

Abstract
CD22 (Siglec-2) is a critical regulator of B cell activation and survival. CD22−/− mice generate significantly impaired antibody (Ab) responses to T cell-independent type 2 (TI-2) antigens (Ag) including haptenated-Ficoll and pneumococcal polysaccharides—Ags which elicit poor T-cell help and activate BCR signaling via multivalent epitope crosslinking. This has been proposed to be due to impaired marginal zone B (MZB) cell development/maintenance in CD22−/− mice. However, mice expressing a mutant form of CD22 unable to bind sialic acid ligands generated normal TI-2 Ab responses, despite significantly reduced MZB cells. Moreover, mice treated with CD22 ligand binding blocking mAbs which deplete MZB cells had little effect on TI-2 Ab responses. We therefore investigated the effects of CD22 deficiency on B-1b cells—an innate-like B cell population that plays a key role in TI-2 Ab responses. B-1b cells from CD22−/− mice had impaired BCR-induced proliferation and significantly increased [Ca++]i responses following BCR crosslinking. Ag-specific B-1b cell expansion and plasmablast differentiation following TI-2 Ag immunization was significantly impaired in CD22−/− mice, consistent with reduced TI-2 Ab responses. We generated CD22−/− mice with reduced CD19 levels (CD22−/−CD19+/−) to test the hypothesis that augmented B-1b cell BCR signaling in CD22−/− mice contributes to impaired TI-2 Ab responses. BCR-induced proliferation and [Ca++]i responses were normalized in CD22−/−CD19+/− B-1b cells. Consistent with this, TI-2 Ag-specific B-1b cell expansion, plasmablast differentiation, survival, and Ab responses were rescued in CD22−/−CD19+/− mice. Thus, CD22 plays a critical role in regulating TI-2 Ab responses through regulating B-1b cell signaling thresholds.
Introduction
Humoral immune responses to T cell independent type 2 (TI-2) antigens (Ag) are critical for protective immunity to encapsulated bacteria such as Streptococcus pneumoniae, an important cause of localized and systemic life-threatening infections(1). TI-2 Ags, such as pneumococcal polysaccharides, are often carbohydrate structures consisting of repeating epitopes that extensively crosslink Ag-specific B cell receptors (BCR) and can induce Ab production in the absence of major histocompatibility complex class II-restricted T cell help (2). TI-2 Ags present unique challenges to vaccine development (3-5). Thus, a better understanding of the mechanisms regulating TI-2 Ab production is necessary to develop enhanced TI-2 Ag-based vaccines.
Ab responses to TI-2 Ags differ in multiple respects to those elicited by T cell dependent (TD) Ags. B-1b and marginal zone B (MZ B) cells produce Ab responses to classical carbohydrate TI-2 Ags including pneumococcal polysaccharide (6), NP-Ficoll (7), and α1-3 dextran (8), as well as protein-based TI Ags present on clinically relevant pathogens (9-15). In contrast, follicular B cells play a more critical role in TD Ab production. Optimal humoral responses to TI-2 Ags rely heavily on distinct BCR signaling pathways (16, 17) as well as key regulators of these pathways. This includes immunoinhibitory cell surface receptors that regulate BCR signaling, such as CD22 and programmed cell death 1 (PD-1) (18-21).
CD22 (Siglec-2) is a B cell-specific glycoprotein of the sialic acid binding lectin (Siglec) family expressed on the surface of maturing B cells (20-22). CD22 binds α 2-6-linked Neu5Gc/Neu5Ac sialic acid ligands via its extracellular domains and regulates signaling via its intracellular inhibitory ITIMs (immunoreceptor tyrosine-based inhibition motifs) domains. CD22 regulates B cell function via both sialic ligand-dependent and –independent mechanisms. Following BCR ligation in conventional B cells, the tyrosine phosphorylated cytoplasmic domain of CD22 recruits effector molecules that regulate BCR and CD19 signaling, including the protein tyrosine phosphatase SHP-1 which dephosphorylates components of the BCR signalling cascade and hence, dampens BCR signaling (20). Activated SHP-1 targets Vav-1, CD19 and SLP65/BLNK (23-26), each of which promotes intracellular calcium ([Ca2+]i) signaling. CD22 also regulates [Ca2+]i signaling by facilitating SHP-1 association and activation of the plasma membrane calcium-ATPase (PMCA4), which promotes calcium efflux and attenuates BCR signaling (27). In addition to SHP-1 mediated-regulation, CD22, Shc, Grb2 and SHIP-1 have been shown to form a quaternary complex that regulates [Ca2+]i responses (28). Interestingly, work conducted with peritoneal B-1(a) cells suggests CD22 is less critical for [Ca2+]i responses (29) although additional inhibitory receptors, such as Siglec-G have been shown to play a role (30, 31). Less is known regarding the potential of CD22 to regulate B-1b cells. In addition, although decoration of TI-2 Ags with sialic acid ligands suppresses Ab responses (32), less is known regarding the role of CD22 sialic acid binding in regulating responses to non-sialylated TI-2 Ags.
Ab responses to TI-2 Ags are significantly impaired in CD22−/− mice, whereas responses to TD Ags are normal or augmented (33-36, 58). Decreased TI-2 Ab responses observed in CD22−/− mice have been primarily attributed to decreased numbers of MZ B cells in CD22−/− mice (37). However, since the effect of CD22 on B-1b cell functions has not been well-studied, we evaluated the impact of CD22 ligand-dependent and independent functions on regulating B-1b cell signaling, activation, proliferation, survival, and differentiation in response to BCR crosslinking in vitro and TI-2 Ag immunization in vivo. Our results suggest a critical role for CD22 in regulating BCR-induced [Ca2+]i signaling and proliferation as well as B-1b cell expansion, isotype switching, and plasmablast differentiation in response to TI-2 Ag in vivo. Importantly, defects in signaling, proliferation, isotype switching, and plasmablast differentiation in CD22−/− B-1b cells were rescued by reducing CD19 expression, thereby suggesting alterations in BCR and/or CD19 signaling thresholds contribute to impaired TI-2 Ab responses in CD22 deficiency.
Materials & Methods
Mice
Mice were bred in-house and included wild type and CD22−/− mice on a C57BL/6 background (The Jackson Laboratory) and CD22Δ1-2 (38) and CD22−/−CD19+/− mice on a C57BL/6 background. Mice were housed in specific pathogen-free conditions. Studies and procedures were approved by the Wake Forest Animal Care and Use Committee.
Immunizations and ELISAs
TNP-Ficoll-specific B cell expansion experiments were performed on mice immunized intraperitoneally (i.p.) with 50 μg TNP65-Ficoll (Biosearch Technologies, Novato, CA) as previously described (18). For serum Ab analyses, mice were immunized i.p. with 25 μg of TNP65-Ficoll. Ags were diluted in sterile PBS and injected in a final volume of 200 μl. In some experiments, mice were administered 250 μg MB22-10 mAb (39) or control mouse IgG2a (Southern Biotechnology Associates, Birmingham, AL) in 200 μl sterile PBS i.p. the day before immunization.
ELISAs were as previously described (18, 19, 40, 41). Sera were diluted 1:1000 for IgM detection and 1:500 for IgG detection in TBS containing 1% BSA. TNP-specific Ab levels were measured by adding diluted serum samples to Costar plates that had been coated with 5 μg/ml TNP-BSA (Biosearch Technologies) in 0.1 M borate buffered saline overnight at 4°C. PPS-3-specific ELISAs used Nunc Maxisorp plates that were coated at room temperature with 5 μg/ml PPS-3 (ATCC) overnight, blocked with TBS containing 1% BSA, and incubated with serum that had been pre-blocked with 10 μg/ml cell wall polysaccharide (Serum Statens Institute). AP-conjugated polyclonal goat anti-mouse IgM, IgG3, and IgG Abs (all from Southern Biotechnology Associates) and pNPP (Sigma) were used to detect Ag-specific Ab.
B cell phenotyping
Single-cell suspensions (2 × 107/mL) were washed with PBS containing 2% newborn calf serum and then incubated with Fc Block (eBioscience) for 15 minutes, followed by staining with a combination of the following fluorochrome-conjugated Abs: CD5 (53–7.3), B220 (RA3-6B2), CD86 (GL-1), CD11b (M1/70), CD86 (GL1) (BioLegend); CD19 (1D3), CD138 (281-2) (eBioscience) and goat F(ab′)2 anti-mouse IgM (Southern Biotechnology). TNP-reactive B cells were detected as previously described (18). Cells were incubated with 20 µg/ml TNP30-Ficoll-Fluorescein (Biosearch Technologies) in PBS containing 2% normal calf serum for 30 min. at room temperature, followed by subsequent staining with fluorochrome-labeled mAbs on ice for 25 min. Cells were then stained with Annexin V-PE or anti-Caspase-3 according to manufacturer’s instructions. Fluorochrome-labeled isotype controls were used to determine background staining levels. Stained cells were analyzed using a FACS Canto II cytometer (BD Biosciences) with forward light scatter (FSC)-A/FSC-H doublet exclusion. Data were analyzed using FlowJo analysis software (Tree Star).
