mRNA by quantitative reverse transcription polymerase chain reaction (qRT-PCR). Both pp-SLR10 and SLR10 induced comparable levels of IFN-α mRNA in the spleen (Fig. 3A). As expected, the nonphosphorylated OH-SLR10 induced minimal levels of IFN-α mRNA. In a separate experiment, we observed comparable ability of pp-SLR14 and SLR14 to induce IFN-α expression in the spleen (Fig. 3B). Consistent with our in vitro (Fig. 1) and in vivo analyses (Fig. 2), both ppp-SLRs and pp-SLRs induced more robust IFN-α mRNA than poly(I:C) (Fig. 3B). These results indicate that in both the serum (IFN-α protein; Fig. 2) and spleen (IFN-α mRNA; Fig. 3B), SLR and pp-SLR induce robust and comparable IFN-α expression.
Fig. 3. Diphosphorylated SLRs induce IFN-α in vivo.
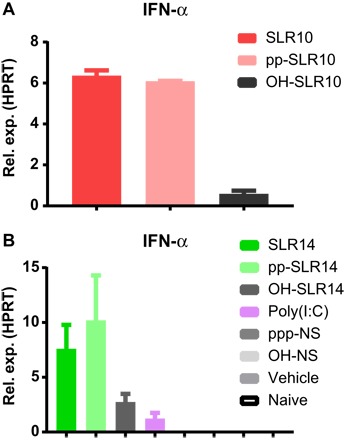
The diphosphorylated counterparts to SLR10 (A) and SLR14 (B) were injected intravenously into C57BL/6 mice, and 5 hours later, RNA was collected from spleens to measure IFN-α by qRT-PCR. Both pp-SLR10 and pp-SLR14 induced comparable IFN-α transcript levels as SLR10 and SLR14.
Type I IFN induction by SLRs is RIG-I–specific
On the basis of structural work and biochemical data (12), SLRs were designed to be a ligand that is specific for RIG-I, as other PRRs (pattern recognition receptors) recognize nucleic acids with different types of features, such as extended length [melanoma differentiation-associated protein 5 (MDA5)] or single-stranded [Toll-like receptor 7 (TLR7)] character. However, it was important to test the comparative PRR specificity of SLR ligands using a diversified set of experiments. Using the IFN-β promoter-luciferase reporter system described above, we found that both SLR10 and pp-SLR10 activate the IFN-β promoter when RIG-I, but not MDA5, is expressed (Fig. 1C). By contrast, poly(I:C) induced IFN-β promoter activation in both RIG-I– and MDA5-expressing reporter cells, consistent with the fact that poly(I:C) contains recognition determinants important for both types of receptors (duplex termini and long RNA duplex regions).
To evaluate RIG-I specificity, we used small interfering RNAs (siRNAs) to knock down RIG-I expression in A549 lung epithelial cells. We then challenged the cells with SLRs and evaluated the effects on IFN-β expression (Fig. 4A). Because knockdown of RIG-I was efficient in these experiments, it was possible to sensitively monitor the influence of challenge ligands on gene expression. Unlike their stimulatory behavior in WT cells, SLRs failed to induce IFN-β in the RIG-I knockdown cells (Fig. 4A). In addition, RIG-I knockdown resulted in diminished expression of ISGs, including Rsad2 (viperin) and Mx1 (Fig. 4A). These experiments show that SLRs are specifically and functionally recognized by RIG-I in human cells.
Fig. 4. SLRs are specifically recognized by RIG-I.
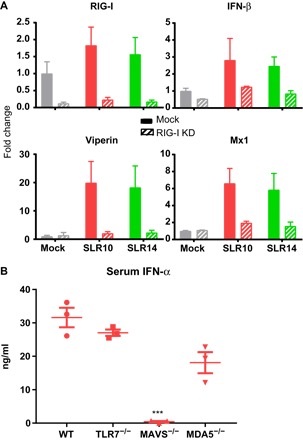
(A) A549 human lung epithelial cells were transfected with mock or RIG-I–targeting siRNAs, stimulated with SLR10 or SLR14, and assayed by qRT-PCR for knockdown efficiency and induction of IFN-β, viperin, and Mx1. Reduction of RIG-I expression resulted in reduced IFN and ISG production. (B) Mice lacking TLR7, MAVS, and MDA5 were injected intravenously with SLR10, and 5 hours later, sera were collected to measure IFN-α. Asterisks indicate significant difference from WT mouse control (***P < 0.0005).
To examine RIG-I specificity in a living animal, and to evaluate the potential contribution of other RNA sensors and signaling adaptors during IFN induction by SLRs, we introduced 25 μg of SLR10 intravenously into WT, Tlr7−/−, Mavs−/−, or Ifih1−/− mice. We were unable to examine the RIG-I–deficient mice due to embryonic lethality (39). We collected sera at 5 hours, and IFN-α levels were assessed by ELISA. These results demonstrate that MAVS, but not TLR7 or MDA5, is critical for IFN-α secretion following SLR injection (Fig. 4B). The fact that SLRs require MAVS, but not MDA5, for IFN-α induction (Fig. 4B) and that RIG-I knockdown eliminates this response in human cells (Fig. 4A) indicates that RIG-I is the receptor for IFN induction by SLRs.
Specific activation of RIG-I by SLR and poly(I:C) reveals a distinct pattern of gene expression
Given the potential for SLR and other RNA agonists for clinical applications, it was of interest to determine the global gene expression profiles following injection of SLRs and other activating RNA ligands. In particular, it was important to explore the landscape of expressed genes in an unbiased manner so that we might be able to identify new processes involved in RIG-I induction. We therefore carried out RNA sequencing (RNA-seq) analysis of expressed genes in the mouse spleen 3 hours after injection of 25 μg of SLRs or poly(I:C). To our knowledge, this is the first time that the profile of RNA-induced gene expression has ever been determined in an animal.
