

Abstract
Fate allocation in the gastrulating embryo is spatially organized as cells differentiate to specialized cell types depending on their positions with respect to the body axes. There is a need for in vitro protocols that allow the study of spatial organization associated with this developmental transition. While embryoid bodies and organoids can exhibit some spatial organization of differentiated cells, these methods do not yield consistent and fully reproducible results. Here, we describe a micropatterning approach where human embryonic stem cells are confined to disk-shaped, sub-millimeter colonies. After 42 hours of BMP4 stimulation, cells form self-organized differentiation patterns in concentric radial domains, which express specific markers associated with the embryonic germ layers, reminiscent of gastrulating embryos. Our protocol takes 3 days; it uses commercial microfabricated slides (CYTOO), human laminin-521 (LN-521) as extra-cellular matrix coating, and either conditioned or chemically-defined medium (mTeSR). Differentiation patterns within individual colonies can be determined by immunofluorescence and analyzed with cellular resolution. Both the size of the micropattern and the type of medium affect the patterning outcome. The protocol is appropriate for personnel with basic stem cell culture training. This protocol describes a robust platform for quantitative analysis of the mechanisms associated with pattern formation at the onset of gastrulation.
Keywords: micropattern, self-organization, hESC, human embryonic stem cell, human stem cell, pluripotent stem cell, PSC, CYTOO chip, spatial pattern, embryo, gastrulating embryo, stem cell fate, BMP4, Wnt, germ layers, gastrulation, Nodal signaling
INTRODUCTION
Within the developing embryo, pluripotent cells of the epiblast undergo a series of cell fate decisions, first differentiating into the three germ layers and eventually to all the cell fates that make up the adult animal. These decisions are under the control of developmental signaling pathways that follow complex spatio-temporal sequences1. While extensive research on model organisms has elucidated the identities and components of these signaling pathways, quantitative, systems-level understanding remains elusive because of the difficulty involved in observing and perturbing embryonic development in vivo. Mammalian development is particularly inaccessible because it takes place in utero, and much of our knowledge of human development is extrapolated from studies on model organisms such as the mouse, despite the substantial differences that are known to exist between species2–4. Many of these issues can be overcome through the use of human embryonic stem cells (hESCs) as a complement to in vivo studies. A challenge of using hESCs to study development is that the differentiation must be made to resemble in vivo development as closely as possible, and, in particular, systems need to be developed in which hESCs differentiate in spatial patterns akin to those in the early embryo. Here we present a method for controlling the spatial organization of hESC differentiation patterns that are associated with embryonic gastrulation.
Development of the method
We initially analyzed the relationship between the TGF-® signaling and cell fate in a murine myoblast cell line, and showed that TGF-βshowed that TGF-cell line, different. We then explored the consequences of this mechanism in cell-fate decisions5,6. We sought to extend similar methods to study signaling and fate decisions in hESCs, but were hampered by the inherent variability between cells. The response of cells to applied ligands varied within the colony and every colony had a different spatial pattern of signaling. As regular hESCs cultures present colonies of different sizes and shapes, we reasoned that variations in colony geometries likely underlie these variable colony-level responses. We therefore sought to control colony geometries. Methods to control the shape of single cells had previously been used to study the biophysics of cell shape, adhesion, and division7,8. Micropatterning technologies to spatially control extra-cellular matrix deposition and thus colony geometries on 2D surfaces had also been applied to hESCs where it was observed that colonies of different size gave rise to different proportions of cell fate upon differentiation9,10, however, spatial differentiation patterns were not observed. In our experiments, we found that micropatterned colonies treated with Bone Morphogenetic Protein 4 (BMP4) responded with particular spatial patterns of signaling that translated into cell fate patterns. These patterns of signaling involved both differential responses to the initial BMP4 stimulus as well as patterns of endogenous Nodal signaling that were shaped by the production of both the Nodal ligand and its feedback inhibitor Lefty11. Wnt signaling likely serves as a required intermediate between BMP4 and Nodal as Wnt ligands are targets of BMP4 signaling both in vivo in the mouse1 and in hESCs differentiated with BMP412.
Comparison to other methods
Here, we describe a protocol that takes advantage of commercially available micropatterned coverslips (Cytoo). These are produced by first covering the culture surface with a cell-repellant substrate such as lysine-grafted polyethylene glycol13 and then selectively removing it using UV light or a plasma etch in a pattern defined by a mask. Homemade solutions using this technique can also yield satisfactory cell confinement14. An alternative method to produce the same results is microcontact printing, which is performed with an embossed stamp coated with an extracellular matrix of interest capable of mediating cellular attachment. When the stamp is pressed onto a slide, it deposits a cell-adherent coating in the desired pattern9,15. The uncoated areas may be backfilled with a passivating material to interfere with nonspecific attachment of cells. The stamps are made by spin coating polydimethylsiloxane (PDMS) elastomer over a negative mold made using standard photolithography or silicon etching. As commercial chips offer only a limited number of designs, it may be necessary to use homemade micropatterned chips for special applications: for example when alternative colony shapes are required or when one want to use a softer substrate than glass.
The main alternative to using two-dimensional micropatterned culture surfaces is to grow a defined number of cells in a three-dimensional aggregate. Various groups recently reported a degree of self-organization in aggregates of mouse ESCs16–18. These methods have the advantage of allowing cell movement in three dimensions, and, in some protocols, the aggregates elongate in a process that mimics gastrulation and convergent extension in the embryo. The cells begin in an approximately spherical orientation and spontaneously break symmetry to position particular germ layers in separate locations. Thus, these systems may represent a promising arena for the investigation of symmetry breaking, and the events that lead to the formation of the body axis from a symmetric embryo. These methods follow from earlier studies showing polarized signaling and differentiation within aggregates of mouse ESCs18 and are similar to methods for growing self-organized organoids19–21, applied to the early embryo as a whole. On the other hand, the initial state of a ball of cells is quite different from the epiblastic disk, and the confined two-dimensional disc of cells in micropatterned colonies may be a better representation of the epiblast that is a disk-shaped epithelium at the onset of gastrulation22 Two-dimensional cultures are also more amenable to imaging. Most importantly, two-dimensional micropatterning allows for reproducible colony geometries that lead to quantitatively reproducible differentiation patterns11. In contrast, quantification of the variability in spatial differentiation patterns has not been performed for three-dimensional aggregate cultures.