B cell in vitro activation assays
CD5− cells were purified from peritoneal cavity lavage by a negative depletion procedure as previously described (18). Macrophages were removed by plate adherence in RPMI containing 5% FCS (1 hr at 37°C, 5% CO2). Nonadherent cells were depleted of Thy1.2+ cells using magnetic bead depletion (Dynal). Thy1.2− cells were further depleted using biotinylated F4/80, GR1, DX5, CD11c, and CD5 mAbs (Biolegend) in conjunction with magnetic depletion using Biotin binder beads (Dynal). CD11b+ B cells were further purified using Miltenyi bead purification. Purities were typically ~85-95% B cells. Purified peritoneal B cells were CFSE-labeled (0.6 μM) using Vybrant’s CFDA SE Cell Tracer Kit (Invitrogen), according to manufacturer’s instructions. Cells (2 × 106/ml) were cultured in complete RPMI 1640 medium containing 10% FCS (Gibco Certified serum, Invitrogen) for 4 days in medium alone or in the presence of 5 μg/ml F(ab′)2 goat anti-mouse IgM (Jackson Immunoresearch, West Grove, PA), or 2.5 μg/ml biotinylated F(ab′)2 goat anti-mouse IgM (Jackson Immunoresearch) along with 5 μg/ml streptavidin (Sigma). Cells were harvested on d4, stained with fluorochrome-labeled mAbs against CD11b and B220, as well 7AAD and Annexin V-PE. An equal number of CD11b+B220+ events were collected using a FACSCantoII instrument and data was analyzed using FlowJo analysis software.
For BCR-induced intracellular calcium flux assays, peritoneal cells were labeled with 2.5 μM Fluo-3 (Invitrogen) according to manufacturer’s instructions, washed, and stained with fluorochrome-labeled mAbs against CD5, CD11b, and B220. Following washing, cells were resuspended in PBS containing 1 mM CaCl2 and 5% FCS (2×106/ml). Following 15 minutes of incubation, cells were analyzed using a FACSCantoII instrument. Baseline readings were taken for 1 min, followed by addition of 5 μg/ml F(ab′)2 goat anti-mouse IgM and collection of cells for 6 additional minutes. Data was analyzed using FlowJo analysis software.
Statistical analysis
Data are shown as means ± SEM. Differences between sample means were assessed using the unpaired Student’s t-test or one-way ANOVA with Tukey’s post-hoc analysis unless otherwise indicated.
Results
CD22 deficiency impairs Ab responses to TI-2, but not TD Ags
As previously reported, CD22−/− mice generate impaired IgM and IgG responses to haptenated Ficoll, including TNP22-Ficoll (Fig. 1A) and TNP52-Ficoll (not shown). Responses to weakly haptenated Ficoll (TNP4-Ficoll) were modest in WT and CD22−/− mice, but were not significantly different (not shown). We found the humoral response to the clinically relevant TI-2 Ag, type 3 pneumococcal polysaccharide (PPS-3), was also significantly impaired in CD22−/− mice (Fig. 1B). In contrast, Ab responses to haptenated KLH (DNP-KLH) were augmented in CD22−/− mice (Fig. 1C) similar to that observed for the TD Ag, NP-CGG (36, 58).
Figure 1. CD22−/− mice generate impaired Ab responses to TI-2 Ags, but augmented responses to a TD Ag.
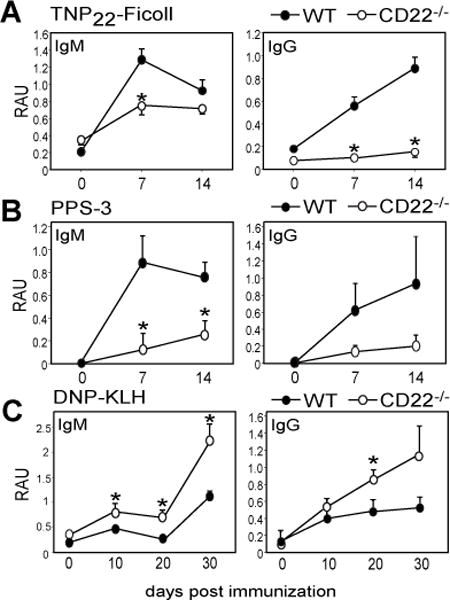
A) TNP-specific serum IgM, and IgG levels in WT and CD22−/− mice after immunization with 25 μg TNP22-Ficoll i.p. (RAU = relative absorbance units). B) PPS3-specific serum IgM and IgG levels in WT and CD22−/− mice after immunization with 1 μg PPS-3 i.p. C) DNP-specific serum IgM and IgG levels in WT and CD22−/− mice after immunization with 100 μg DNP-KLH i.p. on d0 and d21. Asterisks indicate significant differences in means (±SEM) between WT and CD22−/− mice (* p<0.05; n≥4 mice/group).
Mice lacking the CD22 sialic acid ligand binding domain generate normal TI-2 Ab responses
The contribution CD22 sialic acid ligand binding plays in regulating TI-2 Ab responses has not been extensively investigated. One study demonstrated responses to sialylated TI-2 Ags were diminished in the presence of CD22 (32), while another showed mice with a point mutation disrupting CD22 sialic acid ligand binding had near-normal IgM and normal IgG responses to TNP-Ficoll (56). We therefore carried out further studies to assess the extent to which CD22 α2-6-linked sialic acid binding contributes to regulation of TI-2 Ab responses. To do this, we used mice expressing a form of CD22 lacking the 2 amino terminal sialic acid ligand binding domains (CD22Δ1-2). Although CD22Δ1-2 mice have reduced marginal zone B cells (38), they generated normal IgM and IgG responses to TNP-Ficoll delivered i.p. (Fig. 2A) as well as i.v. (Fig. 2B). Similarly, CD22Δ1-2 mice generated normal IgM and IgG responses to PPS-3 (Fig. 2C). Notably, secondary immunization with PPS-3 (Fig. 2C) and TNP-Ficoll (data not shown) did not increase Ab levels in CD22Δ1-2 mice relative to WT mice, suggesting that sialylated Abs proposed to be produced in response to TI-2 Ags (42) do not suppress secondary responses via binding to CD22. Thus, CD22Δ1-2 mice generate normal responses to TI-2 Ags.
Figure 2. CD22Δ1-2 mice generate normal Ab responses to TI-2 Ags.
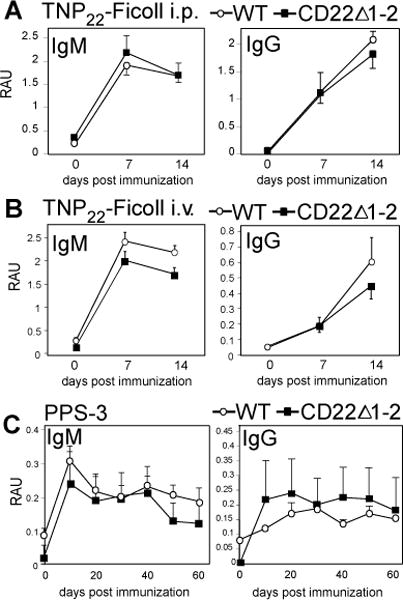
A) TNP-specific serum IgM, and IgG levels in WT and CD22Δ1-2 mice after immunization with 25 μg TNP22-Ficoll i.p. B) TNP-specific serum IgM, and IgG levels in WT and CD22Δ1-2 mice after immunization with 25 μg TNP22-Ficoll i.v. C) PPS3-specific serum IgM and IgG levels in WT and CD22Δ1-2 mice after immunization with 1 μg PPS3 i.p. on d0 and d30.
