

Abstract
Polymeric materials that respond to a variety of endogenous and external stimuli are actively developed to overcome the main barriers to successful systemic delivery of therapeutic nucleic acids. Here, we provide overview of viable stimuli that have been proven to improve systemic delivery of nucleic acids. The main focus is placed on nucleic acid delivery systems (NADS) based on polymers that respond to pathological or physiological changes in pH, redox state, enzyme levels, hypoxia, and reactive oxygen species levels. Additional discussion is focused on NADS suitable for applications which use external stimuli, such as light, ultrasound, and local hyperthermia.
Keywords: Nucleic acids, stimulus-responsive, polyplexes, nanoparticles, in vivo
1. Introduction
Various types of bioactive nucleic acids (NA) have been explored as potentially revolutionary treatments for all major types of diseases, including viral infections [1], hereditary disorders [2], autoimmune disorders, cardiovascular diseases, and cancer [3]. The attractiveness of NA therapies lies in their ability to correct the underlying molecular causes of the disease by intervening at the genomic level [4]. Although NAs have been an indispensable tool for genomic manipulations in vitro, their therapeutic potential remains largely unfulfilled outside of a few disease indications that rely on local administration [5]. The main obstacle to successful clinical translation of NAs has been the lack of suitable delivery methods, especially for applications that require systemic administration. Many of the delivery problems are due to the large size, strong negative charge, and susceptibility to enzymatic degradation of many NAs. Unlike other macromolecular biologics (i.e., monoclonal antibodies) which typically only have to bind plasma membrane receptors for their pharmacologic action, NAs must be delivered inside the target cells [6]. The requirement for delivery to specific intracellular organelles makes pharmaceutical development of NAs extremely challenging as it demands the synthesis of multiple functional features into a single delivery vector that can overcome the multitude of stability issues and intracellular and extracellular barriers. The delivery vectors must provide sufficient stability in the systemic blood circulation and avoid recognition by the immune system, while localizing preferentially at the disease sites. Perhaps the most challenging aspect of the delivery process is that the vectors must have the ability to transport and selectively release the NAs in their correct intracellular location (e.g., cytoplasm, nucleus, mitochondria) without excessive routing and degradation in the lysosomes [7].
Over the years, a growing body of literature has focused on solving these NA delivery problems by developing materials that can alter their properties in response to various stimuli encountered at the different stages of NA delivery [8]. Many of the developed materials are based on cationic polymers, which can electrostatically bind with NAs and self-assemble into nanosized particles called polyplexes. Although polyplexes are simple in concept and effective in vitro, initial attempts to use them for systemic NA delivery in vivo have suffered from problems such as multiple toxic side effects [9], fast clearance due to serum protein (opsonin) binding, low resistance to disassembly, and premature NA release during circulation. Out of these initial failures has arisen a variety of interesting stimulus-responsive design concepts for polyplexes and related NA nanocarriers, including those that offer the possibility of controlling particle charge and size, interactions with cell membranes, and disassembly and release of NAs. It is clear that the development of novel multifunctional nanocarriers, which transform their physicochemical properties in response to various stimuli in a timely and spatially controlled manner, is essential for translating the promise of NA therapies. This progress report surveys state-of-the-art NA delivery vectors that are actively responsive to various physiologically relevant endogenous and exogenous stimuli. We focus most of our attention on polymer-based systems suitable for systemic NA delivery in cancer because they represent the most advanced stimuli-responsive strategies developed based on abnormal tumor microenvironment or aberrant properties of cancer cells. The described NA delivery systems (NADS) are capable of overcoming multiple extracellular and intracellular barriers and achieving controlled NA release in the desired intracellular compartment (Figure 1). We explain the general rationale of sensitivity to a variety of stimuli, both physiological and externally applied, and concentrate on examples of biomedical materials utilizing these design concepts. Furthermore, a brief background discussion of each category and its corresponding mechanism of action are covered, and the latest progress and current challenges are highlighted.
Figure 1.

Overview of various stimuli explored in the design of stimulus-responsive NADS.
2. Delivery systems responsive to endogenous stimuli
2.1. pH-responsive systems
Differences in pH are found at all levels within the body and have been widely explored to improve NA delivery [10]. Cellular organelles, such as the cytoplasm, endosomes, lysosomes, endoplasmic reticulum, Golgi bodies, mitochondria and nuclei, all maintain their own characteristic pH (Figure 2) [10]. Delivery and release of NAs into the cytoplasm (siRNA, mRNA) or nucleus (plasmid DNA) is a crucial step and a major challenge during intracellular delivery [11]. The majority of nanosized particles are internalized by the process of endocytosis. The acidification of endosomes and their subsequent fusion with lysosomes creates a pH gradient that has been utilized for cytoplasmic NA delivery and release. The pH values range from 5–6 in the endosomes to about 4 in lysosomes. Considering this pH gradient, materials with functional groups with pKa between 4.0 and 6.0 can undergo dramatic changes in physicochemical properties during their endo/lysosomal trafficking. Therefore, to avoid the degradation and deactivation of NAs, the design of smart NADS for programmed endosomal escape has been important for successful NA delivery. Polycations have been developed that comprise amino groups with low pKa (4–7), which enable endosomal pH-responsive protonation and the subsequent release of entrapped substances to the cytosol (the so-called proton sponge effect). Polymeric nanoparticles with pH-dependent endosomal escape behavior have been reported to significantly enhance the intracellular bioavailability of NAs [12].
Figure 2.

Intracellular pH at different compartments.
A variety of pathological conditions, including inflammation, infection, and cancer, are characterized by the presence of acidic tissues [10]. In cancer, this decrease in pH is found both in primary and metastatic solid tumors. The pH values typically range from 6.7 to 6.9 in tumor tissues at distances of 100–300 µm from the blood vessels [13]. The reason for this pH difference is that the proliferation of tumor cells is so rapid that the blood vessels of tumors are often insufficient to provide enough nutrients and oxygen for the expansion of tumor cells. This imbalance leads to differences in the metabolic milieu between the solid tumor and the surrounding normal tissue. The lack of oxygen in the tumor leads to hypoxia, which causes the production of lactic acid and the hydrolysis of ATP under energy-deficient conditions, resulting in an acidic microenvironment. This physiological property of solid tumors has been widely used in the design of stimulus-responsive drug and NA delivery systems. For example, pH-triggered charge-conversion of nanoparticles (NPs) was reported to enhance the cellular uptake of NAs [11]. Agmatine- and arginine-chitosan conjugates, CS-DM-Agm [11a] and N-arginine-N-octyl chitosan [11b], which were utilized to transport siRNA or anti-cancer drugs, changed from being negatively charged to positively charged in the slightly acidic extracellular environment of the tumor.
2.1.1. Acid-labile linkers in the design of NADS
Various acid-labile linkers have been incorporated into the structure of stimulus-responsive polyplexes, including imines, orthoesters, acetals/ketals, and maleic acid amides (MAA). The linkers can be placed in the polycation main chain or side chains to prepare biodegradable polyplexes and to enable site-specific release of NAs [14]. Among the reported linkers, we highlight MAA as an example of a linker that has been successfully used for systemic in vivo delivery of NAs.
MAA linkers have recently been used in the design of pH-responsive polymers because of their selective lability in mildly acidic pH (Table 1) [15]. MAA linkers are prepared by the reaction of a primary amine and maleic anhydride derivatives [16]. The obtained linker is stable at neutral pH but hydrolyzes rapidly when exposed to acidic pH. The hydrolysis kinetics of MAA linkers depend on the substituent groups at the double bond of the MAA moiety [15]. In general, MAA from dimethyl maleic acid derivatives shows faster hydrolysis than MAA from monomethyl maleic acid derivatives because of the higher pKa of the carboxylate in dimethyl MAA [17]. As mentioned above, the extracellular pH in tumor tissues is lower than in normal tissues and the bloodstream. The MAA linker chemistry has been used to enable tumor -specific reactions, which can improve tumor uptake by either charge-conversion of polyplexes from negative to positive [18], or by removal of a protective PEG shell to expose the underlying cationic polyplex particles, thereby facilitating cell uptake [19].
Table 1.
Structures of representative pH-responsive polymers
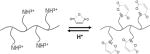
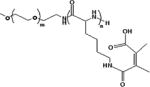
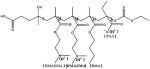
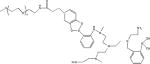
PEG-block-2,3-dimethylmaleic anhydride (DA)-modified poly(L-lysine) (PEG-PLL(DA)) was coated on the surface of polyplexes made from a folate-modified positively charged polypeptide (FK) and a p53 plasmid (FK/p53) through electrostatic interactions to form ternary complexes FK/p53/PEG-PLL(DA) (Table 1) [18a]. The charge-switchable PEG shield (PEG-PLL(DA)) acted as a detachable coating which responded to the lower pH at the tumor site. At the physiological pH of the bloodstream (7.4), PEG-PLL(DA) was able to extend the circulation time by shielding the positively charged FK/p53 complexes. After accumulation of the FK/p53/PEG-PLL(DA) complexes at the tumor site, the acidity-triggered charge switch led to the detachment of PEG-PLL(DA) from the FK/p53 complexes, and resulted in efficient tumor cell entry via folate-mediated uptake and electrostatic binding. The FK/pGL-3/PEGPLL(DA) complexes, which were initially negatively charged, became positively charged within 10 minutes of incubation at pH 6.8, and their zeta potential reached a plateau at around 15 mV after 20 minutes. At pH 7.4, the complexes kept their negative charge even after 100 minutes of incubation. This acid-dependent zeta potential change was mainly attributed to the fact that the rapid hydrolysis of amide bonds in the MAA under acidic conditions led to detachment of the PEG-PLL(DA) shell and exposure of the positively charged surface of the complexes.
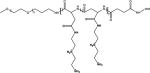
Wang and coworkers reported the use of a MAA linker (Dlinkm) in the synthesis of block copolymers containing PEG, a biodegradable polyester block, and nona-arginine to bind siRNA (Table 1) [19]. Hydrolysis of the MAA linker in the acidic tumor microenvironment caused the detachment of the PEG surface layer, thereby leading to efficient cellular uptake and enhanced gene silencing and therapeutic effect. Cumulative PEG release results revealed that less than 20% of the total PEG was released when the nanocarriers were incubated at pH 7.4 for 24 hours, while at pH 6.5, the release was much more rapid and reached a cumulative total of nearly 60% under otherwise identical conditions.
