

Abstract
Despite their potential, conventional whole-cell cancer vaccines prepared by freeze-thawing or irradiation have shown limited therapeutic efficacy in clinical trials. Recent studies have indicated that cancer cells treated with certain chemotherapeutics, such as mitoxantrone, can undergo immunogenic cell death (ICD) and initiate antitumor immune responses. However, it remains unclear how to exploit ICD for cancer immunotherapy. Here, we present a new material-based strategy for converting immunogenically dying tumor cells into a powerful platform for cancer vaccination and demonstrate their therapeutic potential in murine models of melanoma and colon carcinoma. We have generated immunogenically dying tumor cells surface-modified with adjuvant-loaded nanoparticles. Dying tumor cells laden with adjuvant nanodepots efficiently promote activation and antigen cross-presentation by dendritic cells in vitro and elicit robust antigen-specific CD8α+ T-cells in vivo. Furthermore, whole tumor-cell vaccination combined with immune checkpoint blockade leads to complete tumor regression in ~78% of CT26 tumor-bearing mice and establishes long-term immunity against tumor recurrence. Our strategy presented here may open new doors to “personalized” cancer immunotherapy tailored to individual patient’s tumor cells.
Keywords: Cell engineering, immunogenic cell death, nanoparticle, cancer vaccine, cancer immunotherapy
Graphical Abstract
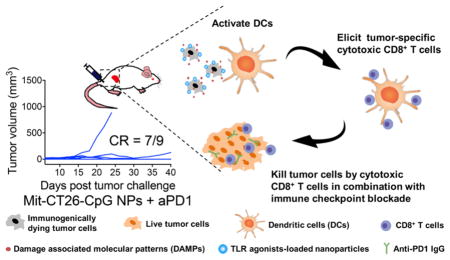
While various technologies, including micelles, lipid vesicles, polymers, and inorganic nanomaterials, have been developed as the delivery platforms for cancer vaccination,1–8 it remains challenging to achieve robust antitumor efficacy with therapeutic potential against established tumors. Cancer vaccines employing defined tumor antigens require extensive antigen discovery and optimization processes, but tumor cells can escape the immune pressure by antigen down-regulation and immunosuppression.9 Notably, recent reports suggest that cancer cells treated with certain anthracyclines undergo immunogenic cell death (ICD),10,11 during which dying tumor cells release immunostimulatory “danger” signals (e.g., high mobility group box 1 (HMGB1),12 calreticulin,13 and ATP14) to break immune tolerance and initiate antitumor immune responses.11,15 While this discovery suggests an entirely new therapeutic approach, it remains unclear how to exploit ICD as a new basis for cancer vaccination. This unmet need is underscored by the limited patient response rates to immune checkpoint blockers (ICBs) that remove immunosuppressive “brakes” on T-cells.16,17 If we can harness the potency of ICD to initiate antitumor immunity against a wide repertoire of antigens released from patients’ dying tumor cells and “liberate” antitumor T-cells with ICBs, this could lead to a powerful and generalizable strategy for “personalized” cancer immunotherapy.
Here, we present a novel synthetic approach for converting immunogenically dying tumor cells into a versatile platform for cancer vaccination and demonstrate their therapeutic potential in multiple murine tumor models. Specifically, we have utilized immunogenically dying tumor cells as the source of both tumor antigens and “danger” signals and amplified their potency by surface-modification of dying tumor cells with adjuvant-loaded nanodepots (Figure 1A). We demonstrate that our whole tumor-cell vaccine approach efficiently promotes activation and antigen presentation by antigen-presenting cells (APCs) and elicits strong antitumor immune responses in murine models of melanoma and colon carcinoma. Importantly, dying tumor cells laden with adjuvants in combination with ICBs exhibited remarkable therapeutic potential, leading to complete tumor regression and long-term protection against tumor recurrence in ~78% of tumor-bearing animals.
Figure 1.