A CD22 ligand blocking mAb that also depletes marginal zone B cells has no effect on TI-2 Ab responses to TNP-Ficoll or PPS3, but impairs Ab responses to PC-Ficoll delivered i.v
We further investigated the role of CD22 sialic acid ligand binding using a CD22 mAb, MB22-10, which blocks interactions with sialic acid ligands and depletes marginal zone B cells and other subpopulations (39, 43). MB22-10 had no effect on IgM or IgG responses to TNP-Ficoll or PPS-3 delivered i.p.(Fig. 3A–B). MB22-10 diminished early (d7) IgM and IgG responses to TNP-Ficoll delivered i.v, but by d14, IgM and IgG responses were normal (Fig. 3C). Notably, Ab responses to PC-Ficoll, known to involve B-1a and marginal zone B cells (6, 13) were normal when Ag was delivered i.p (Fig. 3D), but were significantly impaired when Ag was delivered i.v. (Fig. 3E). This can be explained by the critical role marginal zone B cells play in response to PC delivered i.v.. Marginal zone B cells play a limited role in the Ab response to TNP-Ficoll and PPS-3, as we have previously shown (6, 18), and along with results derived using CD22Δ1-2 mice, the data supports that the sialic acid-binding function of CD22 does not regulate Ab responses to non-sialylated TI-2 Ags.
Figure 3. A CD22 ligand blocking mAb has no effect on Ab responses to TNP-Ficoll or PPS3, but impairs Ab responses to PC-Ficoll delivered i.v.
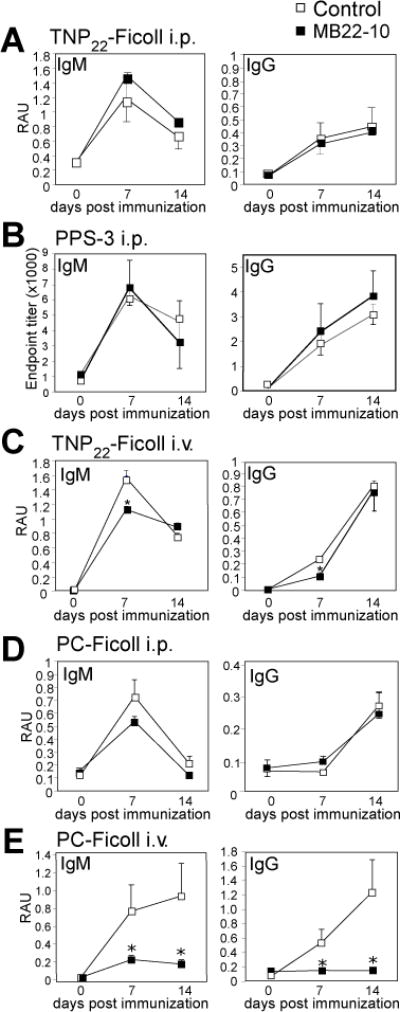
A) TNP-specific serum IgM, and IgG levels in WT mice administered 250 μg MB22-10 or IgG control mAb (i.p.) prior to immunization (d-1) with 25 μg TNP22-Ficoll i.p. B) PPS3-specific serum IgM and IgG levels in WT mice administered 250 μg MB22-10 or IgG control mAb (i.p.) with 1 μg PPS3 i.p. C) TNP-specific serum IgM, and IgG levels in WT mice administered 250 μg MB22-10 or IgG control mAb (i.p.) prior to immunization (d-1) with 25 μg TNP22-Ficoll i.v. D) PC-specific serum IgM, and IgG levels in WT mice administered 250 μg MB22-10 or IgG control mAb (i.p.) prior to immunization with 25 μg PC-Ficoll i.p. E) PC-specific serum IgM, and IgG levels in WT mice administered 250 μg MB22-10 or IgG control mAb (i.p.) prior to immunization with 25 μg PC-Ficoll i.v. Asterisks indicate significant differences in means (±SEM) between WT mice treated with MB22-10 and control IgG (* p<0.05).
CD22 regulates BCR signaling in B-1b cells
Impaired Ab responses to TI-2 Ags in CD22−/− mice have been suggested to be due to reductions and alterations in MZ B cells (37); however, the data generated with the classical TI-2 Ags, TNP-Ficoll and PPS-3 (Figs. 1–3) suggest another mechanism may be at play. Indeed, some mice lacking MZ B cells mount normal Ab responses to highly haptenated Ficoll and PPS-3 (6, 44-48). Given the key role B-1b cells play in these responses (6, 18, 19), we investigated the effect of CD22 deficiency on these cells. CD22−/− mice had significantly increased peritoneal B-1a cell and significantly decreased peritoneal B-2 cell frequencies and numbers, but B-1b cell frequencies and numbers were normal (Fig. 4A). However, BCR-induced proliferation (as measured by CFSE loss) in response to soluble anti-IgM crosslinking (Fig. 4B), or hyper-crosslinking using biotinylated anti-IgM in conjunction with streptavidin (Fig. 4C), was impaired in peritoneal B-1b cells from CD22−/− mice. Proliferative responses to LPS were similar between WT and CD22−/− B-1b cells (Fig. 4D), in contrast to what has been reported for spleen B cells from CD22−/− mice (33, 35). Consistent with a role for CD22 in regulating BCR-induced intracellular calcium ([Ca++]i) responses, peritoneal B-1b cells from CD22−/− mice exhibited significantly increased [Ca++]i following IgM crosslinking (Fig. 4E). These results for B-1b cells contrast to the limited role CD22 has been demonstrated to have in regulating B-1a cell signaling (29). Thus, CD22 dampens B-1b cell BCR-induced calcium signaling and is required for optimal proliferation in response to Ag receptor crosslinking.
Figure 4. CD22 regulates B-1b cell proliferation and calcium signaling.
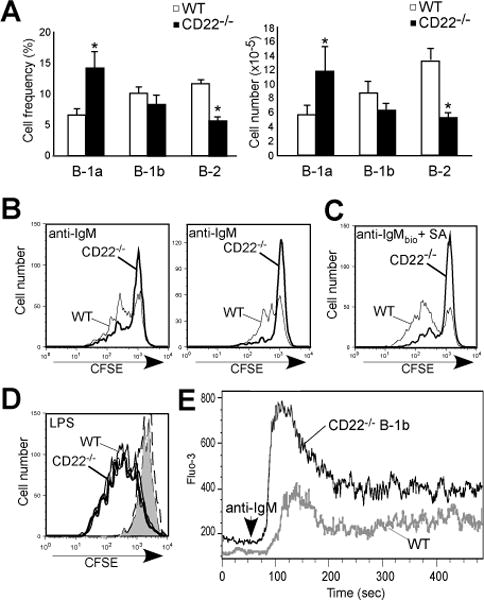
A) Frequencies and numbers of peritoneal B-1a, B-1b, and B-2 cells in WT and CD22−/− mice. Asterisks indicate significant differences in means (±SEM) between WT and CD22−/− mice (* p<0.05; n=9/group). B-C) CFSE-labeled peritoneal B-1b cells from WT and CD22−/− mice were cultured with 5 μg/ml goat anti-mouse IgM F(ab′)2 (B; 2 independent experiments shown) or 2.5 μg/ml goat anti-mouse IgM F(ab′)2 plus 5 μg/ml streptavidin (C) or LPS (D; 5 μg/ml) and analyzed for division by flow cytometry at day 4. Bold and thin lines indicate stimulated CD22−/− and WT B-1b cell proliferation, respectively. In D, the dashed line and gray filled histograms indicate unstimulated CD22−/− and WT B-1b cells. E) BCR-induced [Ca2+]i responses in peritoneal B-1b cells from wild type (WT) and CD22−/− mice. F(ab′)2 goat anti-mouse IgM Ab (5 μg/ml) was added to the cells after 1 min (arrowhead) with relative [Ca2+]i concentrations (indicated by Fluo-3 mean fluorescent intensity) assessed by flow cytometry. Results are representative of those obtained in 3 independent experiments.
CD22 promotes Ag-specific B-1b cell responses to TI-2 Ags
TNP-specific B-1b cells can be identified in the spleen following TNP-Ficoll immunization using CD11b as a marker (18, 49). WT mice show a significant expansion in TNP-specific B-1b cells in both the spleen (6-fold) and peritoneal cavity (2-fold) following immunization (Fig. 5A–B), as we have previously demonstrated (18). In contrast, CD22−/− mice did not demonstrate increases in TNP-specific spleen and peritoneal B-1b cells 3 days following immunization (Fig. 5A–B). TNP-specific splenic and peritoneal B-1b cell responses in CD22−/− mice were also impaired at days 5 (Fig. 5C) and 7 (Supplemental Fig. 1D). We did not detect differences in either the frequencies or numbers of TNP-specific splenic or peritoneal B-1a or B-2 cells in CD22−/− mice relative to WT mice (Fig. 5C and Supplemental Fig. 1D). Of note, there was evidence of activation in TNP-specific B cells from CD22−/− mice, as they had increased size (FSC) and expressed CD86 (Fig. 5D and data not shown), similar to WT cells. Thus, CD22 promotes Ag-specific B-1b cell expansion, but not B cell activation, in response to TNP-Ficoll.