2.1.2. Enhanced endosomal release by pH-responsive NADS
Poly(ethylenimine) (PEI) was one of the first synthetic polycations shown to facilitate effective endosomal escape of polyplexes. Since then, multiple other polycations have been developed that build on the legacy of PEI. For example, highly effective polyaspartamides (PAsp) containing pH-sensitive endosomolytic ethylenediamine pendant groups have been developed [20]. PAsp(DET) combines excellent pH responsiveness with improved safety when compared with PEI. The amines in PAsp(DET) have two distinct pKas (6.2 and 8.9) [21] and thus the ethylenediamine moieties change from the monoprotonated state to the diprotonated state at the endo/lysosomal pH of 4–6. The positive diprotonated PAsp(DET) side chains strongly interact with the vesicular membrane and presumably induce membrane disruption, thereby enabling endosomal escape [21,22]. Ge et al. developed polyplex micelles prepared with PEG-b-PAsp(DET) and plasmid DNA encoding sFlt-1 (Table 1) [23]. The acid-responsive behavior of PAsp(DET) copolymers resulted in selective destabilization of the endosomal membrane and contributed to the significant growth inhibition of subcutaneous pancreatic tumors in mice. In addition, the PAsp main chain was fully biodegradable as a result of a self-catalytic reaction at neutral pH and at 37 °C [24]. This biodegradability, which contributed significantly to the safety and excellent transfection efficiency of PAsp(DET), occurred through an underlying degradation mechanism that has been previously observed in asparagine-containing peptides. Repeated transfection, by administering the polyplexes every 24 hours, showed that PAsp(DET) delivered a continuous increase in transgene expression, while non-degradable control polyplexes showed a decrease in transgene expression after 48 hours, coupled with fluctuations in the expression profiles of endogenous genes, suggesting off-target toxic effects. In vivo intraperitoneal injection of PAsp(DET) induced minimal levels of inflammatory cytokines, comparable to those induced by normal saline.
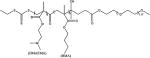
Copolymers of 2-(dimethylamino)ethyl methacrylate (DMAEMA) are another example of a polycation with weakly basic amines that has been shown to improve the endosomal escape of NAs through the presumed proton sponge effect [12b]. Hydrophobic modifications can have beneficial effects on polycationic delivery systems including prolonged serum stability, enhanced membrane binding, improved dissociation in the cytoplasm, and decreased cytotoxicity. A panel of pH-responsive PEG-b-(DMAEMA-co-BMA) diblock copolymers was prepared by reversible addition-fragmentation chain transfer (RAFT) (Table 1) [12b]. The copolymers were then used to create polyplexes with an anti-luciferase siRNA and the gene silencing efficiency was tested in MDA-MB-231 breast cancer cells transduced to constitutively express luciferase. The copolymer with a 50% BMA content (50B) produced polyplexes with the highest luciferase silencing effect (94% reduction in the protein level at 48 hours compared to the negative scrambled control siRNA). Moreover, the 50B polymer had an optimally balanced cationic and hydrophobic content in the core-forming block, which yielded polyplexes with improved resistance against destabilization in the kidneys in vivo and pH-dependent membrane-disrupting activity that was tuned for endosomal escape. The authors also synthesized poly[DMAEMA-b-(BMA-co-PAA-co-DMAEMA)] using RAFT polymerization (Table 1)[25]. These copolymers underwent self-assembly into siRNA-loaded micellar NPs with 40 nm size and positive ζ -potential (+20.2 mV) that were optimized for pH-dependent membrane disruption and tuned for endo-lysosomal escape. These NPs were also incorporated into porous polyester urethane (PEUR) tissue scaffolds. Delivery of siRNA-NPs from these tissue-inductive PEUR scaffolds has the potential to provide a new, tunable platform technology for efficient, local gene silencing.
2.1.3. Phenylboronic acid (PBA)-functionalized polymers
PBA-functionalized polymers have been utilized to target sialic acid (SA) residues on cancer cells [26]. PBA is a Lewis acid which forms reversible borate esters with cis-diols found in various carbohydrates [27]. Moreover, the covalent phenyl boronate ester linkages are thermodynamically stable under neutral or alkaline conditions (at pH >pKa) but are susceptible to cleavage when pH < pKa. PBA can interact with carbohydrates including ribose in RNA [27]. Since ribose ring exists at each 3’-end of double-stranded siRNA, PBA can be chemically conjugated to siRNA without any catalysts and can also form intermolecular cross-links via siRNA bridging, thereby stabilizing polyplexes [27b]. The PBA-siRNA binding is spontaneous and fully reversible, and this can be used to tailor the stability of the complexes to achieve controlled packing/unpacking in specific biological environments with a different pH, such as endosomes and lysosomes. This unique chemical characteristic provides an advantageous base for designing a PBA-mediated SA-targeting drug delivery system for cancer therapy.
The PBA-functionalized copolymer PEG-CPB-PEI (Table 1, PCPP) was used to condense siRNA into polyplexes [28]. This pH-responsive, tumor-specific NADS was constructed to achieve cleavable PEGylation, targeted delivery and effective intracellular release of siRNA. The polyplexes consisted of PEG-catechol (mPEG-Cat) and PBA-grafted low molecular weight PEI (PEI-PBA). The spontaneous formation of boronate linkages between PBA and catechol allowed facile attachment of mPEG-Cat to PEI-PBA under neutral conditions. After intravenous administration, the PEG-shell was detached due to the instability of PBA-catechol linker in the acidic tumor microenvironment. After internalization into the endosomes/lysosomes, the borate ester between PBA and ribose in the cross-linked core could be completely disrupted as the pH further declined to 4.5~5.5. Therefore, the polyplex disassembled and rapidly released siRNA. Meanwhile, the PEI component promoted endosomal/lysosomal escape and allowed the siRNA to be transported into the cytoplasm for gene silencing (Figure 3). In the gene silencing assay, down-regulation of luciferase expression in a pH-dependent manner was observed, and this reduction was more effective at pH 6.5 than at pH 7.4. After cells were incubated with PCPP/FAM-siRNA for 3 hours, the green fluorescence of FAM-siRNA was primarily found to be co-localized with the red signal of LysoTracker, while separate fluorescent signals from FAM-siRNA and LysoTracker were observed after 8 hours of incubation. This suggested that PCPP/FAM-siRNA was initially entrapped within endosomes/lysosomes, but then escaped. When a pH-responsive PCPP-siSur complex was tested in tumor-bearing mice, the tumors in the PCPP-siSur group had a relatively gradual growth profile and the smallest volume, suggesting that the PCPP-siSur nanoparticles effectively inhibited tumor growth in vivo.
Figure 3.

(a) Schematic illustration of the assembly of PCPP/siRNA polyplexes using electrostatic interactions and borate ester formation. (b) Mechanism of action of the dual pH-responsibility and tumor-targeted siRNA delivery by PCPP: (i) avoiding off-target effects by the protection of PEG corona; (ii) accumulation of PCPP/siRNA at the tumor site by EPR effect; (iii) extracellular acidity-activated detachment of PEG-shell and exposure of PBA; (iv) PBA recognition and SA receptor-meditated cellular internalization;(v) endosomal/lysosomal escape and acidity-triggered disassembly of the polyplexes accompanied by the release of siRNA; (vi) gene silencing. Reproduced with permission from[24]. Copyright 2017, Ivy spring.
The slightly lower pH of the cytoplasm compared to the extracellular environment is partly attributed to numerous acidic metabolites residing in the cytosol, e.g., adenosine triphosphate (ATP). The ATP concentration in the cytosol (1–10 mM) is 10-fold higher than that in the extracellular milieu (0.4 mM) [29]. To make a nanocarrier responsive to ATP, the carrier must include functionality capable of differentiating ATP from other components. So far, three types of approaches have been adopted to prepare ATP-responsive nanocarriers: 1) single-stranded DNA aptamers that specifically bind ATP [29a]; 2) enzymes that consume ATP as a source of energy [30]; and 3) PBA motifs, which can bind to diol-containing molecules such as ATP [31]. The main reported ATP-responsive NADS are based on PBA which can form a reversible ester linkage with cis-diol molecules. The Kim group developed a crosslinked polycation to reduce the toxicity of carriers and achieve fast release of loaded NAs in tumor cells in the presence of high concentrations of ATP [32]. In the study, the polycation CrossPEI was constructed from PBA-PEI (PBA-modified PEI) and Gal-PEI (galacturonic acid-modified PEI) (Figure 4). The galacturonic acid contained cis-diols which can form a reversible bond with PBA. Therefore, a crosslinked PEI was formed which encapsulated siRNA better and had a higher gene transfection efficiency than low molecular weight PEI (1.8 kDa). This crosslinked vector successfully delivered siRNA to the tumor and into the cytosol of tumor cells. Furthermore, the crosslinked vector disassembled when exposed to high concentrations of ATP in the cytosol, which resulted in burst release of the siRNA. The ATP-triggered degradation of CrossPEI meant that it had low systemic toxicity compared with high molecular weight PEI. Therefore, this delivery system holds great potential for systemic siRNA delivery and tumor therapy.
Figure 4.

Schematic illustration of anti-angiogenic gene delivery mediated by PBA-PEG-CrossPEI vector, which combines tumor targeting by PBA moiety with intracellular pH- and ATP-responsiveness. Reproduced with permission from[28]. Copyright 2015, Elsevier Ltd.
2.1.4. Calcium phosphate (CaP) particles
CaP-DNA co-precipitates were originally developed as a DNA transfection system by Graham et al. in 1973 [33]. CaP condenses NAs with high efficacy and rapidly dissolves in acidic endosomal or lysosomal environments, leading to endosomal escape and release of NAs into the cytoplasm [34]. However, CaP/NA particles grow quickly to form large agglomerates after preparation, causing reproducibility problems which limit their application in vivo. Moreover, large CaP agglomerates tend to disturb intracellular calcium homeostasis, resulting in cell death [35]. Current efforts are focused on developing stable and pH-responsive CaP-based hybrid nanoparticles. For example, carboxymethyl chitosan grafted with PEG (PEG-CMCS) was used to stabilize CaP particles with siRNA [36]. After cellular uptake, protonation of the amino groups of PEG-CMCS in the acidic environment of the endosome led to the formation of positively charged PEG-CMCS, which then dissociated from the CaP/siRNA particles. The exposed CaP dissolved at the acidic pH, thus releasing the siRNA, and the calcium and phosphate ions increased the osmotic pressure, bursting the endosome. The formed nanoparticles, with a slightly negative zeta potential and an average diameter of ~100 nm, efficiently encapsulated siRNA and had enhanced colloidal and serum stability. Moreover, comprehensive studies of the nanoparticles confirmed their superior safety both in vitro and in vivo. Qiu and coworkers prepared an alendronate-hyaluronan graft polymer (AHA) as the outer shell to stabilize CaP nanoparticles [37]. The inner core of the nanoparticles was formed by the CaP-siRNA co-precipitate, followed by the strong interaction between the calcium ions and the negatively charged phosphate of AHA. A pH-responsive release assay demonstrated that only 5% of the entrapped siRNA was released from CaPAHA10/siRNA NPs after 6 hours of incubation at pH 7.4, while 20% and 85% of the siRNA was released within 2 hours of incubation at pH 6.5 and 5.0, respectively. A lysosomal escape assay indicated that the acidification of lysosomes facilitated the disassembly of CaP-AHA10/siRNA NPs, and the resulting ions sharply increased the inner osmotic pressure, thus enabling the release of siRNA from late lysosomes to the cytoplasm.