Broadly speaking, we observe that SLR14 and SLR10 showed similar gene expression profiles (fig. S4). To a first approximation, we can therefore use SLR14 as a proxy for short, multiphosphorylated RNA duplex ligands and compare them with RNAs that elicit a different profile of expressed genes, such as poly(I:C). We first measured the differentially expressed genes (DEGs) of SLR14 relative to vehicle and compared that with the DEGs of poly(I:C) relative to vehicle. By comparing the profiles, we can assess the relative impact of poly(I:C) and SLR14 on patterns of gene expression.
Both SLR14 and poly(I:C) induced a shared set of genes involved in antiviral response and innate immunity, including Ifit2, Oas3, Csf1, Tlr2, Tlr9, Nod2, and Ddx58 (RIG-I) (fig. S4 and table S1). In addition, genes involved in T cell activation and effector functions (Cd86, Tnf, Ifng, Il1b, and Gzmb) were elevated in response to both SLR14 and poly(I:C) (table S1). Both SLR14 and poly(I:C) induced a marked down-regulation in the expression of genes that belong in a variety of biological pathways, suggesting a shared pathway for co-opting basal cellular function upon induction. Many of these genes encode membrane proteins that are associated with processes such as ion transport, formation of cell junctions, and Wnt signaling (fig. S4 and table S1). In addition, many myeloid-expressed C-type lectins are down-regulated, possibly indicating the loss of dendritic cell subsets from the spleen.
In contrast, the two types of ligands have differential effects on other gene families (Fig. 5 and table S2). A marked divide in the gene expression pattern was seen for type I versus type III IFNs by SLRs and poly(I:C), respectively (Fig. 5C). Type III IFNs, IFN-λ2 and IFN-λ3, were more highly expressed upon introduction of poly(I:C) (Fig. 5C). In the mouse, the IFN-λ1 gene is a pseudogene. In addition, matrix metalloproteinases MMP8 and MMP9 (Fig. 5D and table S2), which degrade extracellular matrix for cell migration and wound repair, are up-regulated to a greater extent by poly(I:C). Similarly, SEMA4F, PLAGL1, and TYRO3 were more up-regulated by poly(I:C) injection than SLRs (Fig. 5, B and D). Conversely, type I IFNs (10 IFN-α genes and IFN-β) were all elevated by injection of SLRs compared to poly(I:C) (Fig. 5C). In addition to type I IFNs, TNFSF4 (OX40L) and several other genes with known roles in immune function (SATB2, VAV3, and VDR) were more strongly up-regulated by SLR14 (Fig. 5 and table S2) (40–42). A divergent noncoding transcript (9130024F11Rik) that shares a promoter with SATB2 is also highly up-regulated (table S2). This suggests a shared mechanism of transcriptional regulation and supports our observations of SATB2 regulation, but the function of these noncoding divergent transcripts is poorly defined.
Fig. 5. Distinct splenic gene expression between mice treated with SLRs and poly(I:C).
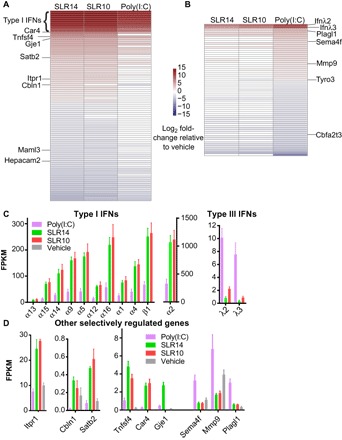
Mice were injected intravenously with SLRs or poly(I:C), and 3 hours later, RNA from spleens was purified for RNA-seq. Heatmaps were generated for genes preferentially regulated by treatment with SLR (A) or poly(I:C) (B). The heatmaps include all genes that are differentially expressed between SLR14 and poly(I:C) [more than twofold change and false discovery rate (FDR) of <0.05]. Highly statistically significant genes (FDR < 1 × 10−5) are labeled. (C) Type I and III IFNs are preferentially expressed following stimulation with SLR and poly(I:C), respectively. (D) Several additional genes, with and without reported immune functions, were differentially induced by SLRs or poly(I:C) (FDR < 1 × 10−5). FPKM, fragments per kilobase of exon per million fragments mapped.
The two treatments also showed differential effects on down-regulated genes, including a Rag1/2 repressor (Zfp608) that was preferentially down-regulated by SLR14 and two repressors of T cell maturation that were preferentially down-regulated by poly(I:C) (Lax1 and Dtx1) (table S2) (43–45). Together, these data indicate that SLR14 and related ligands elicit a potent, type I IFN dominant innate immune response, whereas poly(I:C) induces a stronger type III IFN response. Although we have focused on global trends among groups of known genes, the RNA-seq data are a rich source of information about potentially novel pathways involved in RNA and antiviral response.
To compare the effects of SLR14 relative to those of triphosphorylated, single-stranded RNA, we examined the DEGs of ppp-NS relative to vehicle, again comparing the response to that of SLR14 (fig. S4). DEG responses for ppp-NS and SLR14 showed little overlap, confirming that RIG-I is not activated by triphosphate moieties on single-stranded RNA molecules. Together, it is now possible to map a network of gene regulatory pathways that are selectively modulated by RIG-I and that will be activated upon induction of specific RIG-I ligands, such as synthetic, multiphosphorylated duplexes and viral panhandle RNAs.