Figure 5. CD22 promotes Ag-specific B-1b cell expansion in response to TI-2 Ag.
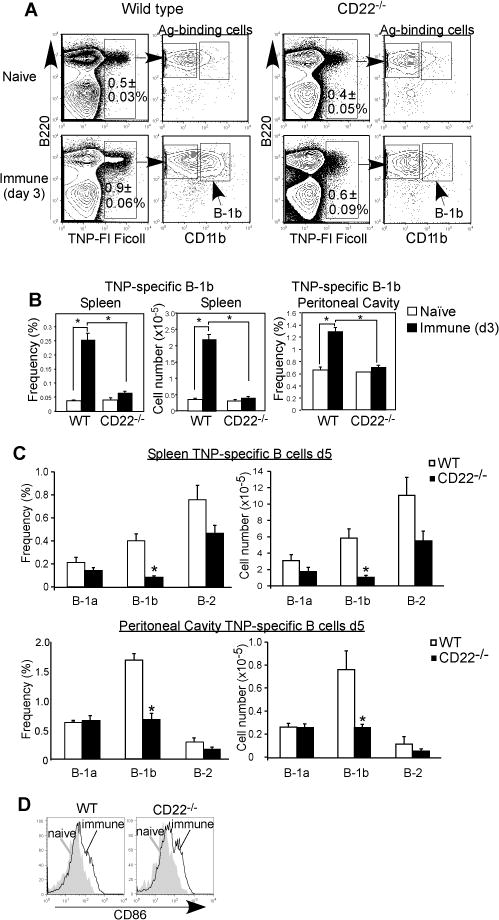
A–B) In vivo expansion of TNP-specific spleen and peritoneal B cells in WT and CD22−/− mice 3 days following immunization with TNP52-Ficoll. A) Representative frequencies of TNP30-FITC-Ficoll-binding by splenic B220+ B cells from WT and CD22−/− mice before and 3 days after immunization. Left panels indicate gating of CD11b+ (B-1b cells). B) Frequencies and numbers of TNP-specific spleen and peritoneal B-1b cells before and 3 days after immunization. C) TNP-specific spleen and peritoneal B cell subset cell frequencies and numbers in WT and CD22−/− mice 5 days following immunization with TNP52-Ficoll as determined by flow cytometry with identification of TNP-specific B-1a (B220+CD5+CD11b+), B-1b (B220+CD11b+CD5−), and B-2 (B220+CD11b−CD5−) cell subsets in WT and CD22−/− mice. D) Representative CD86 staining on TNP-specific spleen B-1b cells from naïve and immune WT and CD22−/− mice. Significant differences in mean values (±SEM) are indicated by asterisks; *, p<0.05 (n=3 naïve and n=5 immune mice/group).
Reduction in CD19 expression rescues BCR-induced proliferation and normalizes BCR-induced [Ca2+]i responses in CD22−/− B-1b cells
The impaired BCR-induced in vitro proliferation and impaired in vivo expansion of B-1b cells from CD22−/− mice, despite activation, lead us to hypothesize that excessive BCR signaling in CD22−/− B-1b cells might contribute to failed proliferation. CD22 plays a key role in modulating B cell activation through regulating both CD19 and BCR signaling (22, 24, 25). We therefore assessed whether reducing CD19 expression in CD22−/− B cells (thereby reducing the B cell activation potential), would affect BCR-induced proliferation and signaling. CD22−/−CD19+/− mice had normal frequencies of B-1b and B-1a cells, but peritoneal B-2 cells and marginal zone B cell frequencies were significantly decreased, similar to CD22−/− mice (Fig. 6A). Spleen and peritoneal cavity cell yields were comparable among CD22−/−CD19+/−, CD22−/−, and WT mice (not shown). Importantly, in contrast to the elevated BCR-induced [Ca++]i responses observed in CD22−/− B-1b cells, [Ca++]i responses were normal in CD22−/−CD19+/− B-1b cells (Fig. 6B). Moreover, in contrast to the impaired BCR-induced proliferation observed for CD22−/− B-1b cells, BCR-induced proliferation in CD22−/−CD19+/− B-1b cells was much more comparable to that observed for WT B-1b cells (Fig. 6C). Thus, reducing CD19 expression in CD22−/− B-1b cells restores BCR-induced proliferation and [Ca2+]i responses to near-normal levels.
Figure 6. Reduction in CD19 expression rescues proliferation and normalizes BCR-induced [Ca2+]i responses in CD22−/− B-1b cells.
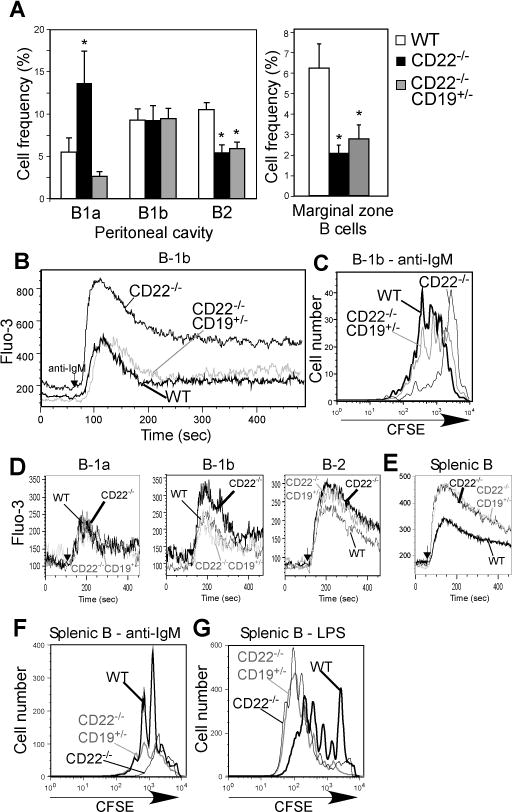
A) Frequencies of peritoneal B-1a, B-1b, and B-2 cells (n=5-6/group) and splenic marginal zone (n>8/group) B cells in WT, CD22−/−, and CD22−/−19+/− mice. Asterisks indicate significant differences in means (±SEM) between WT and CD22−/− mice and between WT and CD22−/−19+/− mice (* p<0.05). B) BCR-induced [Ca2+]i responses in peritoneal B-1b cells from WT, CD22−/− and CD22−/−CD19+/− mice. F(ab′)2 goat anti-mouse IgM Ab (5 μg/ml) was added to the cells after 1 min (arrowhead) with relative [Ca2+]i concentrations (indicated by Fluo-3 mean fluorescent intensity) assessed by flow cytometry. Results are representative of those obtained in 3 independent experiments. C) CFSE-labeled peritoneal B-1b cells from WT, CD22−/−, and CD22−/−CD19+/− mice were cultured with 5 μg/ml goat anti-mouse IgM F(ab′)2 and analyzed for division by flow cytometry at day 4. Results are representative of those obtained in 3 independent experiments. D-E) BCR-induced [Ca2+]i responses in peritoneal B-1a (B220+CD5+CD11b+), B-1b (B220+CD11b+CD5−), and B-2 (B220+CD11b−CD5−) cell subsets (D) and spleen B220+ B cells (E) from WT, CD22−/−, and CD22−/−CD19+/− mice. F(ab′)2 goat anti-mouse IgM Ab (5 μg/ml) was added to the cells after 1 min (arrowhead) with relative [Ca2+]i concentrations (indicated by Fluo-3 mean fluorescent intensity) assessed by flow cytometry. Results are representative of those obtained in 3 or more experiments. F–G) CFSE-labeled CD43neg spleen B cells from WT, CD22−/−, and CD22−/−CD19+/− mice were cultured with 5 μg/ml goat anti-mouse IgM F(ab′)2 (F) or LPS (G) and analyzed for division by flow cytometry at day 4. Results are representative of those obtained in 3-4 independent experiments.