2.2. Redox-responsive systems
2.2.1. Disulfide-containing NADS
Biodegradable polycations that contain disulfide bonds in their structure offer multiple advantages compared to nondegradable and other types of biodegradable polycation (i.e., esters) [38]. The disulfide bonds in such bioreducible polycations are relatively stable under physiological extracellular conditions, but can be cleaved selectively in several intracellular compartments through a thiol–disulfide exchange reaction with the abundant antioxidant glutathione (GSH) [39]. Thus, NADS based on bioreducible polycations are expected to be stable during systemic circulation, but should dissociate rapidly in the reducing environment of the cytosol or nucleus after cellular uptake. Such spatial selectivity allows, in principle, release of NAs in the desired intracellular space. Furthermore, the degradability of bioreducible polycations often leads to decreased cytotoxicity when compared with non-degradable analogs [40]. While there have been many studies published on the topic of bioreducible NADS, the vast majority of these have focused on the synthesis and in vitro examination of NA delivery. Here, we focus on the few selected examples of bioreducible NADS that demonstrate promising activity in vivo.
Branched PEI with molecular weight 25 kDa is the most widely used polycationic NADS due to its highly effective endosomal escape activity, but its in vivo use has been limited by its toxicity [41]. In contrast, low molecular weight PEIs have lower toxicity, but poor transfection efficiency. In order to balance the transfection efficiency and cytotoxicity, low molecular weight PEIs have been crosslinked using disulfide linkers to take advantage of the benefits of high molecular weight PEI. For example, bioreducible PEI (PEI-SS) was prepared and further conjugated with Pluronic copolymers. As expected [42], the PEI-SS NADS showed decreased toxicity in vivo, while DNA delivery and transfection efficiency were enhanced [38a]. Along the same lines, Chung and coworkers prepared PEI-SS using PEI crosslinked with cystamine bisacrylamide (CBA), and they further equipped the polymer with N-acetylglucosamine (GlcNAc) and a targeting ligand for delivery of siRNA to treat liver fibrosis [38b]. The resultant NADS specifically interacted with desmin on activated hepatic stellate cells and showed increased accumulation in the livers of fibrotic mice compared to normal mice. A similar crosslinking approach was reported by Kissel et al. using dithiobis (succinimidyl propionate) (DSP) [43]. Compared to the non-crosslinked 25 kDa PEI polyplexes, these disulfide cross-linked polyplexes showed enhanced resistance against polyanion exchange and high ionic strength, significantly increased colloidal stability, and reduced interactions with major blood components like albumin and erythrocytes. Pharmacokinetic profiles of the PEI-SS/DNA polyplexes in mice after intravenous administration showed higher blood levels for the crosslinked polyplexes, confirming that the stability in the circulation was improved by disulfide crosslinking. In vivo transfection data revealed that unwanted lung transfection was significantly reduced while liver transfection predominated.
Degradable polycationic gene carriers, such as reducible disulfide-containing linear or branched poly(amido amine)s, have been designed that demonstrate similar or enhanced NA delivery efficacy and reduced cytotoxicity when compared to PEI [44]. Bioreducible poly(amido amine)s allow incorporation of a large number of disulfides directly into the polymer backbone by employing Michael-type polyaddition between various primary or secondary amines and a suitable bisacrylamide such as CBA [45]. The high content of disulfides then allows a more pronounced decrease in polycation-related toxicity and better control of NA release after disulfide breakage. Like PEI, poly(amido amine)s can be synthesized to have high buffering capacities in the endosomally favorable pH range of 5–7.4, which may facilitate the endosomal escape of their polyplexes [46]. Zhang et al. reported cholesterol-grafted reducible poly(amidoamine) (rPAA-Chol), which was able to self-assemble into polyplexes with particle sizes of ~100 nm and to act as effective carrier for siRNA. Importantly, in vivo tumor therapy in nude mice bearing MCF-7 tumors further confirmed that the polyplexes containing siRNA to silence EGFR, which were designed to undergo transferrin receptor-mediated targeted delivery, had the greatest inhibitory effect on tumor growth, yet did not activate an immune response or cause significant body weight loss following systemic administration [47].
Dong et al. devised an interesting sheddable micelle architecture (PECssD) for siRNA delivery using disulfide linkages between PEGylated polycaprolactone (PCL) and PDMAEMA (Figure 5a) [48]. In the intracellular microenvironment, the disulfide linkages break and the PDMAEMA/siRNA polyplexes then dissociate from the PEGylated PCL cores of PECssD/siRNA polyplexes, facilitating endosomal escape and efficient release of the siRNA [44]. The distribution of siRNA in vivo was improved and the gene silencing efficiency was increased (Figure 5b). Furthermore, systemic administration of these NPs carrying a specific siRNA against siPlk1 (polo-like kinase 1) induced a tumor-suppressing effect in the HeLa-Luc xenograft model (Figure 5c).
Figure 5.

(a) Preparation of bioreducible PECssD/siRNA micelles that are taken up by cells and the cleavage of disulfide bonds facilitates endosomal escape and siRNA release. (b) Mean fluorescence intensities of PECssD/Cy5-siRNA micelleplexes in isolated organs. (c) Inhibition of tumor growth in HeLa xenograft following i.v. injection of PECssD2/siPlk1. Reproduced with permission from[44]. Copyright 2013, Elsevier Ltd.
Natural polysaccharides, such as chitosan, dextran and hyaluronic acid, offer good biocompatibility and biodegradability when used as components of NADS [49]. These polymers have also been developed for reduction-responsive delivery of NAs. Hyaluronic acid (HA) is among the most commonly used as it offers additional benefits due to its ability to bind with CD44 receptors which are overexpressed in various cancer cells [49–50]. HA-based siRNA polyplexes were prepared by Park et al. using HA grafted with PDMAEMA [51]. The polyplexes were chemically crosslinked via the formation of disulfide bonds under facile conditions. In this system, the hydrophilic HA surface of the polyplexes shielded the siRNA and prolonged its circulation time in the bloodstream. Additionally, the crosslinked polyplexes were able to disassociate through cleavage of the disulfide bonds and enzymatic degradation of HA in the intracellular compartments, thereby efficiently releasing siRNA into the cytosol. In vivo distribution results showed selective accumulation of the polyplexes in tumors after their systemic administration, and excellent gene silencing efficiency was confirmed through in vivo anti-tumor experiments. Recently, Sun and co-workers reported a hybrid nanoparticle based on CaP and crosslinked HA which had dual functions of redox responsiveness and specific tumor-targeting ability [52]. siRNA was efficaciously condensed by CaP via electrostatic interactions, while HA-ss-HA acted as an anionic outer shell to stabilize and compress the particles. Both in vitro and in vivo luciferase gene-silencing studies demonstrated high transfection efficiency. Moreover, equipped with the tumor-targeting component HA, the nanoparticles significantly suppressed the growth of B16F10 xenograft tumors in mice. Gelatin is another natural polymer that has been successfully used in the design of reducible NADS. Thiolated gelatin was used to create a disulfide-crosslinked nanoparticle system for efficient delivery of its NA payload [53]. The results showed improved targeting efficiency, resulting in increased transfection efficiency and tumor growth suppression in vivo.
2.2.2. ROS-responsive NADS
Reactive oxygen species (ROS) include multiple chemical species such as superoxides, hydrogen peroxide, hydroxyl radicals, and peroxyl radicals [54]. The role of ROS can be beneficial (i.e., cell signaling) if their concentrations are low and they are controlled adequately by the homeostasis mechanism; otherwise, it can be harmful (i.e., activation of mechanisms that trigger apoptosis), as in the case of oxidative stress with an abundance of ROS [55]. Many serious human diseases, such as cancer, inflammation, diabetes, Alzheimer's disease, Parkinson's disease, cardiovascular diseases, and arthritis are linked with ROS overproduction [56]. The high ROS levels in these diseases can be exploited in the development of ROS-responsive drug/gene delivery carriers, imaging probes, prodrugs, and theranostic agents.
Several ROS-responsive functional groups have been successfully used in the design of NADS, including boronates, sulfides, thioethers, and thioketals. For systemic NA delivery, boronates and thioethers are the most commonly used examples. They are both capable of being cleaved in the presence of elevated ROS levels and thus can be used to release the NA cargo at the target site. The Murthy group developed thioketal nanoparticles (PPADT) loaded with siRNA against TNF-α. The nanoparticles protected the siRNA from degradation in the gastrointestinal tract and selectively released the siRNA at the target gastrointestinal inflammation sites [57]. siRNA was first complexed with the cationic lipid DOTAP and added to an organic solution containing PPADT to form the ROS-responsive particles. Gastrointestinal inflammation is accompanied by a ten- to hundred-fold increase in mucosal ROS concentration. In a superoxide-rich environment, the thioketal groups are degraded into acetone and thiols. Therefore, the thioketal was cleaved at the target site into acetone and 4-(mercaptomethyl)phenyl methanethiol, leading to quick release of the siRNA. Furthermore, because of the selective nature of the cleavage, the siRNA remained intact in the nanoparticles and traveled through most of the gastrointestinal tract until it reached the target site. This ROS-responsive delivery system achieved successful delivery of siRNA to the intestinal inflammation site and silenced the target gene TNF-α. These results suggest that ROS-responsive materials with a high stimulus specificity can withstand the harsh environments during oral delivery and serve as vehicles for successful delivery of therapeutic NAs in numerous inflammatory gut diseases with elevated levels of oxidative stress.
The Shen group developed a ROS-responsive carrier containing PBA to enable the burst release of DNA in tumor cells [58]. In their study, the NADS contained three functional components: poly(acrylic acid), quaternary ammonium groups, and PBA groups. The negatively charged DNA was condensed by the positively charged polymer under physiological conditions. Upon oxidation of PBA group by ROS, the quaternary ammonium groups release p-quinone methide (p-hydroxylmethylenephenol) and become tertiary amines, which subsequently self-catalyze the fast hydrolysis of the ester groups, producing poly(acrylic acid). Therefore, the positive charge is quickly reversed to a negative charge in the presence of intracellular ROS which leads to the disassembly of the polyplex and burst release of DNA. The resulting ROS-responsive polyplexes show higher gene transfection efficiency than the control PEI. Furthermore, this carrier effectively prevented premature DNA release in the circulation and successfully delivered TRAIL DNA to the tumor. This resulted in up-regulation of the expression of the death ligand TRAIL and achieved a greater anti-tumor efficacy than carriers loaded with anticancer drug doxorubicin only. Another ROS-responsive NADS was developed by the Shen group [59]. They synthesized a new class of disintegrable polysulfoniums with boronic acid/ester moieties that respond to intracellular ROS by degrading into thioether fragments, thus accelerating the release of DNA in tumor cells (Figure 6). The polysulfoniums efficiently complexed DNA into small uniform polyplexes in an aqueous medium. After entering tumor cells, the polysulfoniums broke down into thioether fragments in the presence of ROS, efficiently releasing the DNA and facilitating its transcription. Thus, the polysulfoniums showed high gene transfection efficiency, strong resistance to serum degradation, and low cytotoxicity. In vivo, polysulfoniums successfully delivered the suicide gene pTRAIL and strongly inhibited growth and dissemination of various tumors.