Exploiting ICD for cancer immunotherapy. (A) Immunogenically dying tumor cells surface-decorated with TLR agonist-loaded nanoparticles release tumor antigens and damage-associated molecular patterns, triggering activation of dendritic cells and induction of tumor-specific CD8α+ T-cells that can kill tumor cells. Combination of the whole-cell vaccine with PD1 blockade further improves therapeutic efficacy. (B) The lipid-polymer hybrid nanoparticle (NP) encapsulating the TLR9 agonist CpG was constructed by complexation between cationic liposomes and thiolated HA-SH, an anionic biopolymer, followed by cross-link-mediated stabilization. (C) NP synthesis was optimized by varying the charge ratio between cationic lipids and HA-SH. Particle size and zeta potential were measured by DLS. Results are means ± SEM, n = 3. (D) A representative CpG-loaded NP visualized by transmission electron microscopy with negative staining. Scale bar, 20 nm.
First, we designed a new nanodepot platform (NP) for colocalized delivery of immunostimulatory ligands from immunogenically dying tumor cells. We chose to work with CpG oligonucleotide, a potent Toll-like receptor-9 (TLR9) agonist known to promote antigen cross-presentation and cross-priming of CD8+ T-cell responses.18 We constructed cross-linked lipid-polymer nanodepots by utilizing the charge-mediated complexation between cationic lipid vesicles containing a maleimide-modified lipid and a thiolated anionic biopolymer, hyaluronic acid (HA-SH).19 Subsequent chemical cross-linking led to the formation of multilamellar lipid-polymer hybrid nanodepots (Figure 1B). Briefly, we first synthesized the functional lipid DOBAQ-MAL using the EDC/NHS chemistry (Figure S1A). DOBAQ was successfully modified with a maleimide group, as shown by the results of thin layer chromatography (Figure S1B) and 1H NMR (Figure S2) with the reaction rate of 95% as determined by high-performance liquid chromatography (Figure S1C). Next, we prepared unilamellar liposomes (composed of DOBAQ-MAL, DOTAP, and DOPC) and incubated them with HA-SH and CpG, resulting in stable NPs. Maleimide-sulfhydryl-mediated cross-linking was crucial for the formation of homogeneous NPs, as aggregates were formed when we replaced DOBAQ-MAL (+1 charge) with DOTAP (+1 charge), or when we replaced HASH with HA-bearing the same amount of negative charges (data not shown).
We also optimized nanodepots by varying the amounts of HA-SH. Homogenous NPs with a diameter <300 nm were formed when the charge ratio of cations to anions (from cationic lipids and HA subunits, respectively) was set at =2 (Figure 1C,D). Therefore, we performed the subsequent studies using NPs formulated with 0.63 μmol of total lipids (DOBAQ-MAL/DOTAP/DOPC = 25:50:25, m/m/m) and 100 μg of HA-SH (~0.25 μmol of subunits) with the cation to anion ratio of 1.89. Compared with unilamellar liposomes, these lipid-polymer hybrid NPs exhibited a slightly increased particle size of 250 ± 13 nm with their surface charge converted to −16 ± 0.4 mV (Table S1). The resulting NPs had an average of ~2300 reactive maleimide molecules displayed on each particle, with 28% of lipids exposed on the external surfaces of NPs (as opposed to 45% for unilamellar liposomes) with 81 ± 2% of CpG loading efficiency (Table S1 and S2). These results indicate the successful synthesis of maleimide-displaying, multilamellar NPs loaded with CpG (CpG-NPs).