Thus, two-dimensional micropatterning is the current method of choice for studying signaling and spatial patterning in stem cell colonies, particularly in applications where quantitative reproducibility is essential. Three-dimensional aggregates are a more suitable system for studying the cellular movements involved in gastrulation, and may also allow study of the mechanisms of spontaneous symmetry breaking. In the future, hybrid methods, such as patterning the surface of a gel that the cells can invade upon differentiation may combine the advantages of both methods and also allow for the investigation of mechanical aspects of differentiation that have been shown to play a role in other stem cell systems23.
Limitations
As discussed above, this protocol allows for the quantitative observation of early embryonic signaling and cell fate patterns directly using human cells. It is the only system for examining mammalian patterning in vitro with quantitative reproducibility. The restriction to two dimensions limits cell movements and does not allow the cells to assume an organization identical to that of the embryo in vivo. In particular, gastrulation results in the three germ layers assuming a trilaminar structure with the mesoderm between the ectoderm and endoderm while in micropatterned culture these layers are positioned in the same order one next to the other. Embryos have a well-defined anterior-posterior axis but micropatterned colonies do not and their organization may more closely reflect that of embryos in which the distal visceral endoderm has failed to migrate anteriorly24,25. Finally, while many aspects of in vivo patterning are recapitulated in micropatterned culture, careful comparison to in vivo systems is always required to validate new discoveries.
Experimental design
A single cell suspension of hESCs in Rock-Inhibitor (RI) is used to seed micropatterned colonies at or near confluence. Cells are differentiated with application of BMP4 ligand for the desired period of time. 48 hours of differentiation is sufficient to generate patterns consisting of all germ layers and extraembryonic tissue. Cells can be imaged live during this time or fixed at the conclusion of the protocol. Patterns can be visualized with immunofluorescent staining. Large amounts of imaging data are generated by acquiring tiled images of the entire coverslip. Quantitative analysis of these images provides information on cell fate patterns and the signaling pathways that generate them with single-cell resolution in hundreds to thousands of colonies on a single coverslip. The following details should be considered when planning an experiment with micropatterned hESC.
Cells
The data that we show was obtained using the hESC RUES2 and ESI017 lines. The protocol has also been used successfully with other hESC lines (RUES1, H1) and iPSCs11. Therefore, this protocol may be suitable to induce self-organized differentiation patterns from any high-quality hPSCs.
Culture media
The protocol was originally developed with mouse embryonic fibroblast conditioned media (MEF-CM), and this provides the most robust adhesion to the culture surface. In cases where a defined culture media is essential or for continuity with culturing conditions in labs that used defined media, the protocol can be successfully performed in mTeSR1 culture media as well (see procedure in Box 1 and Figure S1, unpublished results). In MEF-CM, cells have a more spread morphology and tighter adhesion to the culture surface compared to mTeSR1 and use of MEF-CM may require less optimization in other aspects of the protocol to ensure robust adhesion to the culture surface. Due to the differences in surface adhesion and morphology the main challenge with using mTeSR1 is that the cells have a tendency to retract when Rock-Inhibitor is removed following seeding and this tendency is more pronounced in mTeSR1 than in MEF-CM. The retraction can lead to suboptimal filling of the micropatterned area and colonies lifting off the cell surface. These issues can be avoided by initiating differentiation soon after Rock-Inhibitor removal as described in the alternative protocol below. Surface coating. The original protocol involves a two-layer coating of Poly-D-Lysine and matrigel, however, the two step coating protocol increases the complexity and time requirements of the protocol. While standard laminin coating did not yield reproducible adhesion, recombinant laminin-521 (Biolamina) allows for a simpler coating protocol with robust results11. We have observed some batch-to-batch variability that requires optimization of LN521 concentration for each batch.
Cell density
The most important requirement is that the micropatterns be grown to confluence before initiating differentiation. Holes in the colonies create additional borders and can lead to irregular patterns of differentiation that depend on the precise configuration of the gaps. A large range of cell densities is compatible with this requirement and the length scale of differentiation will depend on density. Denser configurations yield smaller rings of the outer layers with a larger inner ectodermal layer. Some optimization of cell density to give the desired outcome is typically necessary.
Immunoflourescence and imaging
Imaging of three antibodies along with a nuclear counterstain is straightforward and follows standard immunofluorescent protocols. Typically, DAPI and Alexa Fluor 488, 555, and 647 are imaged with filters designed for DAPI, GFP, Cy3, and Cy5, although many other permutations are possible. Imaging the entire coverslip greatly enhances the statistical power of the approach, and most microscope controller software have the ability to create tiled montages, for example the Tile explorer function for the open source micromanager software. For some software, additional modules may be required and may require a separate purchase such as the multiposition solution for Olympus cellSens software. Analysis of tiled image sets is discussed in detail below (section Image acquisition and analysis of immunostained micropatterns).
Micropattern design
The protocol use Arena CYTOO chips. These glass coverslips have 19mm×19mm dimensions. Disk shaped micropatterns of 1000, 500, 225, 140 and 80μm diameters are regularly disposed over the full surface. There are respectively 25, 144, 576, 900 and 1296 colonies of the different sizes. Alternatively, one can purchase CYTOO chips comprising of a single colony size.
MATERIALS
REAGENTS
-
We have used human embryonic stem cells, line RUES2 (WiCell) or ESI017 (ESIBIO). Working with hESCs does not require special safety conditions (Biosafety 2 level).
CAUTION: It is essential to regularly test cells for potential Mycoplasma contamination, as it can generate inconsistent results. The hESC lines used in this study repeatedly tested negative for Mycoplasma contamination.