Figure 6.

Design of ROS-responsive polysulfonium polymers and their DNA polyplexes. (a) Mechanism of ROS-triggered degradation into uncharged thioether fragments. (b) Proposed mechanism of uptake and intracellular trafficking of the polyplexes. Reproduced with permission from[55].
2.3. Enzymatically-activated NADS
Enzymes are the catalysts for most biological reactions, and are therefore important components of the biological toolkit. Their remarkable catalytic capacity, site-specific aggregation and requirement for mild reaction conditions means that their use in medical applications is increasing [60]. Many enzymes are overexpressed in tumors or inflammatory sites. Thus, enzyme-responsive materials can be suitable candidates for the design of NADS [61]. An enzyme-responsive material can be defined as a system that undergoes macroscopic changes in its physical/chemical properties upon the catalytic action of an enzyme [62]. The key component of an enzyme-sensitive material is the enzyme-responsive moiety, which is an enzyme substrate or a substrate mimic. Substrates that have emerged as modifications for NADS include enzymatically degradable peptides and esters. A second important factor in the design is the nature of the interactions inside the enzyme-responsive NADS, which lead to biochemical transformation and gene release. Materials can be designed so that they are activated by enzymes to expose their ligands or reverse their charges to facilitate subsequent internalization by, or penetration into, diseased organs [63]. Below, we will illustrate several typical enzyme-responsive materials for NA delivery.
Proteases are enzymes that are indispensable for cell proliferation, invasion and apoptosis. Proteases are also considered as ideal targets because of their up-regulated expression in diseases such as cancer and inflammation [64]. In a recent study, a new class of polyconjugate was developed that depends upon intracellular protease activity for siRNA delivery. These delivery vehicles expand on pH-sensitive dynamic polyconjugates (DPCs) which utilize MAA linkers for reversible masking of the membrane-interacting polyamine. The installed protease-sensitive linkages survive longer in circulation, and allow an increased range of targeting effects for siRNA delivery [65]. Matrix metalloproteinases (MMPs), as one type of protease, have been implicated in extracellular matrix remodeling during development and in inflammation and wound repair processes [66]. MMP-2 and MMP-9 are proteases that mainly engage in the degradation of extracellular matrices in tumors, and they are relatively overexpressed in almost all tumors compared with normal tissues. It has been proven that MMP-2 and MMP-9 responsiveness can be highly sensitive and specific in tumors, which is promising for cancer therapy and imaging. In recent studies, MMP-sensitive nanoparticles were specifically taken up by tumor cells. Tang et al [67] prepared nanoparticles consisting of PEG-block-poly[(1,4-butanediol)-diacrylate-β-5-hydroxyamyl-amine] (PEG-PDHA) and PEI-block-PDHA (PEI-PDHA). The PEG-PDHA and PEI-PDHA polymers were used as the outer layer, since they are amphiphilic, and paclitaxel (PTX) was loaded into the core. Loss of the PEG layer was triggered by the activity of MMP-2 and MMP-9 in the tumor microenvironment, which exposed the positively charged PEI and decreased the particle size. This increased the accumulation of the nanoparticles at the tumor site and enhanced their cellular uptake. Wang et al. also synthesized PEG and poly(ε-caprolactone) (PCL) with an MMP-2-sensitive peptide bridge as an siRNA carrier. The MMP-2-responsive nanoparticles can efficiently enhance siRNA delivery to tumor cells [68].
Esterases are enzymes that usually catalyze hydrolysis of esters into acids and alcohols. The insertion of carboxylic esters into cationic polymers can facilitate cytoplasmic degradation of the polymers by esterases to form negatively charged carboxylate ions, which interrupt the electrostatic interactions between the polymer and the NA cargo. In a recent study, PEI was reacted with p-acetoxybenzyl acrylate and then quaternized to synthesize an esterase-responsive polymer (ERP) as a plasmid DNA carrier (Figure 7). Interestingly, this ERP released the plasmid in tumor cells but not in tumor fibroblasts. The benefit of this carrier is that it selectively kills the tumor cell but avoids fibroblast hyperactivation [69].
Figure 7.

Esterase-responsive charge-reversal polymer (ERP) (a) and its lipid-coated polyplexes with TRAIL plasmid for cancer gene therapy. Reproduced with permission from[65].
Despite the recent progress made in the development of enzyme-responsive drug delivery systems, there are still challenges that need to be addressed. For example, enzyme levels vary in the different types and stages of a disease and a deeper understanding of this issue is required for designing enzyme-responsive materials. Furthermore, the similarities between the substrates of related enzyme families should be taken into account to enhance NA delivery [64].
2.4. Hypoxia
Hypoxia, defined as low oxygen (<1.4%) levels in tissues [70], is a condition in which the whole or a region of the body has insufficient oxygen supply. For example, solid tumors contain hypoxic regions, surrounding areas of impaired circulation and extensive necrosis [71]. Due to the rapid proliferation of tumor cells, new blood vessel formation cannot satisfy the requirement for oxygen, resulting in inadequate blood flow to tumor regions and induction of the hypoxic state [70a,72]. Tumor hypoxia is an important negative prognostic factor and contributes to resistance to radiotherapy and chemotherapy, because of its central role in vasculogenesis, invasiveness, metastasis, resistance to cell death, and altered metabolism [70b]. In addition, hypoxia is also associated with other ischemic diseases such as cardiomyopathy, limb ischemia, ischemic stroke, and neurodegenerative disorders [73]. In human diseases, hypoxia might well be considered a suitable target that can be exploited for improving diagnosis and treatment of various diseases [60].
Several typical bioreductive molecules with a hypoxia-sensitive moiety, such as nitroaromatic compounds, azobenzene (AZO) derivatives, and nitrobenzyl alcohols, have been applied in hypoxia-responsive systems for imaging and the design of hypoxia-sensitive prodrugs and delivery systems [74]. The nitroaromatic derivatives can undergo single-electron reduction to amines via a series of nitro reductases coupled with bioreducing agents that are present in tissues. This single-electron bioreduction is reversible and oxygen-dependent and is thus readily inhibited under normal oxygen levels. However, the nitro group of nitroimidazole can be further reduced to a free radical anion under hypoxic conditions [70]. Similarly, azo groups that can be reduced to amine derivatives under hypoxic conditions with a relatively high sensitivity have been incorporated in the form of bioreductive linkers in the backbone or side chains. Recently, these hypoxia-responsive groups were also utilized in the development of hypoxia-triggered drug release systems for anticancer and diabetes treatment [75]. Perche et al. designed a hypoxia-sensitive polymer containing an azo linkage for hypoxia-induced siRNA uptake and silencing [75c]. This siRNA nanocarrier was capable of hypoxia-activated gene silencing in hypoxic cells in vitro and in tumors after intravenous administration in vivo. In their study, the siRNA nanocarrier PAPD consisted of four components: PEG 2000, PEI, 1,2-dioleyl-sn-glycero-3-phosphoethanolamine (DOPE), and azo bond, which was used as a linker to conjugate PEG to PEI-DOPE units (Figure 8a). The PEI-DOPE conjugates formed complexes with siRNA and promoted the formation of micelles (Figure 8b). The PEG hydrophilic shell not only protected the siRNA from nuclease attack, but also extended the blood circulation time to increase tumor accumulation by the enhanced permeability and retention (EPR) effect. When the PAPD/siRNA micelles accumulated in the hypoxic tumor microenvironment, the PEG groups would be detached due to bioreduction of the azo linker. This exposed the positive charge of PEI in the residual PEI-DOPE/siRNA complexes, which could then be effectively taken up by the tumor cells. In vitro results revealed that the cellular uptake efficiency of siRNA under hypoxia was 3.2-fold higher than that under normoxia. The down-regulation of GFP expression in tumors was also detected after intravenous injection of PAPD/siRNA micelles into A2780/GFP tumors in mice. Moreover, the low oxygen threshold (0.1–1%) of the azo-based probe avoided non-specific activation in normal tissue, thus assuring the biosafety of this siRNA carrier. Such hypoxia-sensitive delivery systems provide a design guideline for cancer targeting and siRNA delivery in vivo.
Figure 8.

Design of hypoxia-sensitive copolymer PEG-Azo-PEI-DOPE (PAPD) (a) and their mechanism of action (b). Reproduced with permission from[71c].
3. Exogenous stimulus-responsive NADS
In addition to the endogenous stimuli described above, there has been a significant growth of interest in the use of physical exogenous stimuli to improve the efficacy of NA delivery [76]. Exogenous stimuli have acquired more attention because of their superior potential to achieve precise spatial and temporal control of the NA delivery. Exogenous stimuli refer to stimuli that are applied externally, such as light or ultrasound. Here, we summarize recent advances in the design of NADS that are able to control the systemic biodistribution of NAs in response to exogenous stimuli [39b].
3.1. Hyperthermia
Temperature change is a property that has the potential for development of stimulus-responsive systems for cancer treatment. Malignant tumors have a slightly higher temperature (40–42 °C) compared with normal tissues (36.5–37.5 °C) due to the Warburg effect [77]. While the natural hyperthermia of solid tumors is well-known, it has not been widely explored for NA delivery. Instead, hyperthermia approaches which utilize external means of increasing the local temperature has attracted wide attention. Hyperthermia can not only induce a direct cytotoxic effect (> 42.5 °C) in tumors but also increases the permeability of tumor vessels to macromolecular drugs [78]. Additionally, hyperthermia is associated with radiation- and chemotherapy-sensitizing properties as well as immunomodulatory effects in cancer therapy [79].