DISCUSSION
RIG-I is a cytosolic sensor that has evolved to detect the presence of RNA molecules that have invaded or have been inappropriately expressed within vertebrate cells. Upon binding and responding to these RNA molecules, RIG-I initiates a signaling cascade that leads to proinflammatory responses and, ultimately, apoptosis. In this way, RIG-I plays a key role in the successful innate immune response to viruses, and its activation is being increasingly linked to antitumor response.
Given the antiviral effects of RIG-I and its potential utility in cancer treatment, it is vital to understand the mechanism of its activation and the molecular structure of its targets, particularly as they are presented in a living animal. This will lead to a better understanding of natural pathways for RIG-I induction, and it will confer the ability to activate and repress RIG-I at will with synthetic RNA molecules and other agonists, giving rise to powerful new therapeutic strategies. Understanding the molecular determinants of RIG-I recognition will also guide the improvement of other therapeutic RNA strategies that elicit unwanted activation of RIG-I, such as siRNA and antisense RNA treatments. Other triphosphorylated ligands can induce IFN production in cells, but the specificity of these ligands is less certain and they have not been tested in vivo (46–48). We therefore set out to identify a potent, minimal ligand for specific RIG-I induction in animals. Building on crystallographic, biochemical, and cell biological data, we created a set of stable, multiphosphorylated RNA duplexes (the SLRs) and performed comparative testing of their influence on IFN induction and overall gene expression in vitro and in vivo. By testing these molecules in knockout and knockdown contexts, we have shown that short (≥10 bp), stable RNA duplexes bearing a 5′-triphosphate or diphosphate are potent and specific activators of RIG-I.
The potency of the SLR ligands is significant because biophysical and cell biological experiments from multiple laboratories have demonstrated that short, polyphosphorylated synthetic RNA duplexes, ranging in length from 10 to 24 bp, can productively bind RIG-I and induce robust RIG-I signaling in cell culture (3, 9, 10, 22, 24, 26). These findings challenged the notion that RIG-I forms functional aggregates on RNA, suggesting that RIG-I/MAVS oligomerization is mediated by other features, such as ubiquitination of the CARD domains (28–30). However, until now, the immune responses and gene expression programs induced by small polyphosphorylated RNA ligands had not been assessed in vivo, leaving open the possibility that a functional RIG-I response in an animal requires larger RNA molecules. Here, we establish that polyphosphorylated RNA stem-loops as short as 10 bp can induce a potent IFN response by RIG-I in vivo. This is significant because SLR10 and SLR14 can only bind a single RIG-I molecule, establishing that RIG-I can perform its function without multimerizing on RNA, and validating the in vivo activity of short dsRNA. Our findings establish that RIG-I activation can be achieved with stable, synthetic RNA agonists that are chemically well defined and structurally characterized, which is important for development as a therapeutic. The stem-loop design is ideal because it permits RIG-I binding at only a single position on the RNA molecule (the polyphosphate terminus), whereas the stabilizing UUCG tetraloop enforces a duplex conformation that is resistant to nucleases and strand dissociation. The SLRs therefore represent a powerful new set of tools for exploring the physical basis of RIG-I signaling and for harnessing the therapeutic potential of this process.
The identification of SLR agonists provides a unique opportunity to explore the gene expression pathways that are specifically controlled by RIG-I and to differentiate them from networks that are initiated by other types of receptors. This has not been possible in the past because RNAs used in previous studies of RIG-I response [such as poly(I:C) or Sendai virus RNA] have large, complex structures that are capable of recruiting many types of receptors (such as MDA5, TLR3, and TLR7) and binding a host of potential coactivator proteins. Furthermore, most studies of RIG-I induction have been conducted in cell culture systems, and there is a lack of transcriptome-wide analysis of RIG-I induction in living animals. We therefore used the SLRs as tools to examine gene expression upon RIG-I activation in vivo and to compare the SLRs with other types of RNA molecules.
As expected, SLRs and poly(I:C) both induce a large number of genes that are associated with antiviral immunity and ISGs. However, SLRs selectively induced elevated levels of genes belonging to type I IFNs, whereas poly(I:C) induced higher levels of type III IFN genes (IFN-λ2 and IFN-λ3). Although the precise mechanisms by which a given RIG-I agonist triggers type I versus type III IFN genes are unknown, recent studies highlight that the intracellular location of MAVS might dictate these differential gene activation pathways (49). Whatever the mechanism might be, our comprehensive transcriptome results have important implications for the use of RNA ligands in humans. For instance, type I IFNs play a key role in initiating antitumor CD8 T cell responses (50–52). Thus, SLRs are expected to better elicit type I IFNs during cancer immunotherapy. On the other hand, systemic recombinant IFN-α therapy using conventional methods is not well tolerated because of significant side effects, so it may be prudent to minimize the type I response, particularly for localized pathologies. For example, hepatocytes express IFN-λR during the antiviral response against hepatitis C virus (HCV) and might benefit from a type III IFN response (53, 54). Thus, poly(I:C) may be a more effective therapy for HCV given its preferential induction of type III IFNs.
Our RNA-seq analysis revealed RIG-I induction of unexpected gene families, including several without annotated immune functions, suggesting potentially novel pathways involved in RIG-I response. For example, genes involved in pH homeostasis (Car4 and Sct) were selectively up-regulated by SLR14. Local acidosis has long been associated with inflammation, and these genes may be indicative of a pathway that functions as a response to this environment (55–57). Furthermore, the maintenance of pH homeostasis could benefit an immune response to tumors with acidic microenvironments. Three other genes preferentially up-regulated by SLR14 have been primarily characterized as neuronal proteins (Gje1, Cbln1, and Itpr1). Further investigation of these neuronal proteins revealed homology with known effectors of immune function (connexins, complement domains, and inositol receptors, respectively), which may suggest a bona fide role for these genes in the immune response (58–61).