We then tethered CpG-NPs on the surfaces of dying tumor cells. We first characterized tumor cells undergoing ICD after treatment with mitoxantrone (Mit), a potent ICD-inducing anthracenedione agent.13 B16F10OVA melanoma cells expressing an exogenous antigen, ovalbumin (OVA), were exposed to 10 μM Mit for 12 h and washed. After 2 days of culture, the majority of tumor cells exhibited signs of apoptosis, as indicated by ~80% of the Annexin V+ cell population (Figure 2A) and released HMGB1 (Figure 2B), a marker of ICD.12 Live as well as immunogenically dying tumor cells have free sulfhydryls on endogenous cell-membrane proteins (as shown by cells stained with Oregon Green-Maleimide, Figure 2C). Thus, we have sought to utilize free sulfhydryls on the surfaces of immunogenically dying tumor cells to attach maleimide-displaying CpG-NPs. A brief treatment of dying tumor cells with 1 mM TCEP, a reducing agent, increased free sulfhydryls on the cell membrane by a 2.5-fold, compared with dying tumor cells without the TCEP treatment (Figure 2C). Preblocking Oregon Green-Maleimide with 100 molar excess of L-cysteine led to ~27-fold reduction in the mean fluorescence intensity (MFI) of cells, showing its specificity toward free thiols on the cells (Figure 2C). Overnight incubation of Mit- and TCEP-treated cells with NPs at 4 °C led to the successful surface-conjugation of NPs in a dose-dependent manner (Figure 2D,E, and Movie S1). When cell-NP conjugates were treated with trypan blue, a cell membrane-impermeable fluorescence-quencher,20,21 ~96% of cell-associated fluorescence signal was quenched (Figure 2F), indicating cell-surface conjugation, rather than internalization, of NPs. In contrast, elevating the incubation temperature from 4 to 37 °C resulted in ~50% of the NPs internalized by tumor cells within 1 h (Figure S3). Mit-treated B16F10OVA cells decorated with NPs displayed the prototypical markers of ICD (Figure S4).
Figure 2.

Surface-modification of immunogenically dying tumor cells with CpG-NPs. (A,B) Mitoxantrone (Mit) induced immunogenic cell death of tumor cells. B16F10OVA cells were treated by 10 μM mitoxantrone for 12 h, followed by media change and cell culture for 2 days. (A) Cell death and (B) cellular release of HMGB1 were measured by Annexin V/PI staining and ELISA, respectively. Numbers in the representative flow cytometry plots indicate the percentage of live, apoptotic, and necrotic cell populations. (C) Mit-treated tumor cells exhibited free thiol groups on their surfaces, and treatment with 1 mM TCEP further increased the level of free thiols, as shown by the representative flow cytometry analysis from two independent experiments. MFI, geometric mean fluorescence intensity. Preblocking of Oregon Green-Maleimide (with 100 molar-excess L-cysteine decreased the MFI. (D) The number of NPs bound on dying tumor cells was quantified after incubation of 106 Mit-treated B16F10 cells at 4 °C for 12 h with varying doses of fluorophore-labeled CpG-NPs. (E) The representative confocal images of a dying tumor cell (green) conjugated with NPs (red) and their 3D reconstruction. Scale bar, 5 μm. (F) In the presence of trypan blue (a membrane-impermeable quencher), fluorescence signal from cell-associated NPs was lost, indicating that NPs were attached externally to the cell membrane. Data in (A,B,D,F) show mean ± SEM (n = 3), representative from 2 to 3 independent experiments. * P < 0.05, *** P < 0.001, analyzed by one-way ANOVA with Bonferroni multiple comparison post-test.
We next asked whether dying tumor cells modified with CpG-NPs can recruit and activate dendritic cells (DCs). In particular, successful cross-priming of CD8α+ T-cells by DCs requires three signals: (1) antigen processing and presentation in the context of major histocompatibility complex (MHC) class I molecule; (2) up-regulation of costimulatory markers, such as CD40, CD80, and CD86; and (3) secretion of TNF-α and IFN-β that mediate inflammatory and innate immune responses,22,23 as well as Th1 cytokines, such as IL-12.24,25 Indeed, we observed that Mit-treated tumor cells significantly increased DC recruitment (a 2.4-fold increase than live tumor cells, P < 0.001, Figure 3A). DCs engulfed Mit-treated tumor cells 4.2-fold more efficiently than live tumor cells (P < 0.001, Figure 3B). We next examined cross-presentation of tumor antigens engulfed by DCs. As shown by staining with a 25-D1.16 monoclonal antibody directed against SIINFEKL-H-2Kb complexes,7 cross-presentation of OVA protein from B16F10OVA cells was significantly enhanced when DCs were cocultured with dying tumor cell-CpG-NP conjugates, compared with tumor cells admixed with the equivalent dose of free CpG or CpG-NPs (P < 0.001, Figure 3C). Moreover, surface-decoration of dying tumor cells with CpG-NPs was important for maturation and up-regulation of CD40 and CD86 on DCs (P < 0.001, Figure 3D,E, Figure S5). Similarly, dying tumor cell-CpG-NP conjugates promoted robust secretion of inflammatory cytokines from DCs, including IL-12p70 (only detected for dying tumor cell-CpG-NP conjugates, Figure 3F), TNF-α, and IFN-β (P < 0.001, Figure 3G,H). Overall, surface-conjugation of CpG-NPs on dying tumor cells, rather than their physical mixture, was crucial for strong DC maturation, antigen cross-presentation, and cytokine secretion.