We next compared intracellular calcium increases following IgM crosslinking in WT, CD22−/−, and CD22−/−CD19+/− peritoneal B-1a, B-1b, and B-2 cells, as well as splenic B cells. Performing these experiments on peritoneal B cells within the same sample indicated that relative to the other subsets, peritoneal B-1a cells exhibited modest increases in [Ca++]i following BCR crosslinking (Fig. 6D and Supplemental Fig. 1A–B). In contrast to results with CD22−/− B-1b cells, no differences were detected among WT, CD22−/−, and CD22−/−CD19+/− peritoneal B-1a cell [Ca++]i responses (Fig. 6D and Supplemental Fig. 1B). BCR-induced [Ca++]i, responses were increased in CD22−/− peritoneal B-2 cells (Fig. 6D, Supplemental Fig. 1B–C). However, in contrast to results with B-1b cells (Fig. 6B), CD19 heterozygosity did not restore BCR-induced [Ca++]i responses to WT levels in CD22−/− peritoneal B-2 cells. In CD22−/−CD19+/− peritoneal B-2 cells, BCR-induced [Ca++]i responses either overlapped (4 out of 7 experiments) those of CD22−/− B-2 cells or were intermediate (3 out of 7 experiments) between CD22−/− and WT B-2 cells, whereas responses of CD22−/−CD19+/− peritoneal B-1b cells were normalized to WT levels (Fig. 6B and D and Supplemental Fig. 1B–C). This could perhaps be due to heterogeneity in the CD11b-negative B cell pool, which is known to contain recirculating (CD11b-negative) B-1 cells or due to the effect of CD11b on CD22-mediated regulation (61). Notably, B-1b cells from CD22Δ1-2 mice exhibited BCR-induced increases in [Ca++]i that were similar to WT mice (Supplemental Fig. 1C).
Consistent with previous studies, splenic CD22−/− B cells (largely B-2 cells) exhibited much higher [Ca++]i responses than WT spleen B cells (Fig. 6E and Supplemental Fig. 1C). In contrast to results with B-1b cells, CD19 heterozygosity did not normalize BCR-induced [Ca++]i responses in splenic CD22−/− B cells. In anti-IgM proliferation assays, CD22−/−CD19+/− splenic B cells exhibited proliferation that was intermediate between WT and CD22−/− mice, indicating CD19 reduction had a modest effect on proliferation of CD22−/− spleen B cells (Fig. 6F). In contrast, LPS-induced proliferation in splenic CD22−/− B cells, as previously shown (33, 35), as well as in CD22−/−CD19+/− B cells was much higher than in WT B cells (Fig. 6G). Thus, in the absence of CD22, B-1b cells are similar to peritoneal and splenic B-2 cells in that they have augmented BCR-induced [Ca++]i responses and reduced BCR-induced proliferation. This is in contrast to B-1a cells, which do not appear to be affected at the level of calcium signaling. However, reducing CD19 levels uniquely normalizes BCR-induced [Ca++]i responses and BCR-induced proliferation in CD22−/− B-1b cells, but is ineffective in normalizing these responses in CD22−/− B-2 cells.
Reduction in CD19 expression restores Ag-specific B-1b cell expansion and ASC differentiation, B cell survival, and Ab production in response to TI-2 Ag in CD22−/− mice
Since reducing CD19 expression in CD22−/− B-1b cells rescued BCR-induced proliferation and [Ca2+]i responses in vitro, we assessed whether responses to TI-2 Ag were also rescued in vivo. TNP-specific B-1b cells from WT, CD22−/−, and CD22−/−CD19+/− mice all became activated following TNP immunization, as evidenced by increased FSC and CD86 expression (Fig. 7A). Interestingly, CD86 expression was significantly higher on TNP-specific B-1b cells from CD22−/− mice relative to WT and CD22−/−CD19+/− mice (Fig. 7A). We did not detect differences in frequencies of naïve TNP-specific peritoneal and splenic B-1b cells among WT, CD22−/−, and CD22−/−CD19+/− mice prior to immunization (Supplemental Fig. 1F). However, following immunization, TNP-specific splenic B-1b cell frequencies and numbers in CD22−/−CD19+/− mice were significantly increased relative to CD22−/− mice and were not significantly different from WT mice (Fig. 7B, and Supplemental Fig. 1F). We did not detect significant differences in splenic TNP-specific B-1a or B-2 cell frequencies or numbers among WT, CD22−/−, and CD22−/− CD19+/− mice, although TNP-specific CD11b-negative B cell frequencies were generally lower, albeit no significantly different in CD22−/− mice. Similar to findings for spleen B-1b cells, TNP-specific peritoneal B-1b cell frequencies and numbers in CD22−/−CD19+/− mice were significantly increased relative to CD22−/− mice and were not significantly different from WT mice (Fig. 7C and Supplemental Fig. 1F). Notably, we did not detect significant differences in TNP-specific peritoneal B-1a or B-2 cell frequencies and numbers among immune WT, CD22−/−, and CD22−/−CD19+/− mice, although Ag-specific B-1a cells were slightly lower in CD22−/−CD19+/− mice (Fig. 7B–C). Thus, reduction in CD19 expression in CD22−/− mice partially to fully rescues Ag-specific peritoneal and splenic B-1b cell expansion in response to TNP-Ficoll.
Figure 7. Reduction in CD19 expression rescues TI-2 Ag-specific B-1b cell expansion and plasmablast differentiation, reduces Ag-specific B cell apoptosis and restores TI-2 Ab responses CD22−/− mice.
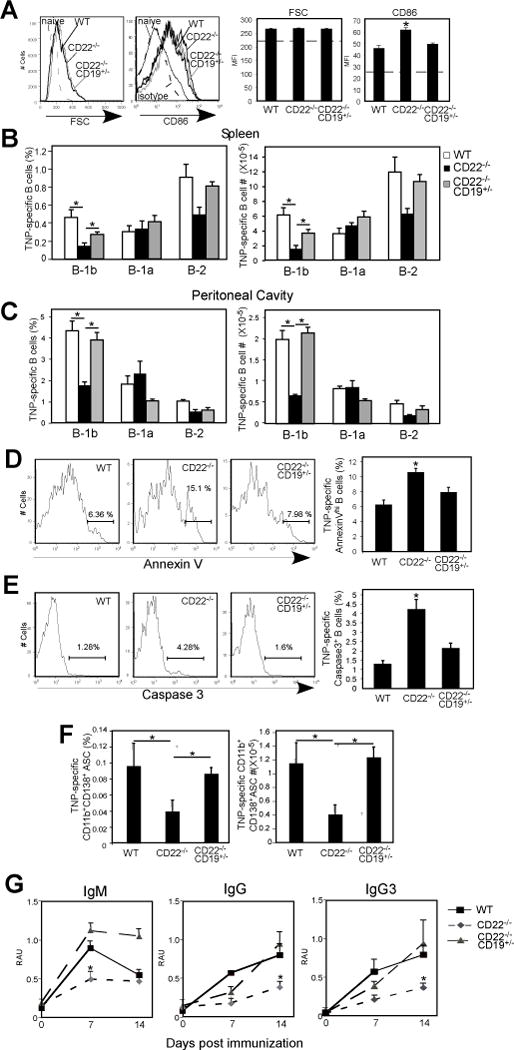
A–C) In vivo activation and expansion of TNP-specific B cells in WT, CD22−/− and CD22−/−CD19+/− mice 5 days following immunization with TNP52-Ficoll. A) Activation of TNP-specific B cells as measured by increased FSC and CD86 expression. Isotype control staining for TNP-specific B cells from immune WT mice is indicated for CD86 staining (dashed line). Dashed horizontal line in the bar graphs indicates naïve baseline values for TNP-specific B cells. Results from 3-4 mice/genotype. B-C) Frequencies and numbers of TNP-specific spleen (B) and peritoneal (C) B-1a, B-1b, and B-2 cells in immune (d5) WT, CD22−/− and CD22−/−CD19+/− mice (n=4-8/group). Populations were identified using the gating strategy shown in Supplemental Fig. 1E. D–E) Frequencies of Annexin Vhi (E) and caspase-3+ (E) cells among TNP-specific spleen B cells in WT, CD22−/− and CD22−/−CD19+/− mice 2 days following immunization (n=5-7 mice/group for Annexin V stain and 3-6 mice/group for caspase-3). F) Frequencies and numbers of CD138+B220+CD11b+ splenic TNP-specific B-1 cells 5 days post TNP-Ficoll immunization in WT, CD22−/− and CD22−/−CD19+/− mice. (G) TNP-specific serum IgM, and IgG levels in WT, CD22−/− and CD22−/−CD19+/− mice after immunization with 25 μg TNP22-Ficoll i.p. (n=4/group). Significant differences in mean values (±SEM) are indicated by asterisks; *, p<0.05, In B–F, one-way ANOVA with Tukey’s post-hoc test was used.