RIG-I induction and antiviral responses are expected to cause restructuring of cellular structure and metabolism, and it is therefore intriguing that specific sets of gene families are down-regulated upon systemic RIG-I activation in the animal. For example, membrane proteins are significantly down-regulated upon stimulation. In particular, genes involved in cell-cell contacts are down-regulated, which is consistent with the migration of cells out of the spleen to peripheral tissues upon immune activation. Furthermore, C-type lectins are down-regulated, including many expressed on dendritic, macrophage, and natural killer cells, suggesting a depletion of these cells in the spleen as they migrate to peripheral tissues (62, 63).
In summary, we have demonstrated that SLRs are highly potent activators of the IFN response in living animals. They provide valuable tools for exploring the molecular determinants of RIG-I activation in the complex environment of the mammalian immune system, and they facilitate characterization of the gene expression pathways that are controlled by the RIG-I receptor. Given their small size and chemically defined composition, SLRs also represent a promising new class of oligonucleotide therapeutics with potential applicability as antivirals, vaccine adjuvants, and antitumor agents.
MATERIALS AND METHODS
RNA synthesis and purification
All triphosphorylated RNA oligonucleotides were synthesized on a MerMade 12 DNA-RNA synthesizer (BioAutomation), using the previously described synthetic procedure (64, 65). The 2′-pivaloyloxymethyl phosphoramidites were obtained from ChemGenes. Synthesized triphosphorylated RNAs were deprotected with ammonium hydroxide as described (65) and purified by polyacrylamide gel electrophoresis. RNA molecules were further analyzed for purity by mass spectrometry (Novatia LLC). Low–molecular weight (LMW) poly(I:C) was purchased from Invivogen and used without further purification.
HEK 293T cell culture and IFN-β induction assays
Cell-based experiments were conducted in HEK 293T cells because they do not express endogenous RLRs (www.proteinatlas.org). Cells were grown and maintained in 15-cm dishes containing Dulbecco’s modified Eagle’s medium (Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and nonessential amino acids (Life Technologies). IFN-β induction assays were conducted in 24-well format. Briefly, 0.5 ml of cells at 100,000 cells/ml was seeded in each well of tissue culture–treated 24-well plates. After 24 hours, each well of cells was transfected with 3 ng of pUNO-hRIG-I or pUNO-hMDA5 (Invivogen), 6 ng of pRL-TK constitutive Renilla luciferase reporter plasmid (Promega), and 150 ng of an IFN-β/firefly luciferase reporter plasmid using the Lipofectamine 2000 transfection reagent (Life Technologies) per the manufacturer’s protocol. Protein expression was allowed to proceed for 24 hours, at which point the cells were challenged by transfection of 1 μg of the indicated RNA or LMW poly(I:C) (six wells per RNA condition), also using the Lipofectamine 2000 reagent. After 12 hours, cells were harvested for luminescence analysis. These experiments were performed in biological triplicate.
Induction of the IFN-β promoter was analyzed using a dual luciferase assay. After aspiration of the growth media, 100 μl of passive lysis buffer (Promega) was added to each well and incubated for 15 min at room temperature. The lysed cells were transferred to a 96-well PCR plate and clarified by centrifugation. Next, 20-μl samples of the supernatant were transferred to a 96-well assay plate for analysis using the Dual-Luciferase Reporter Assay System (Promega). Luminescence was measured using a BioTek Synergy H1 plate reader. The resulting firefly luciferase activity (that is, the induction of IFN-β) was normalized to the activity of the constitutively expressed Renilla luciferase to account for differences in confluency, viability, and transfection efficiency across sample wells.
Mice
C57BL/6NCrl (WT) mice were purchased from Charles River Laboratories. Tlr7 (The Jackson Laboratory; B6.129S1-Tlr7tm1Flv/J) and Mavs knockout mice (The Jackson Laboratory; B6;129-Mavstm1Zjc/J) were backcrossed to C57BL/6 mice for several generations. Ifih1 knockout mice (The Jackson Laboratory; B6.Cg-Ifih1tm1.1Cln/J) were a gift from E. Fikrig. All animal procedures were performed in compliance with Yale Institutional Animal Care and Use Committee protocols.
In vivo injection of SLRs
SLRs were complexed to in vivo-jetPEI (Polyplus-transfection) according to the manufacturer’s instructions, with N/P (nitrogen/phosphate) ratio = 8. Unless otherwise indicated, 25 μg of RNA in a 200-μl volume was injected per mouse intravenously. At various time points, sera were collected and ELISA (PBL Assay Science, eBioscience) was performed according to the manufacturer’s instructions. Spleens were collected into RNAlater (Qiagen) for RNA extraction. A minimum of three mice per group was used per experiment.
RNA extraction from spleen
Tissues collected into RNAlater were blotted dry and transferred into TRIzol (Thermo Fisher Scientific) in a Lysing Matrix D tube (MP Biomedicals) for homogenization. The supernatant was collected for chloroform phase separation, and RNA was precipitated using isopropyl alcohol. The RNA pellet was then washed with 75% ethanol and redissolved in water.
Quantification of gene expression in animals by qRT-PCR
Complementary DNA (cDNA) was synthesized from RNA using the iScript cDNA Synthesis Kit (Bio-Rad). qPCR was performed using iTaq Universal SYBR Green (Bio-Rad) on a CFX Connect instrument (Bio-Rad). Ifna-4 [CTGCTACTTGGAATGCAACTC (forward) and CAGTCTTGCCAGCAAGTTGG (reverse)] and Mx1 [TGTACCCCAGCAAAACATCA (forward) and TTGGAAGCGCTAAAGTGGAA (reverse)] expression was normalized to Hprt [GTTGGATACAGGCCAGACTTTGTTG (forward) and GAGGGTAGGCTGGCCTATTGGCT (reverse)].