Figure 3.

Immunogenically dying tumor cells conjugated with CpG-NPs activate DCs. (A) Mit-treated B16F10OVA tumor cells promoted recruitment of BMDCs as measured by the Transwell migration assay. (B,C) Mit-treated tumor cells were efficiently engulfed and cross-presented by BMDCs. BMDCs were cocultured with Oregon Green-labeled, Mit-treated B16F10OVA cells for 24 h, followed by (B) quantification of tumor antigen-positive BMDCs and (C) SIINFEKL display among antigen-positive BMDCs by flow cytometry. Group legends for panels (B–H) are shown in panel (B). (D–H) Dying tumor cells conjugated with CpG-NPs induced up-regulation of (D) CD40 and (E) CD86 on BMDCs and promoted BMDCs to secrete inflammatory cytokines, including (F) IL-12p70, (G) TNF-α, and (H) IFN-β, as measured by ELISA. ND, not detected. The data show mean ± SEM, from a representative experiment (n = 3) from 2 to 3 independent experiments. ** P < 0.01, *** P < 0.001, analyzed by one-way ANOVA with Bonferroni multiple comparison post-test.
Having shown DC activation in vitro, we examined whether the whole tumor-cell vaccine can elicit antitumor CD8α+ T-cell responses in vivo. We immunized naive C57BL/6 mice subcutaneously (s.c.) at tail base with a single dose of vaccine (4 × 106 Mit-treated B16F10OVA cells with 380 ng CpG per dose). On day 7, the frequency of CD8α+ T-cells against the immunodominant epitope of OVA, SIINFEKL, was measured by the tetramer staining assay on the peripheral blood mononuclear cells (PBMCs) (Figure 4A).26,27 Whereas vaccination with dying tumor cells alone resulted in minimal induction of SIINFEKL-specific CD8α+ T-cells, dying tumor cell-CpG-NP conjugates generated strong antigen-specific CD8α+ T-cell responses (3.2-fold greater than PBS, P < 0.01; and 2.4-fold greater than dying tumor cells, P < 0.05, Figure 4B). Similar results were also found among splenocytes (Figure S6). We also restimulated splenocytes from immunized mice with whole B16F10OVA cells and found that dying tumor cell-CpG-NP conjugates elicited significantly higher levels of IFN-γ+ CD8α+ and CD4 T-cells, compared with dying tumor cells without CpG (P < 0.05, Figure 4C,D). To assess the functionality of these CD8α+ T-cells, we inoculated the animals with 105 B16F10OVA tumor cells s.c. on day 8. A single immunization with dying tumor cell-CpG-NP conjugates protected all animals against tumor initiation (Figure 4E,F). In contrast, vaccination with dying tumor cells alone failed to stop B16F10OVA tumor growth (P < 0.01, Figure 4E) with only 20% survival rate (P < 0.05, Figure 4F). These results demonstrated the potency of the dying tumor cell-CpG-NP conjugates to generate antitumor T-cell immune responses in vivo.
Figure 4.