Excessive BCR-induced activation may lead to impaired survival. Given the strong signaling associated with high valency TI-2 Ags which extensively crosslink the BCR, it is possible that in the absence of CD22, signaling overload and excessive [Ca2+]i levels occur, increasing the potential for apoptosis to occur (50-54). In the absence of survival signals, such as those typically supplied by CD40L-expressing T cells during TD responses, Ag-activated B cells may undergo apoptosis in the absence of CD22, resulting in impaired clonal expansion and ultimately reduced Ab production. Therefore, we examined the extent to which Ag-specific B cells in WT, CD22−/−, and CD22−/−CD19+/− mice had impaired survival. As shown in Fig. 7D, 2 days following TNP-Ficoll immunization, the frequencies of Annexin Vhi TNP-specific splenic B cells from CD22−/− mice were significantly increased relative to TNP-specific splenic B cells from WT and CD22−/−CD19+/− mice. Notably, there was no difference among groups in the frequencies of non-Ag-specific splenic B cells that were Annexin Vhi (WT, 2.9 ± 0.69%; CD22−/−, 2.8 ± 0.65%; CD22−/− CD19+/−, 2.3 ± 0.1%). Moreover, the frequency of caspase-3+ TNP-specific splenic B cells at this time point was increased >3-fold in CD22−/− mice relative to WT mice (Fig. 7E). In contrast, the frequency of caspase-3+ TNP-specific splenic B cells from CD22−/−CD19+/− mice was similar to WT mice. Thus, CD22 deficiency results in impaired B cell survival and expansion during Ag-specific responses to TI-2 Ags, and reduction of CD19 signaling potential restores Ag-specific B cell survival in the context of CD22 deficiency.
We next examined whether restoration of Ag-specific B-1b cell expansion supported increased B-1 cell ASC differentiation and IgG switching in CD22−/−CD19+/− mice. As shown in Fig. 7F, the frequencies and numbers of TNP-specific CD138+ splenic B-1 cell plasmablasts were significantly decreased in CD22−/− mice relative to WT mice, but restored in CD22−/− mice that were heterozygous for CD19 expression. Moreover, TNP-specific IgG3+ B-1 cell frequencies in WT and CD22−/−CD19+/− mice were significantly increased over frequencies in CD22−/− mice (WT, 0.66 ± 0.08%, CD22−/−, 0.18 ± 0.05%; CD22−/−CD19+/−, 0.31 ± 0.01%; naïve WT, 0.036 ± 0.002)). As shown in Fig. 7G, IgM, IgG, and IgG3 responses to TNP-Ficoll were impaired in CD22−/− mice relative to WT mice. However, IgM, IgG, and IgG3 responses to TNP-Ficoll were not significantly different between CD22−/−CD19+/− and WT mice, with the exception of the d14 time point for IgM, which was higher in CD22−/−CD19+/− mice (Fig. 7G). Thus, TI-2 Ag-specific Ab responses are rescued in CD22−/− mice when CD19 expression is reduced. This result is consistent with the critical role that B-1b cells play in TNP-specific antibody responses and with our findings indicating rescue of CD22−/− B-1b cell expansion, plasmablast differentiation, and IgG class switching upon reduction in CD19 expression.
Discussion
CD22 is a critical regulator of B cell function and as such, disruptions in CD22 impact vaccine responses, autoimmunity, and malignancy (20, 21). In this study, we have uncovered an essential role for CD22 in promoting B-1b cell expansion and Ab production in response to TI-2 Ags. To our knowledge, this study is the first to provide an explanation for the impaired TI-2 Ab responses observed in CD22-deficient mice. Furthermore, we have identified this critical CD22-mediated regulation is independent of the CD22 sialic acid ligand binding function. Thus, in contrast to studies which conclude a limited role for CD22 in regulating B-1 (largely B-1a) cells (29), our study clearly demonstrates CD22 is required for optimal functioning of the B-1b cell population.
Our results demonstrate the effects of CD22 on promoting TI-2 Ab responses, B-1b cell development, and B-1b cell BCR-induced calcium signaling are independent of the ability of the 2 amino terminal domains of CD22 to interact with sialic acid ligands. Mice lacking CD22 ligand binding domains as well as wild type mice administered a well-characterized mAb which blocks sialic acid binding and significantly depletes MZ B cells and reduces IL-10 producing regulatory B cell numbers (39, 55) mounted normal Ab responses to PPS-3 and TNP-Ficoll delivered i.p., and near-normal responses to TNP-Ficoll delivered i.v. A similar finding was reported in another strain of mice expressing a mutant form of CD22 unable to bind sialic acid ligands (56). Importantly, additional knockout mice with MZ B cell defects have also been shown to generate normal responses to TI-2 Ags (6, 44-48), thereby demonstrating the importance of non-MZ B cell populations in these responses, especially B-1b cells. In contrast, Ab responses to i.v.-delivered PC-Ficoll, known to be dependent on MZ B cells, were severely impaired following CD22 mAb administration. Notably, it was recently reported that TI-2 Abs are differentially sialylated and that this contributes to regulation of TI-2 Ab responses, including boosting (42). However, our results show TI-2 Ab responses in CD22Δ1-2 mice are refractive to boosting, suggesting that Ab-mediated suppression of boosting does not solely depend upon sialylated Ab interactions with CD22. Thus, while there is a clear role for CD22 sialic acid ligand binding in regulating responses to sialylated Ags (20, 32, 57), our data support a sialic acid ligand-independent role for CD22 in regulating Ab responses to non-sialylated TI-2 Ags.
Sialic acid ligand-dependent and –independent roles have been established for CD22 outside of regulation of TI-2 Ab responses. In vivo studies show CD22−/− and CD22Δ1-2 B cells, as well as B cells in wild type mice treated with mAbs that inhibit CD22 ligand binding, exhibit significantly enhanced B cell turnover due to impaired survival, along with reduced mature recirculating B cells and MZ B cells (33, 35, 38, 39). More recent work with additional knock-in mouse lines suggests a role for the ITIM signaling domains in maintaining recirculating B cell numbers and both signaling and ligand binding domains in maintaining MZ B cell numbers (56). CD22 binds to sialic acids in α2-6 linkages in cis (present on the surface of the same B cell) or in trans (on non-associated molecules). Although we detected normal BCR-induced [Ca++]i responses in B-1b cells from CD22Δ1-2 mice, similar to spleen B cells (38, 39), other work has demonstrated that the loss of sialic acid ligand binding, which contributes to cis-interactions with CD22, leads to hyporeactive B-cells (ie., reduced Ca++ signaling), due to the increased association of CD22 with the BCR (56). The reason for difference is not clear although alterations in CD22 expression levels could contribute. Despite these differences in in vitro responses, CD22Δ1-2 mice and the CD22 mutant mice generated by Muller et al. (56) both approximate wild-type antibody responses to TI-2 Ags. In any case, the data strongly support that CD22 ligand binding is physiologically relevant and is critical for conventional B cell survival and function in vivo, especially in MZ B cells. In contrast, we did not find alterations in naive peritoneal B-1b cell numbers in either CD22−/− or CD22Δ1-2 mice, suggesting these cells may be regulated differently than MZ B cells, at least in the peritoneal environment. CD22 deficiency in mice has been reported to cause significant increases in B-1a cell numbers ((34, 58); Fig. 4) as well as disrupted B-1a cell function during CHS reactions (59), despite the apparent lack of effect CD22 has in regulating CD5+ B-1a cell signaling (29). Nonetheless, Siglec-G appears to be more critical for regulating some aspects of B-1a cell signaling (31). While Siglec-G is also expressed on B-1b cells and may have the potential to regulate TI-2 Ab responses (especially when Ags are decorated with Siglec-G ligands (60)), it is clear from our study that CD22-mediated regulation of B-1b cells during these responses is critical.