RIG-I knockdown experiments
A549 cells (American Type Culture Collection, CCL-185) were propagated in F-12K medium (Kaighn’s modification of Ham’s F-12 medium) containing 10% FBS. Cells were reverse-transfected in a 24-well plate with a RIG-I (DDX58)–specific siRNA sequence (GE Healthcare Dharmacon Inc., catalog no. D-012511-01-005) at a final concentration of 25 nM RNA and 0.5 μl of Lipofectamine 2000 (Thermo Fisher Scientific) per well, according to the manufacturer’s instructions. After 4-hour incubation at 37°C, transfection medium was replaced with fresh growth medium and incubated for another 20 hours at 37°C. Following siRNA treatment, cells were challenged with either mock (transfection reagent only) or exogenous purified RNA (1.7 ng/μl) (SLR14 or SLR10) and 0.5 μl of Lipofectamine 2000 reagent (Thermo Fisher Scientific) per well, according to the manufacturer’s instructions. Cells were transfected for 6 hours at 37°C and then trypsinized, washed in phosphate-buffered saline (PBS), and pelleted for total RNA extraction.
Total RNA from cells was extracted using the RNeasy Mini Kit (Qiagen) protocol. Genomic DNA contamination was removed by DNase I treatment (Qiagen) directly on the Mini kit column. Total RNA was reverse-transcribed into cDNA using oligo(dT) primers and SuperScript III reverse transcriptase enzyme (Thermo Fisher Scientific) according to the manufacturer’s instructions. qRT-PCR was performed using CFX384 Touch Real-Time PCR Detection System (Bio-Rad) and LightCycler 480 SYBR Green I Master (Roche Diagnostics). Each sample was analyzed in technical and biological triplicate and normalized to HPRT expression. Gene expression quantification was performed according to the ΔΔCt method, and fold change values are reported relative to cells without siRNA treatment and mock-challenged with RNA. Primer sequences were as follows: Hprt, TTTCAAATCCAACAAAGTCTGGC (forward) and TGGTCAGGCAGTATAATCCAAAG (reverse); Ddx58, TTCATGTCCACCTTCAGAAGTG (forward) and TCATAGCAGGCAAAGCAAGC (reverse); Ifnb1, GTCACTGTGCCTGGACCATAG (forward) and GTTTCGGAGGTAACCTGTAAGTC (reverse); Rsad2, TCGCTATCTCCTGTGACAGC (forward) and CACCACCTCCTCAGCTTTTG (reverse); and Mx1, AGAGAAGGTGAGAAGCTGATCC (forward) and TTCTTCCAGCTCCTTCTCTCTG (reverse).
RNA-seq library preparation and data analysis
Libraries were prepared using Illumina TruSeq Stranded mRNA sample preparation kits from 500 ng of purified total RNA according to the manufacturer’s protocol. The finished dsDNA libraries were quantified by Qubit fluorometer, Agilent 2200 TapeStation, and qRT-PCR using the Kapa Biosystems library quantification kit according to the manufacturer’s protocols. Uniquely indexed libraries were pooled in equimolar ratios and sequenced on an Illumina NextSeq 500 sequencer with paired-end 75-bp reads by the Dana-Farber Cancer Institute Molecular Biology Core Facilities.
Bioinformatic analysis
Sequenced reads were checked for quality control using the FastQC tool (Babraham Bioinformatics, Babraham Institute, Cambridge, UK). Good quality reads were mapped to the reference genome (GRCm38/mm10) using the software package TopHat version 2.0.6 (66). Default settings were used, except for the mean inner mate distance between the pairs that was set to 150 bp. A maximum of two mismatches and a minimum length of 36 bp per segment were allowed. The BAM files from TopHat were then converted to SAM format by SAMtools (version 1.4) (67), and raw counts were estimated by the Python script HTSeq count (68).
We generated approximately 628 million paired-end sequencing reads by multiplexing 24 samples on four lanes of Illumina NextSeq500 platform. On average, each sample achieved a sequencing depth of 26 million paired-end reads. The overall mapping rate against the reference transcriptome was up to 92%. Normalized gene expression levels, represented as counts per million, were calculated for 39,169 transcripts from the Ensembl database and processed for statistical analysis with EdgeR (69).
The resulting raw counts per gene were processed by the EdgeR program to perform normalization and clustering and to estimate differential expression. EdgeR (Bioconductor release 3.4.2) performs differential abundance analysis using a pairwise design based on the negative binomial model, as an alternative to the Poisson estimates. Normalization of the sequenced libraries was performed to remove effects due to differences in library size. The resulting P values were corrected for multiple testing using the Benjamini-Hochberg FDR approach.
We identified genes uniquely regulated by SLR14 or poly(I:C) using a two-step filtration process. First, each treatment was compared to vehicle, and DEGs that had P < 0.05 and more than twofold change in expression level were selected. This subset of genes was then compared between the SLR14 and poly(I:C) treatments. Genes that were differentially expressed between SLR14 and poly(I:C) with P < 0.05 and more than twofold change in expression level were deemed “significantly more up-/down-regulated by a specific treatment.” All other genes were deemed “mutually up-/down-regulated.” Mutually up-/down-regulated genes were analyzed for functional enrichment using the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 (70).