To date, multiple thermoresponsive materials capable of responding to the hyperthermic conditions have been widely applied in tumor-targeted gene delivery to enhance gene transfection efficiency. The core mechanism involves the polymers undergoing a phase transition when the temperature changes around the upper critical solution temperature or the lower critical solution temperature (LCST) [80]. If the LCST of a thermo-sensitive polymer is near body temperature, the polymer is endowed with inherent sensitivity to physiological temperature. When the temperature rises above the LCST, the thermo-sensitive polymer can bind nucleic acids more compactly and protect them from degradation during the delivery progress. When the temperature is below the LCST, the water solubility of the polymer will increase, resulting in the disaggregation of the polymer/gene complex and facilitating gene release from the complexes. In recent years, poly(N-isopropylacrylamide) (PNIPAM) and its derivatives have been the most widely reported LCST-type thermosensitive polymers for gene delivery [81]. PNIPAM undergoes hydrophilic to hydrophobic phase transition at its LCST of 32°C, and this value can be further tuned by copolymerization with hydrophobic monomers or the introduction of hydrophobic groups [81a]. Zintchenko and coworkers reported a set of PNIPAM/PEI copolymers for thermo-responsive gene delivery by application of local hyperthermia [82].The PEI block was able to condense DNA, and NIPAM, with hydrophilic co-monomers such as acrylamide or vinylpyrrolidone (VP), was used to increase the transition temperature. These copolymers, when complexed with DNA, undergo phase transition and aggregate around the LCST of 42 °C. It was found that gene expression was increased by two orders of magnitude in cells treated with copolymer/DNA under hyperthermic conditions compared with cells transfected at normal temperature. Thermosensitive block copolymers PVP10-BPEI loaded with DNA have also been used for in vivo gene delivery [77]. The gene transfection efficiency in tumors was 10-fold higher in hyperthermia-treated tumors compared with untreated tumors due to the fact that there was increased accumulation of the thermosensitive polymer and aggregation of thermosensitive polyplexes in the hyperthermic tumors. In a similar strategy, an amphiphilic graft copolymer PEI-g-PNIAPM was complexed with DNA to obtain a cationic, thermosensitive nanogel for gastric cancer therapy [83].
Another class of thermoresponsive materials, elastin-like polypeptides (ELPs), has attracted considerable attention as a thermosensitive carrier for gene delivery. ELPs are synthetic polypeptides composed of repeating Val-Pro-Gly-Xaa-Gly (VPGXG) sequences, where “Xaa” is any guest residue other than Pro [84]. The LCST of ELPs is a function of the composition and degree of Xaa substitution, which can be fine-tuned within the range of 0–100 °C for different applications [85]. ELPs have a reversible, rapid response to temperature with an inverse temperature phase transition [86]. Indeed, ELPs are highly soluble in aqueous solutions below the LCST but rapidly aggregate above it. Furgeson et al. designed an ELP-based recombinant diblock copolymer K8-ELP(1–60) for DNA delivery in cancer cells [87]. K8-ELP(1–60) contained a cationic oligolysine (VGK8G) and a thermosensitive ELP block with 60 repetitive pentapeptide units (VPGXG)60. The LCST of K8-ELP(1–60)/DNA was 44.9 °C. K8-ELP(1–60)/DNA complexes successfully transduced MCF-7 cells with qualitative expression of enhanced green fluorescent protein (EGFP) and minimal cytotoxicity compared to branched poly(ethyleneimine) controls.
3.2. Ultrasound-responsive NA delivery
Ultrasound (US) is an effective stimulus for NA delivery because its application can be spatially and temporally controlled at the desired site, therefore preventing unnecessary side effects to healthy tissues. US is also chosen because of its non-invasiveness, the absence of ionizing radiations, and the easy regulation of tissue penetration depth by tuning parameters such as frequency, duty cycles and time of exposure. NADS such as microcapsules with ultrasonic responsiveness are often composed of a cationic polymer or liposome shell and a gas core. Under US exposure, these gas-filled capsules (microbubbles), can rapidly expand, compress and collapse, leading to cavitation [88]. The cavitation then induces the formation of transient pores in the cell plasma membrane. Meanwhile the NAs are released from the carriers and enter the cell through these membrane pores to achieve an enhanced transfection effect. Strategies that have been used in US-triggered delivery systems include the co-administration of: 1) US and NAs; 2) US, microbubbles, and NAs; 3) US, microbubbles, and a separate NADS; 4) microbubble NA carrier constructs; and 5) targeted microbubbles as NA delivery vehicles [76]. Recent advances have mostly used the fourth and fifth strategies. For example, Lu Sun et al. synthesized a cationic microbubble (CMB) with the aim of enhancing its DNA-carrying capacity to improve targeted gene transfection of the ischemic heart for cardiac regeneration. The CMB was composed of octafluoropropane gas core and a lipid shell. It had good biological safety in vivo, and CMB bearing DNA was suitable for administration by intravenous injection [89]. Another study tested the ability of a plasmid-bearing microbubble formulation to target inflamed mesenteric endothelium in a mouse model of Crohn's disease. The US-activated microbubbles, which had a lipid shell and carried a luciferase plasmid, were targeted to sites of disease through functionalization with antibodies against markers of gut endothelial cell inflammation [90].
In addition to the methods described above, a combination of NA therapy and chemotherapy has also been tested in recent research. Yin et al. reported US-responsive nanobubbles encapsulating both the anti-cancer drug paclitaxel (PTX) and siRNA to treat hepatocellular carcinoma. The nanobubbles were developed by assembly of polymeric micelles and liposomes [91]. Yoon et al. reported a hybrid theranostic agent for use with US, namely a microbubble and liposome complex (MB-Lipo) [92]. Tavri et al. developed a novel strategy using stem cells preloaded with DNA-carrying microbubbles for an in vivo transfection. After the stem cells were loaded with the microbubbles, they remained alive, and gene expression could be detected following in vivo delivery and activation by US pulses [93]. For better in vivo transfection, Shapiro et al. focused on optimizing various parameters such as acoustic pressure, microbubble concentration, treatment duration, DNA dosage, and number of treatments in local microbubble-assisted sonoporation for in vivo gene delivery [94]. Mead’s group, using a method in which US, microbubbles and a separate NA carrier were co-administered, developed brain-penetrating nanoparticles (BPN) coated with a dense polyethylene glycol corona for treatment of brain tumors [95]. In another study, Fan’s group improved the gene transfection efficiency in the brain by using DNA-encapsulated microbubbles combined with focused US to disrupt the blood-brain barrier (BBB) [96]. Together, these examples show that with technological progress, US-assisted NA delivery may provide a much-needed therapeutic breakthrough, especially for in vivo applications.
3.3. Light-controlled NA delivery
The mechanisms involved in photochemical reactions can be used to design stimulus-responsive NA vectors. Under light irradiation, this type of vector can stimulate or trigger drug/NA release. Light irradiation also induces the breakage of specific chemical bonds and the generation of ROS, which can promote the release of NAs/drugs from vectors or enhance the endosomal escape of complexes after they are taken up by cells. The ultraviolet, visible, or near-infrared (NIR) regions of the light spectrum can be used to improve NA delivery using appropriately designed NADS. Because of the poor penetration of UV and visible light in tissues due to absorption by endogenous chromophores such as hemoglobin, strategies that depend on NIR light, with its higher penetrability, have attracted much attention. Jayakumar et al. combined the advantages of high-energy UV and NIR irradiation, using up-conversion technology [97]. They prepared core-shell fluorescent up-conversion nanoparticles which, when excited by highly penetrating near-infrared radiation, emitted simultaneously in the ultraviolet and visible ranges. The up-conversion nanoparticles could be used as remote nano-transducers to simultaneously activate endosomal escape and gene knockdown. Gene knockdown was enhanced as much as 30% in vitro compared to the control which was unable to facilitate endosomal escape. A similar gene knockdown trend was observed in vivo. Vankayala et al. reported novel and unique multifunctional gold nanoclusters with nuclear targeting properties. This theranostic nanoplatform was designed to perform simultaneous fluorescence imaging, gene delivery and long (850–1100 nm) NIR light-activated photodynamic therapy for the destruction of cancer cells [98]. The percentage of gene transfection using these multifunctional gold nanoclusters was nearly 3.2-fold higher than that obtained using the most commonly adopted LP2000 liposome gene carrier with NIR light irradiation. Zhang et al. developed smart nanocarriers consisting of gold nanorods as photothermal controllers, thermosensitive polymers as navigators, and Y-motifs as DNA nanorobots to achieve the targeted delivery and controlled release of siRNA and DOX in vivo. In vivo studies showed that a low dosage of nanocarriers synergistically inhibited tumor growth by silencing gene expression and inducing cell apoptosis under mild NIR irradiation [99]. These studies provide good examples of in vivo NA delivery by the NIR light-activated method. With the development of this type of nanotechnology, particularly in the form of multifunctional NAs nanostructures, specific stimuli can be used, providing smarter nanodevices for the personalized treatment of human disease with NAs and drugs.
4. Conclusion
Stimulus-responsive materials have been extensively investigated in a variety of biomedical applications. We have seen great advances in the development of materials that display responsiveness to physiologically relevant stimuli, which opened the door for their use in therapeutic and diagnostic applications. Perhaps in no other biomedical application is their potential as significant as in NA delivery, which has been held back by the lack of sophisticated delivery vectors that can successfully navigate the multitude of barriers encountered during NA delivery. In the last several years, we have seen increasing emphasis on development and evaluation of stimulus-responsive NADS that show activity in vivo. The increased focus on vectors that successfully deliver NAs after systemic administration is a welcomed development as it suggests maturing area of research and increasing realization of the importance of validation in relevant in vivo models. We hope that this progress report demonstrates the practical utility of stimulus-responsive materials in NA delivery and that we will see further advancements towards clinical translation.
Biographies

Minjie Sun is a Professor and Vice-Chair of the Department of Pharmaceutical Sciences at China Pharmaceutical University (CPU). She obtained her Ph.D. in Pharmaceutical Science with Prof. Qineng Ping at School of Pharmacy, CPU. She joined CPU in 2013 as a faculty of School of Pharmacy. She worked as a Visiting Scholar with Prof. Oupický on polymer-drug conjugates at the University of Nebraska Medical Center. Her research is focused on the development of microenvironment responsive drug and gene delivery system.

Kaikai Wang received his Ph.D. in Pharmacy with Prof. Yiqiao Hu and Prof. Jinhui Wu from School of Life Sciences, Nanjing University. He joined China Pharmaceutical University as a postdoc in Prof. Oupický’s group in September 2015. His research currently focuses on the development of multifunctional formulations based on novel polymers and proteins for photodynamic and photothermal therapy applications.

David Oupický is a Parke-Davis Professor of Pharmaceutics at the University of Nebraska Medical Center (UNMC) and Changjiang Scholar Professor at China Pharmaceutical University (CPU) in Nanjing. He received his Ph.D. in Macromolecular Chemistry with Prof. Karel Ulbrich at the Institute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic. He was a postdoc at the University of Birmingham where he worked with Prof. Len Seymour on non-viral gene delivery. After 10 years as a faculty at Wayne State University in Detroit, he joined UNMC in 2013. His research interests include synthesis of novel polymers and development of drug and nucleic acid delivery systems.