Immunogenically dying tumor cells membrane-decorated with CpG-NPs elicit antitumor T-cell responses in vivo. (A) C57BL/6 mice were vaccinated with 4 × 106 Mit-treated B16F10OVA cells, followed by tetramer staining for antigen-specific CD8α+ T-cells on day 7, and intracellular IFN-γ staining or tumor-cell challenge on day 8. (B) The frequency of SIINFEKL-specific CD8α+ T-cells among peripheral blood mononuclear cells (PBMCs) and their representative flow cytometry scatter plots are shown. (C,D) Splenocytes from immunized mice were restimulated with live B16F10OVA cells ex vivo, and the percentages of IFN-γ+ among (C) CD8α+ and (D) CD4+ splenocytes are shown. (E,F) Vaccinated mice were challenged with live B16F10OVA cells, and (E) average tumor volumes and (F) tumor-free percentages are shown. The data show mean ± SEM, (n = 10 for panels (B–D); n = 4–5 for panels (E,F)), analyzed by (B–D) one-way ANOVA, (E) two-way ANOVA with the Bonferroni multiple comparison post-test, or (F) the log-rank (Mantel-Cox) test. (B–D,F) * P < 0.05, ** P < 0.01, *** P < 0.001. (E) ** P < 0.01, *** P < 0.001 versus the PBS control; ## P < 0.01 versus the Mit-B16F10OVA group.
Next, we evaluated our vaccination strategy in a therapeutic setting against established CT26 colon carcinoma (without any exogenous antigen) to provide a more rigorous condition than the prophylactic setting shown above. We confirmed that Mit treatment induced ICD in CT26 cells and that CpG-NPs were successfully conjugated on CT26 cells (Figure S7). BALB/c mice were inoculated at s.c. flank with 2 × 105 CT26 cells, and on day 4 when tumors were palpable, a single dose of vaccine was administered. The dying tumor cell-CpG-NP conjugates significantly inhibited CT26 tumor growth (P < 0.001, compared with PBS or dying tumor cells alone, Figure S8), whereas dying tumor cells physically admixed with the equivalent dose of soluble CpG or CpG-NPs failed to reduce the tumor size. Taken together with the data shown in Figure 3, these results show that surface-conjugation of CpG-NPs on dying tumor cells, rather than their physical mixture, was crucial for potent immune responses and antitumor efficacy.
Lastly, we sought to amplify the therapeutic potential of our whole-cell vaccine by combining with ICBs. This was motivated by the low patient response rates to ICBs,28,29 highlighting the need to improve the outcomes of cancer immunotherapy. We treated CT26 tumor-bearing mice with dying tumor cell-CpG-NP conjugates combined with anti-PD1 IgG therapy (Figure 5A). The combination immunotherapy exerted robust anti-tumor efficacy, leading to potent inhibition of average tumor growth (P < 0.001, Figure 5B) and complete elimination of tumors in ~78% of animals (Figure 5C). In contrast, mice that received anti-PD1 monotherapy or dying tumor-cell vaccine alone failed to stop the average tumor growth in this model. Importantly, 100% of the survivors in the whole tumor-cell vaccine plus anti-PD1 treatment group rejected engraftment of 2 × 105 CT26 tumor cells rechallenged on day 70 (P < 0.001, Figure 5D), demonstrating long-term immunity against tumor recurrence. Moreover, throughout our studies we did not observe any signs of weight loss, toxicity, reactogenicity at the sites of vaccination, nor autoimmunity in animals treated with the combination immunotherapy. Collectively, these studies show that surface-modification of immunogenically dying tumor cells with adjuvant-carrying nanodepots rendered them into a potent vaccine platform with therapeutic potential.
Figure 5.

Immunogenically dying tumor cells conjugated with CpG-NPs exert potent antitumor efficacy in combination with immune checkpoint blockade. (A–D) BALB/c mice were inoculated with 2 × 105 CT26 cells subcutaneously on day 0, then treated with a single dose of the whole-cell vaccine (106 Mit-CT26-CpG-NPs) on day 4 when tumors were palpable, followed by anti-PD1 IgG therapy (clone: RMP1-14, 100 μg) every 3 days from day 5 to day 26. (B) The average tumor growth volumes are shown until day 20 when mice bearing large or ulcerated tumors had to be euthanized. (C) Individual tumor growth curves are shown. CR, complete regression. (D) Tumor growth was monitored after rechallenge of mice cured by the combination therapy (n = 7) with 2 × 105 CT26 tumor cells on day 70. Naive mice (n = 5) were used as control. The data show mean ± SEM from a representative experiment with n = 8–9 for panel (B,C) from two independent experiments, analyzed by (B) two-way ANOVA with Bonferroni multiple comparison post-test, or (D) log-rank (Mantel-Cox) test. (B) * P < 0.05, ** P < 0.01, *** P < 0.001 versus the control group; ## P < 0.01, ### P < 0.001 versus the anti-PD1 group.