Supplementary Material
Acknowledgments
Funding: This study was supported, in part, by the Howard Hughes Medical Institute and awards from the NIH (R01AI054359 and R56AI125504 to A.I.). Author contributions: A.M.P., A.I., M.M.L., and E.S.M. designed the experiments. M.M.L., T.H.D., E.S.M., M.E.F., and O.P. performed the experiments. M.M.L., T.H.D., E.S.M., A.M.P., and A.I. analyzed and interpreted experiments. A.M.P. primarily wrote the manuscript with assistance from A.I., M.M.L., and T.H.D. Competing interests: A.M.P. is an inventor on a patent related to this work that is pending with the U.S. Patent Office (no. 14/776,463, filed 13 March 2013). All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The RNA-seq data discussed in this publication have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus (71) and are accessible through GEO Series accession number GSE106229 (https://ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE106229).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/2/e1701854/DC1
fig. S1. RNA ligands for RIG-I activation.
fig. S2. SLR10 is monomeric in conditions that mimic cell culture and in vivo experiments.
fig. S3. Dose response of SLRs.
fig. S4. SLRs and poly(I:C) regulate a shared set of genes.
table S1. List of genes mutually regulated by SLRs and poly(I:C).
table S2. List of genes preferentially regulated by SLRs or poly(I:C).
Reference (72)
REFERENCES AND NOTES
- 1.Ramos H. J., Gale M. Jr, RIG-I like receptors and their signaling crosstalk in the regulation of antiviral immunity. Curr. Opin. Virol. 1, 167–176 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., Fujita T., The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Devarkar S. C., Wang C., Miller M. T., Ramanathan A., Jiang F., Khan A. G., Patel S. S., Marcotrigiano J., Structural basis for m7G recognition and 2′-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl. Acad. Sci. U.S.A. 113, 596–601 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber M., Sediri H., Felgenhauer U., Binzen I., Bänfer S., Jacob R., Brunotte L., García-Sastre A., Schmid-Burgk J. L., Schmidt T., Hornung V., Kochs G., Schwemmle M., Klenk H.-D., Weber F., Influenza virus adaptation PB2-627K modulates nucleocapsid inhibition by the pathogen sensor RIG-I. Cell Host Microbe 17, 309–319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seth R. B., Sun L., Ea C.-K., Chen Z. J., Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 122, 669–682 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K. J., Yamaguchi O., Otsu K., Tsujimura T., Koh C.-S., Reis e Sousa C., Matsuura Y., Fujita T., Akira S., Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Schoggins J. W., Rice C. M., Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 1, 519–525 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Honda K., Takaoka A., Taniguchi T., Type I inteferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 25, 349–360 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Schlee M., Roth A., Hornung V., Hagmann C. A., Wimmenauer V., Barchet W., Coch C., Janke M., Mihailovic A., Wardle G., Juranek S., Kato H., Kawai T., Poeck H., Fitzgerald K. A., Takeuchi O., Akira S., Tuschl T., Latz E., Ludwig J., Hartmann G., Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31, 25–34 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohlway A., Luo D., Rawling D. C., Ding S. C., Pyle A. M., Defining the functional determinants for RNA surveillance by RIG-I. EMBO Rep. 14, 772–779 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vela A., Fedorova O., Ding S. C., Pyle A. M., The thermodynamic basis for viral RNA detection by the RIG-I innate immune sensor. J. Biol. Chem. 287, 42564–42573 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rawling D. C., Pyle A. M., Parts, assembly and operation of the RIG-I family of motors. Curr. Opin. Struct. Biol. 25, 25–33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duewell P., Steger A., Lohr H., Bourhis H., Hoelz H., Kirchleitner S. V., Stieg M. R., Grassmann S., Kobold S., Siveke J. T., Endres S., Schnurr M., RIG-I-like helicases induce immunogenic cell death of pancreatic cancer cells and sensitize tumors toward killing by CD8+ T cells. Cell Death Differ. 21, 1825–1837 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poeck H., Besch R., Maihoefer C., Renn M., Tormo D., Morskaya S. S., Kirschnek S., Gaffal E., Landsberg J., Hellmuth J., Schmidt A., Anz D., Bscheider M., Schwerd T., Berking C., Bourquin C., Kalinke U., Kremmer E., Kato H., Akira S., Meyers R., Häcker G., Neuenhahn M., Busch D., Ruland J., Rothenfusser S., Prinz M., Hornung V., Endres S., Tüting T., Hartmann G., 5′-triphosphate-siRNA: Turning gene silencing and Rig-I activation against melanoma. Nat. Med. 14, 1256–1263 (2008). [DOI] [PubMed] [Google Scholar]
- 15.van den Boorn J. G., Hartmann G., Turning tumors into vaccines: Co-opting the innate immune system. Immunity 39, 27–37 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Glas M., Coch C., Trageser D., Daßler J., Simon M., Koch P., Mertens J., Quandel T., Gorris R., Reinartz R., Wieland A., Von Lehe M., Pusch A., Roy K., Schlee M., Neumann H., Fimmers R., Herrlinger U., Brüstle O., Hartmann G., Besch R., Scheffler B., Targeting the cytosolic innate immune receptors RIG-I and MDA5 effectively counteracts cancer cell heterogeneity in glioblastoma. Stem Cells 31, 1064–1074 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Goulet M.-L., Olagnier D., Xu Z., Paz S., Belgnaoui S. M., Lafferty E. I., Janelle V., Arguello M., Paquet M., Ghneim K., Richards S., Smith A., Wilkinson P., Cameron M., Kalinke U., Qureshi S., Lamarre A., Haddad E. K., Sekaly R. P., Peri S., Balachandran S., Lin R., Hiscott J., Systems analysis of a RIG-I agonist inducing broad spectrum inhibition of virus infectivity. PLOS Pathog. 9, e1003298 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Probst P., Grigg J. B., Wang M., Muñoz E., Loo Y.