CAUTION: Experiments using hESCs must conform to all relevant governmental and institutional regulations. This work was approved by the Tri-Institutional Stem Cell Initiative Embryonic Stem Cell Oversight Committee (Tri-SCI ESCRO).
CAUTION: The cell lines used in your research should be regularly checked to ensure they are authentic.
GlutaMAX (Life Technologies, cat. no. 35050-061)
Knockout serum replacement (KSR; Life Technologies; cat. no. 10828-028)
MEM non-essential amino acid solution, 100× (Life Technologies, cat. no. 11140-050)
bFGF (AA 1-155) Recombinant Human Protein (Life Technologies, cat. no. PHG0263)
β-Mercaptoethanol, 55 mM (β-ME; Life Technologies, cat. no. 21985-023)
B-27 Supplement (50X), minus vitamin A (Life Technologies, cat. no. 12587-010)
DMEM, high glucose, pyruvate (Life Technologies, cat. no. 11995-065)
CF-1 Mouse Embryonic Fibroblasts (MEF) feeder cells, irradiated, high density (GlobalStem, cat. no. GSC-6101G)
Falcon 875cm2 Rectangular Straight Neck Cell Culture Multi-Flask, 5-layer With Vented Cap (Corning, cat. no. 353144)
35mm tissue culture dishes for chip coating and culture (FALCON, cat. no 353001)
15mL conical centrifuge tubes (Corning, cat. no. 352097)
2mL serological pipettes (FALCON, cat. no. 357507)
5mL serological pipettes (Corning, cat. no. 4487)
10mL serological pipettes (Corning, cat. no. 4488)
25mL serological pipettes (Corning, cat. no. 4489)
10-μl Barrier Pipette tips (Denville Scientific, cat. no. P1096-FR)
20-μl Barrier Pipette tips (ThermoFisher Scientific, cat. no. 2149)
200-μl Barrier Pipette tips (ThermoFisher Scientific, cat. no. 2069GPK)
1000-μl Barrier Pipette tips (ThermoFisher Scientific, cat. no. 2079GPK)
Gelatin, 0.1% in water (STEMCELL Technologies, cat. no. 07903).
Dulbecco’s PBS, calcium, magnesium (DPBS++, Life Technologies, cat. no. 14040-133)
Dulbecco’s PBS, no calcium, no magnesium (DPBS−−, Life Technologies, cat. no. 14190-094)
500mL 0.2-μm sterile filter units (ThermoFisher Scientific, cat. no. 569-0020)
0.2-μm syringe filters (Pall corporation, cat. no. 4612)
3mL syringes (BD, cat. no. 309657)
Laminin-521 (BioLamina, cat. no. LN521-04)
CYTOOchips Arena 500 A (CYTOO, cat. no. 10-024-00-18)
CYTOOchips Arena A (CYTOO, cat. no. 10-020-00-18)
Accutase (Stem Cell Technologies, cat. no. 07920)
mTeSR1 (STEMCELL Technologies, cat. no. 05857)
Rho kinase inhibitor, Y-27632 dihydrochloride (Abcam, cat. no. ab120129)
Penicillin-streptomycin (Life Technologies, cat. no. 15140-148)
Trypan Blue
Recombinant Human BMP-4 Protein (R&D Systems, cat. no. 314-BP-050)
-
Hydrochloric Acid (Fisher Scientific, cat. no. A144S-500)
-
◆
CAUTION: Irritant. Use in fume hood.
-
◆
-
Paraformaldehyde 4% In Phosphate Buffered Saline pH 7.4 (Poly Scientific R&D Corp, cat. no. S2303)
-
◆
CAUTION: Irritant. Use in fume hood.
-
◆
Triton X-100 detergent (BioRad, cat. no. 161-0407)
Fluoromount-G mounting medium (Southern Biotech, cat. no. 0100-01)
Bovine Serum Albumin (Sigma-Aldrich, cat. no. A4503-100g)
Normal Donkey Serum (Jackson ImmunoResearch, cat. no. 017-000-121).
Tween-20 (Sigma-Aldrich, cat. no. P1379-500ML).
DAPI (Cell Signaling Technologies, cat. no. 4083S).
- Primary antibodies:
Antigen Commercial information Dilution POU5F1 BD Biosciences 611203 1:400 SOX2 Cell Signaling 3579 1:200 NANOG R & D Systems AF1997 1:200 BRA R & D systems AF2085 1:200 SOX17 R & D Systems AF1924 1:300 CDX2 Abcam Ab15258 1:50 - Secondary antibodies:
Antibody Commercial information Dilution Donkey anti-Mouse Alexa488 ThermoFisher Scientific A-21202 1:500 Donkey anti-Goat Alexa555 ThermoFisher Scientific A-21432 1:500 Donkey anti-Rabbit Alexa647 ThermoFisher Scientific A-31573 1:500
EQUIPMENT
Inverted fluorescence microscope (for example we used an Olympus IX83) with X-Y motorized stage for tiling and digital imaging capture system (for example we used an Andor Zyla 4.2 C-Mos camera).
Inverted laser scanning confocal microscope (for example we used an Zeiss LSM780) for high-resolution optical slicing.
Inverted tissue culture microscope with phase contrast (for example we used an Olympus CKX41)
Biosafety cabinet for cell culture (e.g. SterilGuard III Advance SG403, The Baker Company)
CO2 incubator with controlling and monitoring system for CO2, humidity and temperature (e.g. HeraCell 150i, Thermo Fisher Scientific, cat. no. 51026282)
Cell culture centrifuge (e.g. Sorvall Legend X1R, Thermo Fisher Scientific, cat. no. 75004261)
Glass hemocytometer (e.g. Electron Microscopy Sciences, cat. no. 63511-11)
Cell culture disposables: Petri dishes, multiwell plates, centrifuge tubes, pipettes, pipette tips, filter units etc.