In summary, we induced ICD of tumor cells by treating them with mitoxantrone and exploited immunogenically dying tumor cells as the platform for codelivery of tumor antigens and immunostimulatory agents. To promote immune activation, we have tethered CpG-NPs onto the surfaces of dying tumor cells via sulfhydryl-maleimide chemistry, a facile cell-engineering approach that has been utilized on therapeutic T-cells.30,31 Notably, our strategy based on nanodepots achieve codelivery of adjuvants without their premodification with cell-anchoring structures,32,33 or prelabeling of cells with metabolic ligands or antibodies34,35 and could deliver a single or potentially multiple synergistic TLR agonists.36 Here, we have demonstrated that immunogenically dying tumor cells decorated with CpG-NPs recruited DCs (Figure 3A), promoted DC maturation (Figure 3D–H), uptake of tumor antigens (Figure 3B), and subsequent antigen cross-presentation (Figure 3C), thereby triggering robust antigen-specific T-cell responses with antitumor efficacy in vivo (Figure 4). Importantly, by employing a combination approach with anti-PD1 therapy, we achieved regression of established CT26 tumors in ~78% of mice and protected them against future tumor relapse (Figure 5). Furthermore, a single vaccine dose employed in this study would simplify the immunization scheme and increase the translational potential of our strategy. To the best of our knowledge, we demonstrate for the first time that immunogenically dying tumor cells engineered to release exogenous adjuvants can exert potent prophylactic as well as therapeutic antitumor efficacy in multiple murine tumor models. We are currently exploring ways to codeliver ICD inducers and immunostimulatory molecules to tumors in vivo for generating whole tumor-cell vaccines in situ without ex vivo manipulations. Looking forward, we provide a general framework for exploiting ICD of tumor cells for elicitation of immunity against a wide repertoire of antigens found in whole tumor cells without a priori knowledge of antigens. Our strategy may open new avenues to “personalized” cancer immunotherapy tailored to individual patient’s tumor cells.
Supplementary Material
Acknowledgments
We acknowledge the NIH Tetramer Core Facility (contract HHSN272201300006C) for provision of MHC-I tetramers. We thank Dr. Jutaek Nam and Mengying Zhang for technical contributions. This work was supported in part by NIH (R01EB022563, R01CA210273, U01CA210152), MTRAC for Life Sciences Hub, UM Forbes Institute for Cancer Discovery Pilot Grant, and Emerald Foundation. J.J.M. is a Young Investigator supported by the Melanoma Research Alliance (348774), DoD/CDMRP Peer Reviewed Cancer Research Program (W81XWH-16-1-0369), and NSF CAREER Award (1553831). Y.F. is supported in part by the UM Rackham Predoctoral Fellowship. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense.
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nano-lett.7b03218.