Our data supports that CD22 promotes Ab responses to TI-2 Ags through regulating Ag receptor and/or CD19 signaling in B-1b cells, which is likely to be intense when triggered by TI-2 Ags displaying multiple epitopes and C3-bound fragments that stimulate CD21/35-CD19 signaling. Despite evidence of BCR-induced activation in CD22−/− B-1b cells in vitro (ie., strong increases in [Ca2+]i and blasting), BCR-induced proliferation was nonetheless impaired. Consistent with this finding, Ag-specific B-1b cells in CD22−/− mice became activated following TI-2 Ag immunization, but showed reduced expansion, isotype switching, and differentiation to CD138+ plasmablasts. Given the strong signaling associated with high valency TI-2 Ags which extensively crosslink the BCR, it is possible that in the absence of CD22, signaling overload and excessive [Ca2+]i levels increase the potential for apoptosis to occur (50-54). Of note, increased BCR-induced apoptosis also occurs in conventional B cells lacking CD22 on a B6 background (35, 62). This effect is due to regulation of c-myc by a modifier gene, EndoU (63). However, BCR-induced survival and proliferation can be rescued in these B cells by signals supplied through CD40 and therefore, augmented Ab responses in CD22−/− mice likely involve rescue by T cell-derived CD40L. In contrast, during TI-2 Ab responses exogenous survival factors may be more limiting, and hence, survival of activated B-1b cells may be more dependent upon CD22 for attenuation of excessive BCR signaling. Interestingly, CD11b, which is typically selectively expressed on B-1 cells including Ag-activated B-1b cells outside of the peritoneal cavity (18, 19, 41, 49), has been shown to associate with CD22 and promote its inhibitory effects on BCR signaling (61). It is presently unclear as to whether the lack of association of CD11b with CD22 in CD22−/− B-1b cells contributes to dysregulation that is unique to B-1b cells. Further work is needed to understand the function of CD11b on B-1b cells, especially as it relates to CD22.
Reduction in CD19 expression restored BCR-induced calcium responses and proliferation in CD22−/− B-1b cells in vitro, and partially to fully rescued Ag-specific B-1b cell expansion, ASC differentiation, isotype switching, as well as Ag-specific B cell survival and Ab production in vivo. This effect may have been due to a general reduction in the strength of BCR-induced intracellular signaling in CD19+/−CD22−/− B-1b cells, as CD19 can associate with the BCR to enhance Ag receptor signaling, including [Ca++]i signaling independently of the CD21/35 coreceptor (46, 64-68). In response to strong TI-2 Ags, CD19 may intensify Ag receptor signaling and contribute to enhanced BCR-induced apoptosis in the absence of survival factors, thereby inhibiting TI-2 Ab responses (50). In support of this, CD19−/− mice generate normal to augmented Ab responses to strong TI-2 Ags (6, 44, 45, 69). Thus, CD19 is not required for B-1b cells to productively respond to TI-2 Ags in vivo and excessive CD19 expression, as occurs in the absence of CD21/35 expression, results in impaired TI-2 Ab responses (6, 46). It is possible that CD22 inhibits CD19 activity in B-1b cells by targeting CD19 dephosphorylation, as has been shown for conventional B cells (25), although one group has suggested CD19 phosphorylation is regulated differently in B-1b cells than in conventional splenic B cells (70, 71). However, these studies found B-1b cells were largely nonresponsive to BCR crosslinking, in contrast to our current and previous results (46) as well as those of Dal Porto et al. (72). This discrepancy could be the result of differences in activation conditions or B-1b cell purification given the heterogeneity in the B-1b cell population (49). In addition to our in vivo data demonstrating a selective effect of CD22 deficiency on B-1b cell responses to TI-2 Ags, our in vitro data further support that B-1b cells may be uniquely regulated by CD22 and CD19 as CD19 heterozygosity normalized proliferation as well as BCR-induced Ca++ signaling in CD22−/− B-1b cells, but did not achieve this effect in B-2 cells. This supports a unique role for CD22 and CD19 in regulating B-1b cells. Indeed, the rescue of CD22−/− B-1b cells, but not B-2 cells, by lowered CD19 expression could be due to the fact that B-1b cells exhibit weaker BCR signaling relative to B-2 cells, and are thus more sensitive to the effects of CD19 heterozygosity in dampening responses. Alternatively, the selective effect on CD22−/− B-1b cells could be due to altered role CD19 plays in regulating B-1b cells versus B-2 cells (6, 46, 70-71).
Our findings demonstrating a role for CD22 in regulating B-1b cell responses to TI-2 Ags, including bacterial polysaccharides, may have implications for human health. We have identified a population of B-1b-like cells (CD22-expressing) in nonhuman primates that responds to TI-2 Ags in a manner quite similar to mice (73). This supports the likelihood of a CD22-regulated B-1b-like population in humans. Anti-CD22 mAb treatment for malignancies and autoimmunity is associated with CD22 surface downregulation (74) and could therefore have implications for quality of TI-2 Ab responses in these patients. Nonetheless, CD19 levels following anti-CD22 treatment are also reduced in patients (39, 74), and this may ultimately rescue these responses based on the results of our current study. In further support of this, CD22 mAb treatment in mice induces partial downregulation of CD22 (39), yet our results demonstrates this had minimal effect on TI-2 Ab responses, with the exception of responses to i.v.-administered PC-Ficoll, which requires MZ B cells. Evaluation of responses to TI-2 Ags, such as responses to Pneumovax23, in patients treated with CD22 mAb therapies is nonetheless warranted.
Supplementary Material
References
- 1.Mehr S, Wood N. Streptococcus pneumoniae–a review of carriage, infection, serotype replacement and vaccination. Paediatric Resp Rev. 2012;13:258–64. doi: 10.1016/j.prrv.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Vos Q, Lees A, Wu ZQ, Snapper CM, Mond JJ. B-cell activation by T-cell-independent type 2 antigens as an integral part of the humoral immune response to pathogenic microorganisms. Immunol Rev. 2000;176:154–170. doi: 10.1034/j.1600-065x.2000.00607.x. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez-Fernandez A, Faro J, Fernandez C. Immune responses to polysaccharides: lessons from humans and mice. Vaccine. 2008;26:292–300. doi: 10.1016/j.vaccine.2007.11.042. [DOI] [PubMed] [Google Scholar]
- 4.Weller S, Reynaud CA, Weill JC. Vaccination against encapsulated bacteria in humans: paradoxes. Trends Immunol. 2005;26:85–89. doi: 10.1016/j.it.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Klein Klouwenberg P, Bont L. Neonatal and infantile immune responses to encapsulated bacteria and conjugate vaccines. Clin Dev Immunol. 2008;2008:628963. doi: 10.1155/2008/628963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haas KM, Poe JC, Steeber DA, Tedder TF. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity. 2005;23:7–18. doi: 10.1016/j.immuni.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Hsu MC, Toellner KM, Vinuesa CG, Maclennan IC. B cell clones that sustain long-term plasmablast growth in T-independent extrafollicular antibody responses. Proc Natl Acad Sci USA. 2006;103:5905–5910. doi: 10.1073/pnas.0601502103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foote JB, Kearney JF. Generation of B cell memory to the bacterial polysaccharide alpha-1,3 dextran. J Immunol. 2009;183:6359–6368. doi: 10.4049/jimmunol.0902473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alugupalli KR, Leong JM, Woodland RT, Muramatsu M, Honjo T, Gerstein RM. B1b lymphocytes confer T cell-independent long-lasting immunity. Immunity. 2004;21:379–390. doi: 10.1016/j.immuni.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 10.Gil-Cruz C, Bobat S, Marshall JL, Kingsley RA, Ross EA, Henderson IR, Leyton DL, Coughlan RE, Khan M, Jensen KT, Buckley CD, Dougan G, MacLennan IC, Lopez-Macias C, Cunningham AF. The porin OmpD from nontyphoidal Salmonella is a key target for a protective B1b cell antibody response. Proc Natl Acad Sci. 2009;106:9803–9808. doi: 10.1073/pnas.0812431106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colombo MJ, Alugupalli KR. Complement factor H-binding protein, a putative virulence determinant of Borrelia hermsii, is an antigenic target for protective B1b lymphocytes. J Immunol. 2008;180:4858–64. doi: 10.4049/jimmunol.180.7.4858. [DOI] [PubMed] [Google Scholar]