Pipette controller (Accujet pro, BrandTech, cat. no. 26333)
1000-μl pipette (Pipetman Classic, Gilson, cat. no. F123602)
200-μl pipette (Pipetman Classic, Gilson, cat. no. F123601)
20-μl pipette (Pipetman Classic, Gilson, cat. no. F123600)
10-μl pipette (Pipetman Classic, Gilson, cat. no. F144802)
2-μl pipette (Pipetman Classic, Gilson, cat. no. F144801)
Coverslip Forceps (Fine Science Tools, cat. no. 11251-33)
REAGENT SETUP
Preparation of FM10 media Prepare 500 ml of FM10 media by mixing 439 ml of DMEM, 50ml of FBS, 10ml of GlutaMAX, and 1 ml of β-mercaptoethanol. Filter the medium with a 0.22-μm filter unit, and store for up to 4 weeks at 4°C.
Preparation of HUESM media Prepare 500 ml of HUESM medium by mixing 379 ml of DMEM medium with 100 ml of KSR, 5 ml of GlutaMAX, 5 ml of NEAA, 1 ml of β-mercaptoethanol, and 10ml of B27 supplement without VitaminA. Filter the medium with a 0.22-μm filter unit, and store for up to 4 weeks at 4°C.
Preparation of MEF-conditioned media (MEF-CM) Coat a 5-layered flask (875 cm2) with 0.1% gelatin for 20 min at 37°C. Thaw 9 vials of irradiated MEFs (60×106 cells), and resuspend into 125 ml of FM10 media. Aspirate the gelatin from the flask and add the MEF/FM10 mixture. Incubate overnight. The next day, remove the media and replace with 150ml of HUESM. Incubate overnight to condition the medium. After 24hrs, harvest the conditioned medium into 50ml conicals, and replace with fresh HUESM. Repeat HUESM collection for up to 9 additional days. Freeze down MEF-CM aliquots at −80°C after collection and store for up to 6 months. When ready to use, add fresh bFGF at 20 ng/ml.
Preparation of BMP4 Prepare a 4mM HCl solution containing 0.1% (wt/vol) BSA in a sterile tube and use it to dissolve the lyophylized BMP4 to a final concentration of 50 μg/ml. Prepare 20 μl aliquots in microcentrifuge tubes, and store them at −80°C for up to 6 months. Thawed aliquots can be stored at 4°C for 2 weeks.
Preparation of bFGF Resuspend the lyophylized bFGF in PBS containing 0.1% (wt/vol) BSA to a final concentration of 20 μg/ml. Prepare 100-μl aliquots in microcentrifuge tubes, and store them at −80°C for up to 6 months. Thawed aliquots can be stored at 4°C for 2 weeks.
Preparation of blocking solution Add 10 μl of Triton X-100 and 300 μl of Normal Donkey Serum to 10 ml of PBS–. Gently mix by inversion. Do not vortex to avoid foaming.
Preparation of washing solution Add 20 μl of Tween-20 to 20 ml of PBS–. Gently mix by inversion. Do not vortex to avoid foaming.
Preparation of DAPI stock Reconstitute the 1mg DAPI in 10ml of deionized water to obtain a 0.1 mg/ml solution. Make 100 ul aliquots and freeze at −20°C for up to 2 years.
Preparation of LN-521 solution Thaw an aliquot of LN-521 at 4°C. Dilute 100 μg/ml LN-521 solution to the required concentration in DPBS++ (containing Ca++ and Mg++). LN-521 solution can be stored at 4°C for 1 month. CRITICAL Avoid repeated freeze-thaw cycles of LN-521.
PROCEDURE
Coating the CYTOO chip with LN-521 – TIMING 3 h
CRITICAL There is some batch-to-batch variability in the quality of LN521 and optimal concentrations may need to be determined empirically in the range of 5-20 μg/ml. We typically use a final concentration of 5 μg/ml. For one CYTOO chip use 2 ml of LN-521 solution.
-
Use tweezers to place the CYTOO chip face-up in a 35 mm tissue culture dish. Pipet 2 ml of LN-521 on top of the chip.
◆ CRITICAL STEP The side of the CYTOO chip on which the CYTOO label is written in the forward direction is the patterned surface.
◆ CRITICAL STEP The chip should remain submerged during the coating procedure. If necessary press the borders of the chip with the tweezers to keep it at the bottom of the dish. Be careful to touch only the borders, and not to scratch the internal patterned surface when using tweezers.
Incubate the chip at 37°C for 2 h or overnight at 4°C
-
Pre-warm 34 ml of DPBS++ to 37°C. Pipet 4 ml of DPBS++ into the dish. Remove 3 ml of the DPBS++ from the dish.
◆ CRITICAL STEP Minimize the time the chip is exposed to air during transfer to prevent drying of the chip and damage to the Laminin matrix.
-
Wash the chip 5X with DPBS by adding 6 ml to the dish and removing 6 ml from the dish.
◆ CRITICAL STEP The chip should remain submerged under DPBS throughout the all wash cycles to avoid drying the surface. If necessary press the borders of the chip with the tweezers to keep it submerged during the washes. Be careful to touch only the borders, and not to scratch the internal patterned surface when using tweezers.
◆ PAUSE POINT The coated chip can be stored under DPBS++ for at least 1 week at 4°C.
Single-cell passage and seeding of hESCs onto LN-521 coated CYTOO chips – TIMING 3 h
-
◆
CRITICAL One CYTOO chip requires 5 × 105 – 1 × 106 cells grown in MEF-CM. Cells should be passaged from a dish that is between 60 – 80% confluent. A 35 mm dish that is between 60 – 80% confluent should contain 1-2 × 106 cells. The volumes referred to in step 5- 14 will be that required when using one 35mm dish.
-
5
Before passaging, prepare media by adding bFGF (20 ng/ml), Y-27632 (10 μM), and Penicillin Streptomycin (Pen/Strep) (1%) to MEF-CM. Warm media to room temperature (18 – 25°C). Prepare 2 ml of media for each 35 mm dish to be passaged and 2 ml of media for each CYTOO chip to be seeded.
-
6
Rinse the dish containing hESCs with DPBS−−. Add enough volume of Accutase to the dish to cover the cell layer. For one 35 mm dish use 1 ml of Accutase. Incubate at room temperature for 5 – 7 min or until cells detach from the culture surface.