Experimental protocols and supporting figures and tables (PDF)
Representative confocal 3D reconstruction of a dying tumor cell conjugated with NPs (MOV)
References
- 1.Moon JJ, Huang B, Irvine DJ. Adv Mater. 2012;24(28):3724–46. doi: 10.1002/adma.201200446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Irvine DJ, Swartz MA, Szeto GL. Nat Mater. 2013;12(11):978–90. doi: 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sahdev P, Ochyl LJ, Moon JJ. Pharm Res. 2014;31(10):2563–82. doi: 10.1007/s11095-014-1419-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, Van Egeren DS, Park C, Irvine DJ. Nature. 2014;507(7493):519–22. doi: 10.1038/nature12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan Y, Moon JJ. Vaccines (Basel, Switz) 2015;3(3):662–85. doi: 10.3390/vaccines3030662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Nat Rev Cancer. 2016;16(4):219–33. doi: 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- 7.Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Nat Mater. 2016;16(4):489–496. doi: 10.1038/nmat4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang C, Ye Y, Hu Q, Bellotti A, Gu Z. Adv Mater. 2017;29:1606036. doi: 10.1002/adma.201606036. [DOI] [PubMed] [Google Scholar]
- 9.Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, Mellstedt H. Nat Rev Clin Oncol. 2014;11(9):509–24. doi: 10.1038/nrclinonc.2014.111. [DOI] [PubMed] [Google Scholar]
- 10.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Annu Rev Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 11.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Nat Rev Immunol. 2016;17(2):97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 12.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L. Nat Med. 2007;13(9):1050–9. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 13.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Nat Med. 2007;13(1):54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 14.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, Uhl M, Genin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, Andre F, Lidereau R, McLaughlin NM, Haynes NM, Smyth MJ, Kroemer G, Zitvogel L. Nat Med. 2009;15(10):1170–8. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 15.Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, Coutant F, Metivier D, Pichard E, Aucouturier P, Pierron G, Garrido C, Zitvogel L, Kroemer G. J Exp Med. 2005;202(12):1691–701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teng MW, Ngiow SF, Ribas A, Smyth MJ. Cancer Res. 2015;75(11):2139–45. doi: 10.1158/0008-5472.CAN-15-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zou W, Wolchok JD, Chen L. Sci Transl Med. 2016;8(328):328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krieg AM. Annu Rev Immunol. 2002;20:709–60. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 19.Fan Y, Sahdev P, Ochyl LJJJA, Moon JJ. J Controlled Release. 2015;208:121–129. doi: 10.1016/j.jconrel.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Amersfoort ES, Van Strijp JA. Cytometry. 1994;17(4):294–301. doi: 10.1002/cyto.990170404. [DOI] [PubMed] [Google Scholar]
- 21.Patino T, Soriano J, Barrios L, Ibanez E, Nogues C. Sci Rep. 2015;5:11371. doi: 10.1038/srep11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balkwill F. Nat Rev Cancer. 2009;9(5):361–71. doi: 10.1038/nrc2628. [DOI] [PubMed] [Google Scholar]
- 23.Yang X, Zhang X, Fu ML, Weichselbaum RR, Gajewski TF, Guo Y, Fu YX. Cancer Cell. 2014;25(1):37–48. doi: 10.1016/j.ccr.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trinchieri G. Nat Rev Immunol. 2003;3(2):133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 25.Zhu J, Yamane H, Paul WE. Annu Rev Immunol. 2010;28:445–89. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klenerman P, Cerundolo V, Dunbar PR. Nat Rev Immunol. 2002;2(4):263–72. doi: 10.1038/nri777. [DOI] [PubMed] [Google Scholar]
- 27.Ochyl LJ, Moon JJ. J Visualized Exp. 2015;(98):e52771. doi: 10.3791/52771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, Blank CU, Hamid O, Mateus C, Shapira-Frommer R, Kosh M, Zhou H, Ibrahim N, Ebbinghaus S, Ribas A. N Engl J Med. 2015;372(26):2521–32. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 29.Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, Aren Frontera O, Havel L, Steins M, Garassino MC, Aerts JG, Domine M, Paz-Ares L, Reck M, Baudelet C, Harbison CT, Lestini B, Spigel DR. N Engl J Med. 2015;373(2):123–35. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ. Nat Med. 2010;16(9):1035–41. doi: 10.1038/nm.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang B, Abraham WD, Zheng Y, Bustamante Lopez SC, Luo SS, Irvine DJ. Sci Transl Med. 2015;7(291):291ra94. doi: 10.1126/scitranslmed.aaa5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu H, Kwong B, Irvine DJ. Angew Chem, Int Ed. 2011;50(31):7052–5. doi: 10.1002/anie.201101266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tom JK, Mancini RJ, Esser-Kahn AP. Chem Commun. 2013;49(83):9618–20. doi: 10.1039/c3cc45468a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mongis A, Piller F, Piller V. Bioconjugate Chem. 2017;28(4):1151–1165. doi: 10.1021/acs.bioconjchem.7b00042. [DOI] [PubMed] [Google Scholar]
- 35.Ahmed KK, Geary SM, Salem AK. J Controlled Release. 2017;248:1–9. doi: 10.1016/j.jconrel.2016.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Nat Immunol. 2005;6(8):769–76. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.