References
- 1.a) Morrissey DV, Lockridge JA, Shaw L, Blanchard K, Jensen K, Breen W, Hartsough K, Machemer L, Radka S, Jadhav V. Nat. Biotechnol. 2005;23:1002. doi: 10.1038/nbt1122. [DOI] [PubMed] [Google Scholar]; b) Okumura A, Pitha PM, Harty RN. Proc. Natl. Acad. Sci. U. S. A. 2008;105:3974. doi: 10.1073/pnas.0710629105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alnylam RNAi Roundtable: Conjugate Delivery. 2012 [Google Scholar]
- 3.a) Ferrari M, Sun T, Shen H. Cancer Gene Ther. 2012;19 doi: 10.1038/cgt.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Davis ME, Zuckerman JE, Choi CHJ, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Nature. 2010;464:1067. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lemieux P, Vinogradov S, Gebhart C, Guerin N, Paradis G, Nguyen H-K, Ochietti B, Suzdaltseva Y, Bartakova E, Bronich T. J. Drug Targeting. 2000;8:91. doi: 10.3109/10611860008996855. [DOI] [PubMed] [Google Scholar]
- 5.Molla MR, Levkin PA. Adv. Mater. 2016;28:1159. doi: 10.1002/adma.201502888. [DOI] [PubMed] [Google Scholar]
- 6.Li Y, Maciel D, Rodrigues J, Shi X, Tomas H. Chem. Rev. 2015;115:8564. doi: 10.1021/cr500131f. [DOI] [PubMed] [Google Scholar]
- 7.a) Du F, Wang Y, Zhang R, Li Z. Soft Matter. 2010;6:835. [Google Scholar]; b) Piskin E. Expert Rev. Med. Devices. 2005;2:501. doi: 10.1586/17434440.2.4.501. [DOI] [PubMed] [Google Scholar]
- 8.Soliman M, Allen S, Davies MC, Alexander C. Chem. Commun. 2010;46:5421. doi: 10.1039/c0cc00794c. [DOI] [PubMed] [Google Scholar]
- 9.Dobrovolskaia MA, Mcneil SE. J. Controlled Release. 2013;172:456. doi: 10.1016/j.jconrel.2013.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerweck LE, Seetharaman K. Cancer Res. 1996;56:1194. [PubMed] [Google Scholar]
- 11.a) Li Y, Yang J, Xu B, Gao F, Wang W, Liu W. ACS Appl. Mater. Interfaces. 2015;7:8114. doi: 10.1021/acsami.5b00851. [DOI] [PubMed] [Google Scholar]; b) Sun M, Li J, Zhang C, Xie Y, Qiao H, Su Z, Oupický D, Ping Q. Adv. Healthcare Mater. 2017;6 doi: 10.1002/adhm.201600693. [DOI] [PubMed] [Google Scholar]
- 12.a) Li H, Yu SS, Miteva M, Nelson CE, Werfel T, Giorgio TD, Duvall CL. Adv. Funct. Mater. 2013;23:3040. doi: 10.1002/adfm.201202215. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nelson CE, Kintzing JR, Hanna A, Shannon JM, Gupta MK, Duvall CL. ACS Nano. 2013;7:8870. doi: 10.1021/nn403325f. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Karimi M, Ghasemi A, Sahandi Zangabad P, Rahighi R, Moosavi Basri SM, Mirshekari H, Amiri M, Shafaei Pishabad Z, Aslani A, Bozorgomid M, Ghosh D, Beyzavi A, Vaseghi A, Aref AR, Haghani L, Bahrami S, Hamblin MR. Chem. Soc. Rev. 2016;45:1457. doi: 10.1039/c5cs00798d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Molavian HR, Kohandel M, Milosevic M, Sivaloganathan S. Cancer Res. 2009;69:9141. doi: 10.1158/0008-5472.CAN-09-2112. [DOI] [PubMed] [Google Scholar]
- 14.Cohen JL, Schubert S, Wich PR, Cui L, Cohen JA, Mynar JL, Fréchet JM. Bioconjugate Chem. 2011;22:1056. doi: 10.1021/bc100542r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takemoto H, Miyata K, Nishiyama N, Kataoka K. Adv. Genet. 2014;88:289. doi: 10.1016/B978-0-12-800148-6.00010-9. [DOI] [PubMed] [Google Scholar]
- 16.Dixon H, Perham R. Biochem. J. 1968;109:312. doi: 10.1042/bj1090312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Maeda Y, Pittella F, Nomoto T, Takemoto H, Nishiyama N, Miyata K, Kataoka K. Macromol. Rapid Commun. 2014;35:1211. doi: 10.1002/marc.201400049. [DOI] [PubMed] [Google Scholar]; b) Rozema DB, Ekena K, Lewis DL, Loomis AG, Wolff JA. Bioconjugate Chem. 2003;14:51. doi: 10.1021/bc0255945. [DOI] [PubMed] [Google Scholar]
- 18.a) Chen S, Rong L, Lei Q, Cao P-X, Qin S-Y, Zheng D-W, Jia H-Z, Zhu J-Y, Cheng S-X, Zhuo R-X. Biomaterials. 2016;77:149. doi: 10.1016/j.biomaterials.2015.11.013. [DOI] [PubMed] [Google Scholar]; b) Han L, Tang C, Yin C. Biomaterials. 2015;44:111. doi: 10.1016/j.biomaterials.2014.12.020. [DOI] [PubMed] [Google Scholar]; c) Jiang Q, Nie Y, Chen X, He Y, Yue D, Gu Z. Adv. Funct. Mater. 2017 [Google Scholar]
- 19.a) Xu C-F, Zhang H-B, Sun C-Y, Liu Y, Shen S, Yang X-Z, Zhu Y-H, Wang J. Biomaterials. 2016;88:48. doi: 10.1016/j.biomaterials.2016.02.031. [DOI] [PubMed] [Google Scholar]; b) Sun C-Y, Shen S, Xu C-F, Li H-J, Liu Y, Cao Z-T, Yang X-Z, Xia J-X, Wang J. J. Am. Chem. Soc. 2015;137:15217. doi: 10.1021/jacs.5b09602. [DOI] [PubMed] [Google Scholar]
- 20.a) Seo K, Kim D. Acta Biomater. 2010;6:2157. doi: 10.1016/j.actbio.2009.11.016. [DOI] [PubMed] [Google Scholar]; b) Zhang M, Liu M, Xue Y-N, Huang S-W, Zhuo R-X. Bioconjugate Chem. 2009;20:440. doi: 10.1021/bc800214u. [DOI] [PubMed] [Google Scholar]; c) Kim HJ, Ishii A, Miyata K, Lee Y, Wu S, Oba M, Nishiyama N, Kataoka K. J. Controlled Release. 2010;145:141. doi: 10.1016/j.jconrel.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 21.Miyata K, Oba M, Nakanishi M, Fukushima S, Yamasaki Y, Koyama H, Nishiyama N, Kataoka K. J. Am. Chem. Soc. 2008;130:16287. doi: 10.1021/ja804561g. [DOI] [PubMed] [Google Scholar]
- 22.Uchida H, Miyata K, Oba M, Ishii T, Suma T, Itaka K, Nishiyama N, Kataoka K. J. Am. Chem. Soc. 2011;133:15524. doi: 10.1021/ja204466y. [DOI] [PubMed] [Google Scholar]
- 23.Ge Z, Chen Q, Osada K, Liu X, Tockary TA, Uchida S, Dirisala A, Ishii T, Nomoto T, Toh K. Biomaterials. 2014;35:3416. doi: 10.1016/j.biomaterials.2013.12.086. [DOI] [PubMed] [Google Scholar]
- 24.Itaka K, Ishii T, Hasegawa Y, Kataoka K. Biomaterials. 2010;31:3707. doi: 10.1016/j.biomaterials.2009.11.072. [DOI] [PubMed] [Google Scholar]
- 25.Nelson CE, Kim AJ, Adolph EJ, Gupta MK, Yu F, Hocking KM, Davidson JM, Guelcher SA, Duvall CL. Adv. Mater. 2014;26:607. doi: 10.1002/adma.201303520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) Deshayes S, Cabral H, Ishii T, Miura Y, Kobayashi S, Yamashita T, Matsumoto A, Miyahara Y, Nishiyama N, Kataoka K. J. Am. Chem. Soc. 2013;135:15501. doi: 10.1021/ja406406h. [DOI] [PubMed] [Google Scholar]; b) Ellis GA, Palte MJ, Raines RT. J. Am. Chem. Soc. 2012;134:3631. doi: 10.1021/ja210719s. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ji M, Li P, Sheng N, Liu L, Pan H, Wang C, Cai L, Ma Y. ACS Appl. Mater. Interfaces. 2016;8:9565. doi: 10.1021/acsami.5b11866. [DOI] [PubMed] [Google Scholar]; d) Wang X, Tang H, Wang C, Zhang J, Wu W, Jiang X. Theranostics. 2016;6:1378. doi: 10.7150/thno.15156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a) Singh N, Willson RC. J. Chromatogr. A. 1999;840:205. doi: 10.1016/s0021-9673(98)01080-2. [DOI] [PubMed] [Google Scholar]; b) Naito M, Ishii T, Matsumoto A, Miyata K, Miyahara Y, Kataoka K. Angew. Chem. Int. Ed. 2012;51:10751. doi: 10.1002/anie.201203360. [DOI] [PubMed] [Google Scholar]
- 28.Fan B, Kang L, Chen L, Sun P, Jin M, Wang Q, Bae YH, Huang W, Gao Z. Theranostics. 2017;7:357. doi: 10.7150/thno.16855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.a) Mo R, Jiang T, Disanto R, Tai W, Gu Z. Nat. Commun. 2014;5:3364. doi: 10.1038/ncomms4364. [DOI] [PubMed] [Google Scholar]; b) Zhang Y, Lu Y, Wang F, An S, Zhang Y, Sun T, Zhu J, Jiang C. Small. 2017;13 doi: 10.1002/smll.201602494. [DOI] [PubMed] [Google Scholar]; c) Leist M, Single B, Castoldi AF, Kühnle S, Nicotera P. J. Exp. Med. 1997;185:1481. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biswas S, Kinbara K, Niwa T, Taguchi H, Ishii N, Watanabe S, Miyata K, Kataoka K, Aida T. Nat. Chem. 2013;5:613. doi: 10.1038/nchem.1681. [DOI] [PubMed] [Google Scholar]
- 31.Naito M, Ishii T, Matsumoto A, Miyata K, Miyahara Y, Kataoka K. Angew. Chem. 2012;51:10751. doi: 10.1002/anie.201203360. [DOI] [PubMed] [Google Scholar]
- 32.Jinhwan K, Yeong Mi L, Hyunwoo K, Dongsik P, Jihoon K, Won Jong K. Biomaterials. 2015;75:102. [Google Scholar]
- 33.Graham FL, Aj VDE. Virology. 1973;54:536. doi: 10.1016/0042-6822(73)90163-3. [DOI] [PubMed] [Google Scholar]