-
7
Gently break up colonies into single cells by pipetting with a 1 ml tip.
? TROUBLESHOOTING
-
8
Add cell suspension to the same volume of media. Centrifuge the suspension for 4 min at 300g and discard the supernatant. Resuspend the pellet in a volume of media that brings the concentration of cells to 1 – 3 × 106 ml−1 (about 1 ml). Pipet suspension gently with a 1 ml tip to break up any aggregates that formed during centrifugation.
-
9
Mix 5 – 10 μl of the cell suspension 1:1 with Trypan Blue and count cells using a hemocytometer.
-
10
Add the volume of the cell suspension containing 5 × 105 – 1 × 106 cells to additional media to bring the total volume to 2.5 ml.
-
11
Aspirate the DPBS++ from the dish containing the CYTOO chip and add the cell suspension.
◆ CRITICAL STEP Minimize the amount of time that the coated chip is exposed to air. Drying can damage the Laminin matrix.
-
12
Incubate the chip at 37°C and 5% CO2 for 2 h.
-
13
Prepare MEF-CM with bFGF (20 ng/ml) and Pen/Strep (1%) without Y-27632 and warm to 37 °C.
-
14
Aspirate media and wash chip once with 2 ml pre-warmed DPBS++. Add 2 ml of MEF-CM media without Y-27632 prepared in step 13.
? TROUBLESHOOTING
BOX 1 – ALTERNATIVE PROTOCOL USING mTeSR1 TIMING 6 h
Perform procedure steps 5-12 replacing MEF-CM media containing bFGF and Y-27632 with mTeSR1 containing Y-27632 (10 μM). In step 10, use 1-1.5 × 106 cells
Warm mTeSR1 medium without Y-27632 to 37°C, Aspirate the media and add 2ml of prewarmed mTeSR1 without Y-27632
Incubate for 3 hours at 37°C and 5% CO2
Prepare mTeSR1 with BMP4 (50 ng/ml) and warm to 37°C. Aspirate the media from the chip and add 2ml of fresh media containing BMP4.
-
Continue the remainder of the protocol beginning with step 17.
CRITICAL We have observed some retraction of the cells the day following BMP4 treatment so that at approximately 12-24 hours the cell colony may not reach to the edge of the micropatterned circle, however, the cells then recover to cover the entire colony and form extremely reproducible patterns 2 days after seeding.
Differentiation of hESCs into organized germ layers on LN-521 coated CYTOO chips – TIMING 2 – 3 days
-
15
Incubate seeded chip overnight at 37°C and 5% CO2 for 12 – 18 h.
? TROUBLESHOOTING
-
16
Prepare MEF-CM with bFGF (20 ng/ml), BMP4 (50 ng/ml), and Pen/Strep (1%) warm to 37 °C. Aspirate media from the chip and add 2 ml fresh media containing BMP4.
-
17
Incubate the chip at 37°C and 5% CO2 for 48 h.
Immunofluorescence staining of hESCs on CYTOO chips
-
18
Wash chip once with 2 ml DPBS– and move chip patterned surface to a new 35 mm dish.
-
19
Fix with 2 ml of 4% (wt/vol) paraformaldehyde for 20 min at room temperature.
◆ CAUTION The pipettes that contact paraformaldehyde should be discarded as chemical waste.
-
20
Remove the paraformaldehyde solution, wash the chip 2X with 2 ml DPBS–.
◆ CAUTION The paraformaldehyde solution should be discarded as chemical waste.
◆ PAUSE POINT The chip can be stored under 2 ml PBS for 1 week.
-
21
Prepare block solution and filter using a 0.2 μm pore size. Aspirate DPBS– and add 2 ml block solution. Incubate for 30 min at room temperature.
-
22
Remove block and add primary antibodies in block solution. Use 500 μl of block solution with antibodies for one chip in a 35 mm dish. Keep dish covered to prevent drying.
◆ CAUTION Different primary antibodies may require specific immunostaining conditions
-
23
Incubate at room temperature for 2 h or at 4°C for at least 8 h
? TROUBLESHOOTING
-
24
Remove block solution with primary antibodies and wash 3X with 1 ml washing solution.
-
25
Dilute secondary antibodies 1:500 and add 1 μg/ml DAPI to block solution. Remove washing solution and add 500 μl of block solution with secondary antibodies and DAPI. Cover and incubate for 30 min at room temperature.
-
26
Remove block solution with secondary antibodies and DAPI. Wash 2X with 1 ml wash solution followed by one wash with DPBS−−.
◆ PAUSE POINT The chip can be stored under 2 ml DPBS−− for 1 week.
-
27
To mount chip on a microscope slide dab the edge of the chip on a paper towel to remove excess PBS. Apply 30 – 50 μl of mounting media to the patterned surface and lay chip patterned surface down on a clean microscope slide.
◆ CRITICAL STEP Lower chip slowly onto the microscope slide to avoid trapping bubbles in the sample.
-
28
Allow sample to dry overnight protected from light.
Image acquisition and analysis of immunostained micropatterns
CRITICAL all steps can be accomplished using the open source software ImageJ26, or integrated in a more dedicated custom framework.
-
29
Acquisition. Acquire images and correct for uneven illumination using flatfield correction. Subtract background intensity from the images.
-
30
Stitching. Stitch the individual images to obtain a larger field of view with several colonies. CRITICAL STEP Combining all the individual tiles into a large one will result in very large image sizes. It is therefore advisable to only store a down-sampled stitched image and the positions relative to each other of the individual tiles.
-
31
Colony identification. Using the down-sampled stitched image from step 30, apply morphological closing to remove small-scale noise. Optionally, apply a Gaussian filter with a large standard deviation to further smoothen the image. Then, obtain the individual colonies by segmentation of the image, e.g. using Otsu’s thresholding, to decide whether pixels belong to the background or a colony. Classify the connected components obtained after thresholding as colonies, and inspect visually for correct identification.