-M., Ireton R. C., Gale M. Jr, Iadonato S. P., Bedard K. M., A small-molecule IRF3 agonist functions as an influenza vaccine adjuvant by modulating the antiviral immune response. Vaccine 35, 1964–1971 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ireton R. C., Gale M. Jr, RIG-I like receptors in antiviral immunity and therapeutic applications. Viruses 3, 906–919 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herbst-Kralovetz M. M., Pyles R. B., Quantification of poly(I:C)-mediated protection against genital herpes simplex virus type 2 infection. J. Virol. 80, 9988–9997 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hochheiser K., Klein M., Gottschalk C., Hoss F., Scheu S., Coch C., Hartmann G., Kurts C., Cutting edge: The RIG-I Ligand 3pRNA potently improves CTL cross-priming and facilitates antiviral vaccination. J. Immunol. 196, 2439–2443 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Luo D., Ding S. C., Vela A., Kohlway A., Lindenbach B. D., Pyle A. M., Structural insights into RNA recognition by RIG-I. Cell 147, 409–422 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo D., Kohlway A., Vela A., Pyle A. M., Visualizing the determinants of viral RNA recognition by innate immune sensor RIG-I. Structure 20, 1983–1988 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kowalinski E., Lunardi T., McCarthy A. A., Louber J., Brunel J., Grigorov B., Gerlier D., Cusack S., Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147, 423–435 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Kolakofsky D., Kowalinski E., Cusack S., A structure-based model of RIG-I activation. RNA 18, 2118–2127 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang F., Ramanathan A., Miller M. T., Tang G.-Q., Gale M., Patel S. S., Marcotrigiano J., Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 479, 423–427 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Louber J., Kowalinski E., Bloyet L.-M., Brunel J., Cusack S., Gerlier D., RIG-I self-oligomerization is either dispensable or very transient for signal transduction. PLOS ONE 9, e108770 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang X., Kinch L. N., Brautigam C. A., Chen X., Du F., Grishin N. V., Chen Z. J., Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 36, 959–973 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peisley A., Wu B., Xu H., Chen Z. J., Hur S., Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 509, 110–114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeng W., Sun L., Jiang X., Chen X., Hou F., Adhikari A., Xu M., Chen Z. J., Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 141, 315–330 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peisley A., Wu B., Yao H., Walz T., Hur S., RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol. Cell 51, 573–583 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Patel J. R., Jain A., Chou Y.-y., Baum A., Ha T., García-Sastre A., ATPase-driven oligomerization of RIG-I on RNA allows optimal activation of type-I interferon. EMBO Rep. 14, 780–787 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goubau D., Schlee M., Deddouche S., Pruijssers A. J., Zillinger T., Goldeck M., Schuberth C., Van der Veen A. G., Fujimura T., Rehwinkel J., Iskarpatyoti J. A., Barchet W., Ludwig J., Dermody T. S., Hartmann G., Reis e Sousa C., Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 514, 372–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kulkarni R. R., Rasheed M. A. U., Bhaumik S. K., Ranjan P., Cao W., Davis C., Marisetti K., Thomas S., Gangappa S., Sambhara S., Murali-Krishna K., Activation of the RIG-I pathway during influenza vaccination enhances the germinal center reaction, promotes T follicular helper cell induction, and provides a dose-sparing effect and protective immunity. J. Virol. 88, 13990–14001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szabo A., Fekete T., Koncz G., Kumar B. V., Pazmandi K., Foldvari Z., Hegedus B., Garay T., Bacsi A., Rajnavolgyi E., Lanyi A., RIG-I inhibits the MAPK-dependent proliferation of BRAF mutant melanoma cells via MKP-1. Cell. Signal. 28, 335–347 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Wang Y., Wang X., Li J., Zhou Y., Ho W., RIG-I activation inhibits HIV replication in macrophages. J. Leukoc. Biol. 94, 337–341 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rawling D. C., Fitzgerald M. E., Pyle A. M., Establishing the role of ATP for the function of the RIG-I innate immune sensor. eLife 4, e09391 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rawling D. C., Kohlway A. S., Luo D., Ding S. C., Pyle A. M., The RIG-I ATPase core has evolved a functional requirement for allosteric stabilization by the Pincer domain. Nucleic Acids Res. 42, 11601–11611 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato H., Sato S., Yoneyama M., Yamamoto M., Uematsu S., Matsui K., Tsujimura T., Takeda K., Fujita T., Takeuchi O., Akira S., Cell type-specific involvement of RIG-I in antiviral response. Immunity 23, 19–28 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Dobreva G., Dambacher J., Grosschedl R., SUMO modification of a novel MAR-binding protein, SATB2, modulates immunoglobulin μ gene expression. Genes Dev. 17, 3048–3061 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roth S., Bergmann H., Jaeger M., Yeroslaviz A., Neumann K., Koenig P.-A., Prazeres da Costa C., Vanes L., Kumar V., Johnson M., Menacho-Márquez M., Habermann B., Tybulewicz V. L., Netea M., Bustelo X. R., Ruland J., Vav proteins are key regulators of Card9 signaling for innate antifungal immunity. Cell Rep. 17, 2572–2583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu P. T., Stenger S., Li H., Wenzel L., Tan B. H., Krutzik S. R., Ochoa M. T., Schauber J., Wu K., Meinken C., Kamen D. L., Wagner M., Bals R., Steinmeyer A., Zügel U., Gallo R. L., Eisenberg D., Hewison M., Hollis B. W., Adams J. S., Bloom B. R., Modlin R. L., Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311, 1770–1773 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Zhang F., Thomas L. R., Oltz E. M., Aune T. M., Control of thymocyte development and recombination-activating gene expression by the zinc finger protein Zfp608. Nat. Immunol. 