- 34.a) Bisht S, Bhakta G, Mitra S, Maitra A. Int. J. Pharm. 2005;288:157. doi: 10.1016/j.ijpharm.2004.07.035. [DOI] [PubMed] [Google Scholar]; b) Li J, Chen Y-C, Tseng Y-C, Mozumdar S, Huang L. J. Controlled Release. 2010;142:416. doi: 10.1016/j.jconrel.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neumann S, Kovtun A, Dietzel ID, Epple M, Heumann R. Biomaterials. 2009;30:6794. doi: 10.1016/j.biomaterials.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 36.Xie Y, Qiao H, Su Z, Chen M, Ping Q, Sun M. Biomaterials. 2014;35:7978. doi: 10.1016/j.biomaterials.2014.05.068. [DOI] [PubMed] [Google Scholar]
- 37.Qiu C, Wei W, Sun J, Zhang H-T, Ding J-S, Wang J-C, Zhang Q. Nanoscale. 2016;8:13033. doi: 10.1039/c6nr04034a. [DOI] [PubMed] [Google Scholar]
- 38.a) Ma XF, Sun J, Qiu C, Wu YF, Zheng Y, Yu MZ, Pei XW, Wei L, Niu YJ, Pang WH, Yang ZJ, Wang JC, Zhang Q. J. Controlled Release. 2016;235:99. doi: 10.1016/j.jconrel.2016.05.051. [DOI] [PubMed] [Google Scholar]; b) Park TG, Jeong JH, Kim SW. Adv. Drug Delivery Rev. 2006;58:467. doi: 10.1016/j.addr.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 39.a) Takemoto H, Miyata K, Nishiyama N, Kataoka K. Adv. Genet. 2014;88:289. doi: 10.1016/B978-0-12-800148-6.00010-9. [DOI] [PubMed] [Google Scholar]; b) Kim TI, Kim SW. React. Funct. Polym. 2011;71:344. doi: 10.1016/j.reactfunctpolym.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.a) Shim MS, Kwon YJ. Adv. Drug Delivery Rev. 2012;64:1046. doi: 10.1016/j.addr.2012.01.018. [DOI] [PubMed] [Google Scholar]; b) Movahedi F, Hu RG, Becker DL, Xu C. Nanomedicine. 2015;11:1575. doi: 10.1016/j.nano.2015.03.006. [DOI] [PubMed] [Google Scholar]; c) Chen G, Wang K, Hu Q, Ding L, Yu F, Zhou Z, Zhou Y, Li J, Sun M, Oupicky D. ACS Appl. Mater. Interfaces. 2017;9:4457. doi: 10.1021/acsami.6b14184. [DOI] [PubMed] [Google Scholar]
- 41.a) Guo X, Huang L. Acc. Chem. Res. 2012;45:971. doi: 10.1021/ar200151m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yamazaki Y, Nango M, Matsuura M, Hasegawa Y, Hasegawa M, Oku N. Gene Ther. 2000;7:1148. doi: 10.1038/sj.gt.3301217. [DOI] [PubMed] [Google Scholar]
- 42.a) Zhang L, Zhang Y, Chen Z, He Y. Int. J. Nanomed. 2016;11:5245. doi: 10.2147/IJN.S94995. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kim SJ, Ise H, Kim E, Goto M, Akaike T, Chung BH. Biomaterials. 2013;34:6504. doi: 10.1016/j.biomaterials.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 43.Neu M, Germershaus O, Mao S, Voigt KH, Behe M, Kissel T. J. Controlled Release. 2007;118:370. doi: 10.1016/j.jconrel.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 44.Wu C, Li J, Zhu Y, Chen J, Oupicky D. Biomaterials. 2013;34:8843. doi: 10.1016/j.biomaterials.2013.07.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.a Li J, Zhu Y, Hazeldine ST, Li CY, Oupicky D. Angew. Chem., Int. Ed. 2012;51:8740. doi: 10.1002/anie.201203463. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li J, Oupicky D. Biomaterials. 2014;35:5572. doi: 10.1016/j.biomaterials.2014.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin C, Zhong ZY, Lok MC, Jiang XL, Hennink WE, Feijen J, Engbersen JFJ. Bioconjugate Chem. 2007;18:138. doi: 10.1021/bc060200l. [DOI] [PubMed] [Google Scholar]
- 47.Gao LY, Liu XY, Chen CJ, Wang JC, Feng Q, Yu MZ, Ma XF, Pei XW, Niu YJ, Qiu C, Pang WH, Zhang Q. Biomaterials. 2014;35:2066. doi: 10.1016/j.biomaterials.2013.11.046. [DOI] [PubMed] [Google Scholar]
- 48.Lin D, Jiang Q, Cheng Q, Huang Y, Huang P, Han S, Guo S, Liang Z, Dong A. Acta Biomater. 2013;9:7746. doi: 10.1016/j.actbio.2013.04.031. [DOI] [PubMed] [Google Scholar]
- 49.Li YL, Maciel D, Rodrigues J, Shi XY, Tomas H. Chem. Rev. 2015;115:8564. doi: 10.1021/cr500131f. [DOI] [PubMed] [Google Scholar]
- 50.Au KM, Satterlee A, Min YZ, Tian X, Kim YS, Caster JM, Zhang LZ, Zhang T, Huang L, Wang AZ. Biomaterials. 2016;82:178. doi: 10.1016/j.biomaterials.2015.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoon HY, Kim HR, Saravanakumar G, Heo R, Chae SY, Um W, Kim K, Kwon IC, Lee JY, Lee DS, Park JC, Park JH. J. Controlled Release. 2013;172:653. doi: 10.1016/j.jconrel.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 52.Zhou ZW, Li HP, Wang KK, Guo Q, Li CZ, Jiang HL, Hu YQ, Oupicky D, Sun MJ. ACS Appl. Mater. Interfaces. 2017;9:14576. doi: 10.1021/acsami.6b15347. [DOI] [PubMed] [Google Scholar]
- 53.Xu J, Singh A, Amiji MM. BMC Cancer. 2014;14 doi: 10.1186/1471-2407-14-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.a) Chen X, Wang F, Hyun J, Wei T, Qiang J, Ren X, Shin I, Yoon J. Chem. Soc. Rev. 2016;45:2976. doi: 10.1039/c6cs00192k. [DOI] [PubMed] [Google Scholar]; b) Dröge W. Physiol. Rev. 2002;82:47. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]; c) Tapeinos C, Pandit A. Adv. Mater. 2016;28:5334. doi: 10.1002/adma.201670187. [DOI] [PubMed] [Google Scholar]
- 55.a) Hotamisligil GS. Nature. 2007;444:860. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]; b) Alfadda AA, Sallam RM. BioMed Res. Int. 2012;2012 [Google Scholar]; c) Mangge H, Becker K, Fuchs D, Gostner JM. World journal of cardiology. 2014;6:462. doi: 10.4330/wjc.v6.i6.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lü R. J. Mol. Cell. Cardiol. 2017 [Google Scholar]
- 57.Wilson DS, Dalmasso G, Wang L, Sitaraman SV, Merlin D, Murthy N. Nat. Mater. 2010;9:923. doi: 10.1038/nmat2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu X, Xiang J, Zhu D, Jiang L, Zhou Z, Tang J, Liu X, Huang Y, Shen Y. Adv. Mater. 2016;28:1743. doi: 10.1002/adma.201504288. [DOI] [PubMed] [Google Scholar]
- 59.Zhu D, Yan H, Liu X, Xiang J, Zhou Z, Tang J, Liu X, Shen Y. Adv. Funct. Mater. 2017;27 [Google Scholar]
- 60.Lu Y, Aimetti AA, Langer R, Gu Z. Nat. Rev. Mater. 2016;2:16075. [Google Scholar]
- 61.Ghadiali JE, Stevens MM. Adv. Mater. 2008;20:4359. [Google Scholar]
- 62.Takemoto H, Miyata K, Nishiyama N, Kataoka K. Adv. Genet. 2014;88:289. doi: 10.1016/B978-0-12-800148-6.00010-9. [DOI] [PubMed] [Google Scholar]
- 63.Karimi M, Ghasemi A, Sahandi Zangabad P, Rahighi R, Moosavi Basri SM, Mirshekari H, Amiri M, Shafaei Pishabad Z, Aslani A, Bozorgomid M, Ghosh D, Beyzavi A, Vaseghi A, Aref AR, Haghani L, Bahrami S, Hamblin MR. Chem. Soc. Rev. 2016;45:1457. doi: 10.1039/c5cs00798d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hu Q, Katti PS, Gu Z. Nanoscale. 2014;6:12273. doi: 10.1039/c4nr04249b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rozema DB, Blokhin AV, Wakefield DH, Benson JD, Carlson JC, Klein JJ, Almeida LJ, Nicholas AL, Hamilton HL, Chu Q, Hegge JO, Wong SC, Trubetskoy VS, Hagen CM, Kitas E, Wolff JA, Lewis DL. J. Controlled Release. 2015;209:57. doi: 10.1016/j.jconrel.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 66.a) Callmann CE, Barback CV, Thompson MP, Hall DJ, Mattrey RF, Gianneschi NC. Adv. Mater. 2015;27:4611. doi: 10.1002/adma.201501803. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Olson ES, Jiang T, Aguilera TA, Nguyen QT, Ellies LG, Scadeng M, Tsien RY. Proc. Natl. Acad. Sci. U. S. A. 2010;107:4311. doi: 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tang S, Meng Q, Sun H, Su J, Yin Q, Zhang Z, Yu H, Chen L, Chen Y, Gu W. Adv. Funct. Mater. 2016;26:6033. [Google Scholar]
- 68.Wang HX, Yang XZ, Sun CY, Mao CQ, Zhu YH, Wang J. Biomaterials. 2014;35:7622. doi: 10.1016/j.biomaterials.2014.05.050. [DOI] [PubMed] [Google Scholar]
- 69.Qiu N, Liu X, Zhong Y, Zhou Z, Piao Y, Miao L, Zhang Q, Tang J, Huang L, Shen Y. Adv. Mater. 2016;28:10613. doi: 10.1002/adma.201603095. [DOI] [PubMed] [Google Scholar]