-
32
Segmentation of nuclei on individual colonies. Use a nuclear marker (such as DAPI) to segment the individual cell nuclei. Examples of standard methods for this are graph cut algorithms FARsight27, seeded watershed transformations such as ImageJ’s 3D Watershed28, machine learning such as Ilastik (ilastik.org), or line-of-sight decomposition29. Combine the resulting pixel values for each segmented nucleus in an array and store the list of arrays corresponding to each nucleus. If performing the analysis using ImageJ, launch the “Trainable Weka Segmentation” plugin. Define 3 classes, background, nuclei, and cell-cell contacts, and train the classifier. After segmentation, get the probability maps and select the image corresponding to the “nuclei” class. Convert this image to a binary mask, and fill holes, erode, dilate, watershed. Use the “Analyze particles“ class to identify individual nuclei.
-
33
Application of nuclear data to other channels. Having obtained the pixel information of the nuclei in the step 32, apply this to the other channels to obtain expression levels in the respective channels. Subtract background and treat channels for unequal illumination, if necessary. If a 3D reconstruction f the nuclear data is available from optical sectioning using a confocal microscope, sum all pixels that belong to a nucleus in the respective channels. If only 2D data is available, correct for nuclei having different sizes and being in different positions relative to the focal plane by normalizing the integrated intensity of the nuclear marker. If using ImageJ, apply the pixel information from the segmented DAPI image by using the “Set Measurements” function and selecting an open image to “Redirect to”. Save the resulting list of intensities.
TIMING
Step 1-3, coating the chip with LN-521: 2 h (or overnight)
Steps 4, washing the chip: 15 min
Steps 5–11, single cell passage onto chip: 30 min
Steps 12-14, incubation and washing: 2 h
Steps 15-17, differentiation: variable (~ 2 days)
Steps 18-28, immunostaining: variable (~ 2 days)
Steps 29-33, image acquisition and analysis: variable (~2-3 days)
BOX 1 – ALTERNATIVE PROTOCOL USING mTeSR1 TIMING 6 h
?TROUBLESHOOTING
See Table 1 for troubleshooting guidance.
Table 1.
TROUBLESHOOTING TABLE
| Step | Problem | Possible Reason | Solution |
|---|---|---|---|
| 14 | Cells attach outside of the micropatterned colonies | Too high concentration of LN-521 | Find the working dilution of LN-521 for each batch; test in the range of 1:5 to 1:20 |
| Substrate dried up during coating/washing steps | Ensure that the chips are kept immersed in liquid | ||
| Substrate was not washed properly | Properly wash the substrate according to the protocol | ||
| Cells were left in ROCK inhibitor too long | Ensure that cells are exposed to Y-27632 for the appropriate time | ||
| Too many cells seeded | Adjust the number of cells used in Step 8 such that surface coverage of the colonies is 95-100% 2 hours after seeding | ||
| 14 | Uneven seeding | Poor mixing | Gently mix the cells when seeding the chip, taking care not to swirl as this will concentrate cells in the center of the dish. |
| Cells not reduced to a single-cell suspension | Single cells are critical for accurate counting and seeding; if colonies are difficult to break up into single colonies, incubate longer with Accutase | ||
| Too many or too few cells seeded | Adjust the number of cells used in Step 8 such that surface coverage of the colonies is 95-100% 2 hours after seeding | ||
| 17 | Holes forming in colonies upon removal of ROCK inhibitor | Poor seeding | Adjust the number of cells used in Step 8 such that surface coverage of the colonies is 100% 2 hours after seeding |
| 17 | Cells or colonies detaching from the chip | Problems with coating | Try higher concentration of LN-521 or longer coating time |
| Cell density is too high | Try lowering the cell concentration |
ANTICIPATED RESULTS
Upon seeding, hESCs will only attach to the permissive areas of the CYTOO chip (Fig. 1) and will form circular colonies with tightly packed cells within a couple of hours (Fig. 1).
Figure 1. Timeline of the morphology of micropatterned colonies.
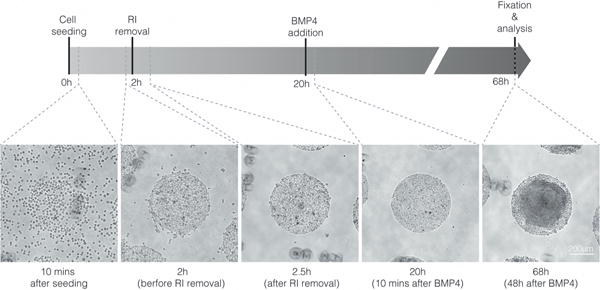
The images show the morphology of the micropatterned hESC colonies under phase microscopy 10 minutes after seeding (Step 11), before removal of ROCK inhibitor (Step 13), after removal of ROCK inhibitor (Step 14), 10 minutes after BMP4 addition (Step 16), and 48 hours after BMP4 addition (Step 17). RI=ROCK inhibitor. ESCRO institutional regulatory board permission was obtained to perform these experiments. Scale bar = 200 μm, applies to all panels.
In the absence of added morphogens and cultured under pluripotency conditions, hESCs on patterns maintain expression of the pluripotency markers SOX2, NANOG, and OCT4 for at least 24 h (Fig. 2A). Although all cells appear pluripotent, in colonies with diameters 500 μm or larger, the immunofluorescence intensity of these markers increases from the center of the colony to the edge. This effect may be interpreted as a consequence of edge-sensing in the signaling pathways, as the levels of signal transducers (e.g. SMAD 1/5/8 in the BMP pathway) are also elevated at the colony borders11. Colonies 250 μm or smaller may therefore be considered equivalent to the edges of large colonies.
Figure 2. Immunostaining of micropatterns: pluripotency and differentiation.