7, 1309–1316 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Zhu M., Granillo O., Wen R., Yang K., Dai X., Wang D., Zhang W., Negative regulation of lymphocyte activation by the adaptor protein LAX. J. Immunol. 174, 5612–5619 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Izon D. J., Aster J. C., He Y., Weng A., Karnell F. G., Patriub V., Xu L., Bakkour S., Rodriguez C., Allman D., Pear W. S., Deltex1 redirects lymphoid progenitors to the B cell lineage by antagonizing Notch1. Immunity 16, 231–243 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Davis W. G., Bowzard J. B., Sharma S. D., Wiens M. E., Ranjan P., Gangappa S., Stuchlik O., Pohl J., Donis R. O., Katz J. M., Cameron C. E., Fujita T., Sambhara S., The 3′ untranslated regions of influenza genomic sequences are 5′PPP-independent ligands for RIG-I. PLOS ONE 7, e32661 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nallagatla S. R., Hwang J., Toroney R., Zheng X., Cameron C. E., Bevilacqua P. C., 5′-Triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science 318, 1455–1458 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Kim D.-H., Longo M., Han Y., Lundberg P., Cantin E., Rossi J. J., Interferon induction by siRNAs and ssRNAs synthesized by phage polymerase. Nat. Biotechnol. 22, 321–325 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Odendall C., Dixit E., Stavru F., Bierne H., Franz K. M., Durbin A. F., Boulant S., Gehrke L., Cossart P., Kagan J. C., Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 15, 717–726 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spaapen R. M., Leung M. Y. K., Fuertes M. B., Kline J. P., Zhang L., Zheng Y., Fu Y.-X., Luo X., Cohen K. S., Gajewski T. F., Therapeutic activity of high-dose intratumoral IFN-β requires direct effect on the tumor vasculature. J. Immunol. 193, 4254–4260 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Fuertes M. B., Kacha A. K., Kline J., Woo S.-R., Kranz D. M., Murphy K. M., Gajewski T. F., Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J. Exp. Med. 208, 2005–2016 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gajewski T. F., Fuertes M. B., Woo S.-R., Innate immune sensing of cancer: Clues from an identified role for type I IFNs. Cancer Immunol. Immunother. 61, 1343–1347 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pagliaccetti N. E., Robek M. D., Interferon-λ in HCV infection and therapy. Viruses 2, 1589–1602 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller D. M., Klucher K. M., Freeman J. A., Hausman D. F., Fontana D., Williams D. E., Interferon lambda as a potential new therapeutic for hepatitis C. Ann. N. Y. Acad. Sci. 1182, 80–87 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Menkin V., Warner C. R., Studies on inflammation: XIII. Carbohydrate metabolism, local acidosis, and the cytological picture in inflammation. Am. J. Pathol. 13, 25–44.1 (1937). [PMC free article] [PubMed] [Google Scholar]
- 56.De Backer D., Lactic acidosis. Intensive Care Med. 29, 699–702 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Kominsky D. J., Campbell E. L., Colgan S. P., Metabolic shifts in immunity and inflammation. J. Immunol. 184, 4062–4068 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glass A. M., Snyder E. G., Taffet S. M., Connexins and pannexins in the immune system and lymphatic organs. Cell. Mol. Life Sci. 72, 2899–2910 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tom Tang Y., Hu T., Arterburn M., Boyle B., Bright J. M., Palencia S., Emtage P. C., Funk W. D., The complete complement of C1q-domain-containing proteins in Homo sapiens. Genomics 86, 100–111 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Gerwick L., Reynolds W. S., Bayne C. J., A precerebellin-like protein is part of the acute phase response in rainbow trout, Oncorhynchus mykiss. Dev. Comp. Immunol. 24, 597–607 (2000). [DOI] [PubMed] [Google Scholar]
- 61.Vig M., Kinet J.-P., Calcium signaling in immune cells. Nat. Immunol. 10, 21–27 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart, M. E. Etzler, R. P. McEver, Essentials of Glycobiology (Cold Spring Harbor Laboratory Press, ed. 2, 2009). [PubMed] [Google Scholar]
- 63.Kelley J., Walter L., Trowsdale J., Comparative genomics of natural killer cell receptor gene clusters. PLOS Genet. 1, e27 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zlatev I., Manoharan M., Vasseur J.-J., Morvan F., Solid-phase chemical synthesis of 5′-triphosphate DNA, RNA, and chemically modified oligonucleotides. Curr. Protoc. Nucleic Acid Chem. Chapter 1, Unit1.28 (2012). [DOI] [PubMed] [Google Scholar]
- 65.Thillier Y., Decroly E., Morvan F., Canard B., Vasseur J.-J., Debart F., Synthesis of 5′ cap-0 and cap-1 RNAs using solid-phase chemistry coupled with enzymatic methylation by human (guanine-N7)-methyl transferase. RNA 18, 856–868 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trapnell C., Pachter L., Salzberg S. L., TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anders S., Pyl P. T., Huber W., HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang D. W., Sherman B. T., Lempicki R. A., Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 71.Edgar R., Domrachev M., Lash A. E., Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Freier S. M., Kierzek R., Jaeger J. A., Sugimoto N., Caruthers M. H., Neilson T., Turner D. H., Improved free-energy parameters for predictions of RNA duplex stability. Proc. Natl. Acad. Sci. U.S.A. 83, 9373–9377 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/2/e1701854/DC1
fig. S1. RNA ligands for RIG-I activation.
fig. S2. SLR10 is monomeric in conditions that mimic cell culture and in vivo experiments.
fig. S3. Dose response of SLRs.
fig. S4. SLRs and poly(I:C) regulate a shared set of genes.
table S1. List of genes mutually regulated by SLRs and poly(I:C).
table S2. List of genes preferentially regulated by SLRs or poly(I:C).
Reference (72)