- 70.a) Brown JM, Wilson WR. Nat. Rev. Cancer. 2004;4:437. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]; b) Wilson WR, Hay MP. Nat. Rev. Cancer. 2011;11:393. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 71.a) Ryan RM, Green J, Lewis CE. BioEssays. 2006;28:84. doi: 10.1002/bies.20336. [DOI] [PubMed] [Google Scholar]; b) Oelschlaeger TA. Bioengineered. 2010;1:146. doi: 10.4161/bbug.1.2.11248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.a) Coleman CN. J. Natl. Cancer Inst. 1988;80:310. doi: 10.1093/jnci/80.5.310. [DOI] [PubMed] [Google Scholar]; b) Harris A. Nat. Rev. Cancer. 2002;2:38. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 73.a) Arany Z, Foo S-Y, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM. Nature. 2008;451:1008. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]; b) Yu J, Zhang Y, Hu X, Wright G, Gu Z. Ann. Biomed. Eng. 2016;44:1931. doi: 10.1007/s10439-016-1578-6. [DOI] [PubMed] [Google Scholar]
- 74.a) Liu JN, Bu W, Shi J. Chem Rev. 2017;117:6160. doi: 10.1021/acs.chemrev.6b00525. [DOI] [PubMed] [Google Scholar]; b) Bowers DT, Tanes ML, Das A, Lin Y, Keane NA, Neal RA, Ogle ME, Brayman KL, Fraser CL, Botchwey EA. ACS Nano. 2014;8:12080. doi: 10.1021/nn504332j. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Thambi T, Park JH, Lee DS. Chem Commun (Camb) 2016;52:8492. doi: 10.1039/c6cc02972h. [DOI] [PubMed] [Google Scholar]; d) Do QN, Ratnakar JS, Kovács Z, Sherry AD. ChemMedChem. 2014;9:1116. doi: 10.1002/cmdc.201402034. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Guise CP, Mowday AM, Ashoorzadeh A, Yuan R, Lin W-H, Wu D-H, Smaill JB, Patterson AV, Ding K. Chin. J. Cancer. 2014;33:80. doi: 10.5732/cjc.012.10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.a) Lin Q, Bao C, Yang Y, Liang Q, Zhang D, Cheng S, Zhu L. Adv. Mater. 2013;25:1981. doi: 10.1002/adma.201204455. [DOI] [PubMed] [Google Scholar]; b) Thambi T, Deepagan VG, Yoon HY, Han HS, Kim SH, Son S, Jo DG, Ahn CH, Suh YD, Kim K, Kwon IC, Lee DS, Park JH. Biomaterials. 2014;35:1735. doi: 10.1016/j.biomaterials.2013.11.022. [DOI] [PubMed] [Google Scholar]; c) Perche F, Biswas S, Wang T, Zhu L, Torchilin V. Angew. Chem. 2014;126:3430. doi: 10.1002/anie.201308368. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liu H, Zhang R, Niu Y, Li Y, Qiao C, Weng J, Li J, Zhang X, Xiao Z, Zhang X. RSC Adv. 2015;5:20848. [Google Scholar]; e) Liu Y, Liu Y, Bu W, Cheng C, Zuo C, Xiao Q, Sun Y, Ni D, Zhang C, Liu J. Angew. Chem., Int. Ed. 2015;54:8105. doi: 10.1002/anie.201500478. [DOI] [PubMed] [Google Scholar]; f) Qian C, Yu J, Chen Y, Hu Q, Xiao X, Sun W, Wang C, Feng P, Shen QD, Gu Z. Adv. Mater. 2016;28:3313. doi: 10.1002/adma.201505869. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Qian C, Feng P, Yu J, Chen Y, Hu Q, Sun W, Xiao X, Hu X, Bellotti A, Shen QD. Angew. Chem., Int. Ed. 2017;56:2588. doi: 10.1002/anie.201611783. [DOI] [PubMed] [Google Scholar]; h) Wang Y, Xie Y, Li J, Peng ZH, Sheinin Y, Zhou J, Oupicky D. ACS Nano. 2017;11:2227. doi: 10.1021/acsnano.6b08731. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Feng L, Cheng L, Dong Z, Tao D, Barnhart TE, Cai W, Chen M, Liu Z. ACS Nano. 2017;11:927. doi: 10.1021/acsnano.6b07525. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Yu J, Zhang Y, Ye Y, DiSanto R, Sun W, Ranson D, Ligler FS, Buse JB, Gu Z. Proc. Natl. Acad. Sci. U. S. A. 2015;112:8260. doi: 10.1073/pnas.1505405112. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Yu J, Qian C, Zhang Y, Cui Z, Zhu Y, Shen Q, Ligler FS, Buse JB, Gu Z. Nano Lett. 2017;17:733. doi: 10.1021/acs.nanolett.6b03848. [DOI] [PubMed] [Google Scholar]
- 76.Rychak JJ, Klibanov AL. Adv. Drug Delivery Rev. 2014;72:82. doi: 10.1016/j.addr.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schwerdt A, Zintchenko A, Concia M, Roesen N, Fisher K, Lindner LH, Issels R, Wagner E, Ogris M. Hum. Gene Ther. 2008;19:1283. doi: 10.1089/hum.2008.064. [DOI] [PubMed] [Google Scholar]
- 78.a) Kong G, Anyarambhatla G, Petros WP, Braun RD, Colvin OM, Needham D, Dewhirst MW. Cancer Res. 2000;60:6950. [PubMed] [Google Scholar]; b) Lindner LH, Eichhorn ME, Eibl H, Teichert N, Schmitt-Sody M, Issels RD, Dellian M. Clin. Cancer Res. 2004;10:2168. doi: 10.1158/1078-0432.ccr-03-0035. [DOI] [PubMed] [Google Scholar]; c) Gaber MH, Wu NZ, Hong K, Huang SK, Dewhirst MW, Papahadjopoulos D. Int. J. Radiat. Oncol. Biol. Phys. 1996;36:1177. doi: 10.1016/s0360-3016(96)00389-6. [DOI] [PubMed] [Google Scholar]
- 79.Schlemmer M, Lindner LH, Abdel-Rahman S, Issels RD. Radiologe. 2004;44:301. doi: 10.1007/s00117-004-1044-6. [DOI] [PubMed] [Google Scholar]
- 80.a) Roy D, Brooks WL, Sumerlin BS. Chem. Soc. Rev. 2013;42:7214. doi: 10.1039/c3cs35499g. [DOI] [PubMed] [Google Scholar]; b) Li Y, Gao J, Zhang C, Cao Z, Cheng D, Liu J, Shuai X. Top Curr Chem (Cham) 2017;375:27. doi: 10.1007/s41061-017-0119-6. [DOI] [PubMed] [Google Scholar]
- 81.a) Yoshida R, Uchida K, Kaneko Y, Sakai K, Kikuchi A, Sakurai Y, Okano T. Nature. 1995;374:240. [Google Scholar]; b) Hinrichs W, Schuurmans-Nieuwenbroek N, Van De Wetering P, Hennink W. J. Controlled Release. 1999;60:249. doi: 10.1016/s0168-3659(99)00075-9. [DOI] [PubMed] [Google Scholar]; c) Kurisawa M, Yokoyama M, Okano T. J. Controlled Release. 2000;69:127. doi: 10.1016/s0168-3659(00)00297-2. [DOI] [PubMed] [Google Scholar]; d) Du F-S, Wang Y, Zhang R, Li Z-C. Soft Matter. 2010;6:835. [Google Scholar]
- 82.Zintchenko A, Ogris M, Wagner E. Bioconjugate Chem. 2006;17:766. doi: 10.1021/bc050292z. [DOI] [PubMed] [Google Scholar]
- 83.Cao P, Sun X, Liang Y, Gao X, Li X, Li W, Song Z, Li W, Liang G. Nanomedicine (Lond) 2015;10:1585. doi: 10.2217/nnm.15.20. [DOI] [PubMed] [Google Scholar]
- 84.a) McPherson DT, Xu J, Urry DW. Protein Expression Purif. 1996;7:51. doi: 10.1006/prep.1996.0008. [DOI] [PubMed] [Google Scholar]; b) Meyer DE, Chilkoti A. Nat. Biotechnol. 1999;17:1112. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- 85.Girotti A, Reguera J, Arias FJ, Alonso M, Testera AM, Rodríguez-Cabello JC. Macromolecules. 2004;37:3396. [Google Scholar]
- 86.Urry D. J. Phys. Chem. B. 1997;101:11007. [Google Scholar]
- 87.Chen TH, Bae Y, Furgeson DY. Pharm. Res. 2008;25:683. doi: 10.1007/s11095-007-9382-5. [DOI] [PubMed] [Google Scholar]
- 88.Mura S, Nicolas J, Couvreur P. Nat. Mater. 2013;12:991. doi: 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]
- 89.Sun L, Huang CW, Wu J, Chen KJ, Li SH, Weisel RD, Rakowski H, Sung HW, Li RK. Biomaterials. 2013;34:2107. doi: 10.1016/j.biomaterials.2012.11.041. [DOI] [PubMed] [Google Scholar]
- 90.Tlaxca JL, Rychak JJ, Ernst PB, Konkalmatt PR, Shevchenko TI, Pizarro TT, Rivera-Nieves J, Klibanov AL, Lawrence MB. J. Controlled Release. 2013;165:216. doi: 10.1016/j.jconrel.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yin T, Wang P, Li J, Wang Y, Zheng B, Zheng R, Cheng D, Shuai X. Biomaterials. 2014;35:5932. doi: 10.1016/j.biomaterials.2014.03.072. [DOI] [PubMed] [Google Scholar]
- 92.Yoon YI, Kwon YS, Cho HS, Heo SH, Park KS, Park SG, Lee SH, Hwang SI, Kim YI, Jae HJ, Ahn GJ, Cho YS, Lee H, Lee HJ, Yoon TJ. Theranostics. 2014;4:1133. doi: 10.7150/thno.9945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tavri S, Vezeridis A, Cui W, Mattrey RF. Radiology. 2015;276:518. doi: 10.1148/radiol.15141380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shapiro G, Wong AW, Bez M, Yang F, Tam S, Even L, Sheyn D, Ben-David S, Tawackoli W, Pelled G, Ferrara KW, Gazit D. J. Controlled Release. 2016;223:157. doi: 10.1016/j.jconrel.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mead BP, Mastorakos P, Suk JS, Klibanov AL, Hanes J, Price RJ. J. Controlled Release. 2016;223:109. doi: 10.1016/j.jconrel.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fan CH, Chang EL, Ting CY, Lin YC, Liao EC, Huang CY, Chang YC, Chan HL, Wei KC, Yeh CK. Biomaterials. 2016;106:46. doi: 10.1016/j.biomaterials.2016.08.017. [DOI] [PubMed] [Google Scholar]
- 97.Jayakumar MK, Bansal A, Huang K, Yao R, Li BN, Zhang Y. ACS Nano. 2014;8:4848. doi: 10.1021/nn500777n. [DOI] [PubMed] [Google Scholar]
- 98.Vankayala R, Kuo CL, Nuthalapati K, Chiang CS, Hwang KC. Adv. Funct. Mater. 2015;25:5934. [Google Scholar]
- 99.Zhang P, Wang C, Zhao J, Xiao A, Shen Q, Li L, Li J, Zhang J, Min Q, Chen J, Chen HY, Zhu JJ. ACS Nano. 2016;10:3637. doi: 10.1021/acsnano.5b08145. [DOI] [PubMed] [Google Scholar]