(A) Immunofluorescence staining of hESCs grown on a 1000 μm micropattern 24 hours after seeding on a CYTOO chip. Pluripotency markers are expressed in all cells of the colony. DAPI (top left, gray); OCT4 (top right, green); NANOG (bottom left, red); SOX2 (bottom right, blue). Scale bar = 200 μm. (B) The central portion of a CYTOO chip following 24 hours of BMP4 (50 ng/ml) differentiation. Nuclei are marked by DAPI staining. Scale bar = 2 mm. Colonies of different sizes (1000, 500, 250, 125, and 80 μm) can be identified. (C) Immunofluorescence staining of hESCs grown on a 1000 μm micropattern 48 hours after BMP4 (50 ng/ml) treatment. DAPI (top left, gray); CDX2 (top right, green); BRA (bottom left, red); SOX2 (bottom right, blue). Scale bar = 200 μm. ESCRO institutional regulatory board permission was obtained to perform these experiments.
Following a 48 h stimulation of colonies with 50 ng/ml BMP4, hESCs differentiate into organized and radially symmetric rings resembling embryonic patterning (Fig. 2B,C). The cells at the center of the colonies express SOX2, marking the prospective ectoderm, followed by concentric rings of cells expressing BRA, SOX17, and CDX2, marking the emergence of, respectively, mesoderm, endoderm, and extraembryonic trophoblast11 (Fig. 2C). Patterned differentiation is also evident in a morphological change of the colony, marked by a dense buildup of cells at the center and an enlargement and wider spreading of cells at the border (Fig. 1).
The spatial organization of fates is affected by the size of colonies, such that the smaller colonies do not exhibit central fates (Fig. 3). This observation indicates that, in the case of BMP4, fates are acquired from the edges of the colonies inward. Another factor that affects the outcome of patterning with BMP4 is the initial seeding density of cells. Namely, the spatial organization arises only if the cell density is sufficiently high. Therefore, if attempting alternative differentiation strategies, the size of confined colonies and the initial seeding density should be carefully considered.
Figure 3. Effect of colony size on cell fates.
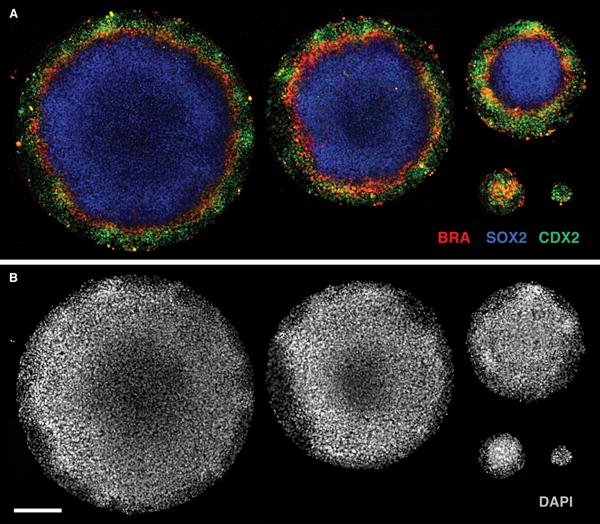
(A) As the colony radius decreases from 1000 μm to 80 μm, the central fate is gradually lost. Radius length of the colonies shown are 1000, 800, 500, 200, and 80 μm. Immunofluorescence staining of hESCs 48 hours after BMP4 (50 ng/ml) treatment. CDX2 (green); BRA (red); SOX2 (blue). (B) DAPI image for colonies in (A). Scale bar = 200 μm, applies to all panels. ESCRO institutional regulatory board permission was obtained to perform these experiments.
Similar results are obtained using the chemically defined medium mTeSR instead of conditioned media (unpublished results, Fig. S1). Furthermore, in this paper, we have utilized two different cell lines RUES2 and ESI017, and we have previously shown11, that RUES1 and H1 cells can be differentiated into spatially organized patterns using the protocol described here.
Supplementary Material
Supplementary Figure 1. BMP4 differentiation in micropatterned hESC colonies grown in mTeSR. Immunofluorescence staining of ESI017 hESCs 48 hours after BMP4 (50 ng/ml) treatment. Shown are colonies of 1000, 800, and 500 μm seeded and grown following the alternative protocol described in BOX 1. CDX2 (green); BRA (red); SOX2 (blue). Scale bars = 250 μm. ESCRO institutional regulatory board permission was obtained to perform these experiments.
Editorial Summary.
This protocol describes how to differentiate and image human embryonic stem cells on micropatterned colonies to create radially organized domains of the germ layers mimicking embryonic gastrulation in vitro.
Acknowledgments
The authors would like to thank members of the Brivanlou, Siggia, and Warmflash laboratories for helpful discussions. This work was supported by NIH/NIGMS R01 GM101653 (AHB, ES), NIH/DHHS R01 HD080699 (AHB, ES), Cancer Prevention Research Institute of Texas (CPRIT) grant RR140073 (AW), NSF grant DGE-1325261 (AY), and NSF grant MCB-1553228 (AW).
Footnotes
Author contributions statements
All authors contributed to the design of the experiments. AD, FE, MCG, IM, JM, AR, MS, AY, and AW performed and analyzed the experiments. All authors wrote the manuscript.
Competing Financial Interests
The authors declare that they have no competing financial interests
References
- 1.Arnold SJ, Robertson EJ. Making a commitment: cell lineage allocation and axis patterning in the early mouse embryo. Nature reviews Molecular cell biology. 2009;10:91–103. doi: 10.1038/nrm2618. [DOI] [PubMed] [Google Scholar]
- 2.Behringer RR, Wakamiya M, Tsang TE, Tam PP. A flattened mouse embryo: leveling the playing field. Genesis. 2000;28:23–30. doi: 10.1002/1526-968x(200009)28:1<23::aid-gene30>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 3.Dobreva MP, Pereira PN, Deprest J, Zwijsen A. On the origin of amniotic stem cells: of mice and men. Int J Dev Biol. 2010;54:761–777. doi: 10.1387/ijdb.092935md. [DOI] [PubMed] [Google Scholar]
- 4.Rossant J. Mouse and human blastocyst-derived stem cells: vive les differences. Development. 2015;142:9–12. doi: 10.1242/dev.115451. [DOI] [PubMed] [Google Scholar]
- 5.Warmflash A, et al. Dynamics of TGF-beta signaling reveal adaptive and pulsatile behaviors reflected in the nuclear localization of transcription factor Smad4. Proc Natl Acad Sci U S A. 2012;109:E1947–1956. doi: 10.1073/pnas.1207607109. doi:1207607109 [pii] 10.1073/pnas.1207607109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sorre B, Warmflash A, Brivanlou AH, Siggia ED. Encoding of temporal signals by the TGF-beta pathway and implications for embryonic patterning. Dev Cell. 2014;30:334–342. doi: 10.1016/j.devcel.2014.05.022. doi:S1534-5807(14)00342-6 [pii] 10.1016/j.devcel.2014.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thery M, et al. Anisotropy of cell adhesive microenvironment governs cell internal organization and orientation of polarity. Proc Natl Acad Sci U S A. 2006;103:19771–19776. doi: 10.1073/pnas.0609267103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thery M, Jimenez-Dalmaroni A, Racine V, Bornens M, Julicher F. Experimental and theoretical study of mitotic spindle orientation. Nature. 2007;447:493–496. doi: 10.1038/nature05786. [DOI] [PubMed] [Google Scholar]
- 9.Peerani R, Bauwens C, Kumacheva E, Zandstra PW. Patterning mouse and human embryonic stem cells using micro-contact printing. Methods Mol Biol. 2009;482:21–33. doi: 10.1007/978-1-59745-060-7_2. [DOI] [PubMed] [Google Scholar]
- 10.Lee LH, et al. Micropatterning of human embryonic stem cells dissects the mesoderm and endoderm lineages. Stem Cell Res. 2009;2:155–162. doi: 10.1016/j.scr.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 11.Warmflash A, Sorre B, Etoc F, Siggia ED, Brivanlou AH. A method to recapitulate early embryonic spatial patterning in human embryonic stem cells. Nat Methods. 2014;11:847–854. doi: 10.1038/nmeth.3016. doi:nmeth.3016 [pii] 10.1038/nmeth.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurek D, et al. Endogenous WNT signals mediate BMP-induced and spontaneous differentiation of epiblast stem cells and human embryonic stem cells. Stem Cell Reports. 2015;4:114–128. doi: 10.1016/j.stemcr.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azioune A, Storch M, Bornens M, Thery M, Piel M. Simple and rapid process for single cell micro-patterning. Lab Chip. 2009;9:1640–1642. doi: 10.1039/b821581m. [DOI] [PubMed] [Google Scholar]
- 14.Ma Z, et al. Self-organizing human cardiac microchambers mediated by geometric confinement. Nat Commun. 2015;6:7413. doi: 10.1038/ncomms8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thery M, Piel M. Adhesive micropatterns for cells: a microcontact printing protocol. Cold Spring Harb Protoc. 2009;2009 doi: 10.1101/pdb.prot5255. pdb prot5255. [DOI] [PubMed] [Google Scholar]
- 16.van den Brink SC, et al. Symmetry breaking, germ layer specification and axial organisation in aggregates of mouse embryonic stem cells. Development. 2014;141:4231–4242. doi: 10.1242/dev.113001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poh YC, et al. Generation of organized germ layers from a single mouse embryonic stem cell. Nat Commun. 2014;5:4000. doi: 10.1038/ncomms5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.ten Berge D, et al. Wnt signaling mediates self-organization and axis formation in embryoid bodies. Cell Stem Cell. 2008;3:508–518. doi: 10.1016/j.stem.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eiraku M, et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature. 2011;472:51–56. doi: 10.1038/nature09941. [DOI] [PubMed] [Google Scholar]
- 20.Lancaster MA, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 2013;340:1190–1194. doi: 10.1126/science.1234852. [DOI] [PubMed] [Google Scholar]
- 22.Deglincerti A, et al. Self-organization of the in vitro attached human embryo. Nature. 2016;533:251–254. doi: 10.1038/nature17948. [DOI] [PubMed] [Google Scholar]
- 23.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 24.Migeotte I, Omelchenko T, Hall A, Anderson KV. Rac1-dependent collective cell migration is required for specification of the anterior-posterior body axis of the mouse. PLoS Biol. 2010;8:e1000442. doi: 10.1371/journal.pbio.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nowotschin S, et al. The T-box transcription factor Eomesodermin is essential for AVE induction in the mouse embryo. Genes Dev. 2013;27:997–1002. doi: 10.1101/gad.215152.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Kofahi Y, Lassoued W, Lee W, Roysam B. Improved automatic detection and segmentation of cell nuclei in histopathology images. IEEE Trans Biomed Eng. 2010;57:841–852. doi: 10.1109/TBME.2009.2035102. [DOI] [PubMed] [Google Scholar]
- 28.Gniadek TJ, Warren G. WatershedCounting3D: a new method for segmenting and counting punctate structures from confocal image data. Traffic. 2007;8:339–346. doi: 10.1111/j.1600-0854.2007.00538.x. [DOI] [PubMed] [Google Scholar]
- 29.Mathew B, et al. Robust and automated three-dimensional segmentation of densely packed cell nuclei in different biological specimens with Lines-of-Sight decomposition. BMC Bioinformatics. 2015;16:187. doi: 10.1186/s12859-015-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amat F, et al. Efficient processing and analysis of large-scale light-sheet microscopy data. Nat Protoc. 2015;10:1679–1696. doi: 10.1038/nprot.2015.111. [DOI] [PubMed] [Google Scholar]
- 31.Amat F, et al. Fast, accurate reconstruction of cell lineages from large-scale fluorescence microscopy data. Nat Methods. 2014;11:951–958. doi: 10.1038/nmeth.3036. doi:nmeth.3036 [pii] 10.1038/nmeth.3036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. BMP4 differentiation in micropatterned hESC colonies grown in mTeSR. Immunofluorescence staining of ESI017 hESCs 48 hours after BMP4 (50 ng/ml) treatment. Shown are colonies of 1000, 800, and 500 μm seeded and grown following the alternative protocol described in BOX 1. CDX2 (green); BRA (red); SOX2 (blue). Scale bars = 250 μm. ESCRO institutional regulatory board permission was obtained to perform these experiments.