- 12.Alugupalli KR. A distinct role for B1b lymphocytes in T cell-independent immunity. Curr Top Microbiol Immunol. 2008;319:105–130. doi: 10.1007/978-3-540-73900-5_5. [DOI] [PubMed] [Google Scholar]
- 13.Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 2001;14:617–629. doi: 10.1016/s1074-7613(01)00129-7. [DOI] [PubMed] [Google Scholar]
- 14.Cole LE, Yang Y, Elkins KL, Fernandez ET, Qureshi N, Shlomchik MJ, Herzenberg LA, Vogel SN. Antigen-specific B-1a antibodies induced by Francisella tularensis LPS provide long-term protection against F. tularensis LVS challenge. Proc Natl Acad Sci U S A. 2009;106:4343–4348. doi: 10.1073/pnas.0813411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol. 2011;11:34–46. doi: 10.1038/nri2901. [DOI] [PubMed] [Google Scholar]
- 16.Patterson HC, Kraus M, Kim YM, Ploegh H, Rajewsky K. The B cell receptor promotes B cell activation and proliferation through a non-ITAM tyrosine in the Iga cytoplasmic domain. Immunity. 2006;25:55–65. doi: 10.1016/j.immuni.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 17.Fruman DA, Satterthwaite AB, Witte ON. Xid-like phenotypes: a B cell signalsome takes shape. Immunity. 2000;13:1–3. doi: 10.1016/s1074-7613(00)00002-9. [DOI] [PubMed] [Google Scholar]
- 18.Haas KM. Programmed cell death 1 suppresses B-1b cell expansion and long-lived IgG production in response to T cell-independent type 2 antigens. J Immunol. 2011;187:5183–5195. doi: 10.4049/jimmunol.1101990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKay JT, Egan RP, Yammani RD, Chen L, Shin T, Yagita H, Haas KM. PD-1 Suppresses Protective Immunity to Streptococcus pneumoniae through a B Cell-Intrinsic Mechanism. J Immunol. 2015;194:2289–99. doi: 10.4049/jimmunol.1401673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poe JC, Tedder TF. CD22 and Siglec-G in B cell function and tolerance. Trends Immunol. 2012;33:413–20. doi: 10.1016/j.it.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nitschke L. CD22 and Siglec-G regulate inhibition of B-cell signaling by sialic acid ligand binding and control B-cell tolerance. Glycobiology. 2014;24:807–17. doi: 10.1093/glycob/cwu066. [DOI] [PubMed] [Google Scholar]
- 22.Tedder TF, Poe JC, Haas KM. CD22: a multi-functional receptor that regulates B lymphocyte survival and signal transduction. Adv Immunol. 2005;88:1–50. doi: 10.1016/S0065-2776(05)88001-0. [DOI] [PubMed] [Google Scholar]
- 23.Gerlach J, Ghosh S, Jumaa H, Reth M, Wienands J, Chan AC, Nitschke L. B cell defects in SLP65/BLNK-deficient mice can be partially corrected by the absence of CD22, an inhibitory coreceptor for BCR signaling. Eur J Immunol. 2003;33:3418–3426. doi: 10.1002/eji.200324290. [DOI] [PubMed] [Google Scholar]
- 24.Sato S, Jansen PJ, Tedder TF. CD19 and CD22 expression reciprocally regulates tyrosine phosphorylation of Vav protein during B lymphocyte signaling. Proc Natl Acad Sci, USA. 1997;94:13158–13162. doi: 10.1073/pnas.94.24.13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujimoto M, Bradney AP, Poe JC, Steeber DA, Tedder TF. Modulation of B lymphocyte antigen receptor signal transduction by a CD19/CD22 regulatory loop. Immunity. 1999;11:191–200. doi: 10.1016/s1074-7613(00)80094-1. [DOI] [PubMed] [Google Scholar]
- 26.Fujimoto M, Poe JC, Jansen PJ, Sato S, Tedder TF. CD19 amplifies B lymphocyte signal transduction by regulating Src-family protein tyrosine kinase activation. J Immunol. 1999;162:7088–7094. [PubMed] [Google Scholar]
- 27.Chen J, McLean PA, Neel BG, Okunade G, Shull GE, Wortis HH. CD22 attenuates calcium signaling by potentiating plasma membrane calcium-ATPase activity. Nat Immunol. 2004;5:651–657. doi: 10.1038/ni1072. [DOI] [PubMed] [Google Scholar]
- 28.Poe JC, Fujimoto M, Jansen PJ, Miller AS, Tedder TF. CD22 forms a quaternary complex with SHIP, Grb2 and Shc. A pathway for regulation of B lymphocyte antigen receptor-induced calcium flux. J Biol Chem. 2000;275:17420–17427. doi: 10.1074/jbc.M001892200. [DOI] [PubMed] [Google Scholar]
- 29.Lajaunias F, Nitschke L, Moll T, Martinez-Soria E, Semac I, Chicheportiche Y, Parkhouse RM, Izui S. Differentially regulated expression and function of CD22 in activated B-1 and B-2 lymphocytes. J Immunol. 2002;168:6078–6083. doi: 10.4049/jimmunol.168.12.6078. [DOI] [PubMed] [Google Scholar]
- 30.Hutzler S, Ozgor L, Naito-Matsui Y, Klasener K, Winkler TH, Reth M, Nitschke L. The ligand-binding domain of Siglec-G is crucial for its selective inhibitory function on B1 cells. J Immunol. 2014;192:5406–14. doi: 10.4049/jimmunol.1302875. [DOI] [PubMed] [Google Scholar]
- 31.Hoffmann A, Kerr S, Jellusova J, Zhang J, Weisel F, Wellmann U, Winkler TH, Kneitz B, Crocker PR, Nitschke L. Siglec-G is a B1 cell-inhibitory receptor that controls expansion and calcium signaling of the B1 cell population. Nat Immunol. 2007;8:695–704. doi: 10.1038/ni1480. [DOI] [PubMed] [Google Scholar]
- 32.Duong BH, Tian H, Ota T, Completo G, Han S, Vela JL, Ota M, Kubitz M, Bovin N, Paulson J, Nemazee D. Decoration of T-independent antigen with ligands for CD22 and Siglec-G can suppress immunity and induce B cell tolerance in vivo. J Exp Med. 2010;207:173–187. doi: 10.1084/jem.20091873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otipoby KL, Andersson KB, Draves KE, Klaus SJ, Farr AG, Kerner JD, Perlmutter RM, Law CL, Clark EA. CD22 regulates thymus-independent responses and the lifespan of B cells. Nature. 1996;384:634–637. doi: 10.1038/384634a0. [DOI] [PubMed] [Google Scholar]
- 34.Sato S, Miller AS, Inaoki M, Bock CB, Jansen PJ, Tang MLK, Tedder TF. CD22 is both a positive and negative regulator of B lymphocyte antigen receptor signal transduction: altered signaling in CD22-deficient mice. Immunity. 1996;5:551–562. doi: 10.1016/s1074-7613(00)80270-8. [DOI] [PubMed] [Google Scholar]
- 35.Nitschke L, Carsetti R, Ocker B, Kohler G, Lamers MC. CD22 is a negative regulator of B-cell receptor signaling. Curr Biol. 1997;7:133–143. doi: 10.1016/s0960-9822(06)00057-1. [DOI] [PubMed] [Google Scholar]
- 36.Onodera T, Poe JC, Tedder TF, Tsubata T. CD22 regulates time course of both B cell division and antibody response. J Immunol. 2008;180:907–13. doi: 10.4049/jimmunol.180.2.907. [DOI] [PubMed] [Google Scholar]
- 37.Samardzic T, Marinkovic D, Danzer CP, Gerlach J, Nitschke L, Wirth T. Reduction of marginal zone B cells in CD22-deficient mice. Eur J Immunol. 2002;32:561–567. doi: 10.1002/1521-4141(200202)32:2<561::AID-IMMU561>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 38.Poe JC, Fujimoto Y, Hasegawa M, Haas KM, Miller AS, Sanford IG, Bock CB, Fujimoto M, Tedder TF. CD22 regulates B lymphocyte function in vivo through both ligand-dependent and ligand-independent mechanisms. Nat Immunol. 2004;5:1078–1087. doi: 10.1038/ni1121. [DOI] [PubMed] [Google Scholar]
- 39.Haas KM, Sen S, Sanford IG, Miller AS, Poe JC, Tedder TF. CD22 ligand binding regulates normal and malignant B lymphocyte survival in vivo. J Immunol. 2006;177:3063–3073. doi: 10.4049/jimmunol.177.5.3063. [DOI] [PubMed] [Google Scholar]
- 40.McKay JT, Haro MA, Daly CA, Yammani RD, Pang B, Swords WE, Haas KM. PD-L2 Regulates B-1 Cell Antibody Production against Phosphorylcholine through an IL-5-Dependent Mechanism. J Immunol. 2017;199:2020–2029. doi: 10.4049/jimmunol.1700555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haas KM, Blevins MW, High KP, Pang B, Swords WE, Yammani RD. Aging Promotes B-1b Cell Responses to Native, but Not Protein-Conjugated, Pneumococcal Polysaccharides: Implications for Vaccine Protection in Older Adults. The JJ Infect Dis. 2014;209:87–97. doi: 10.1093/infdis/jit442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hess C, Winkler A, Lorenz AK, Holecska V, Blanchard V, Eiglmeier S, Schoen AL, Bitterling J, Stoehr AD, Petzold D, Schommartz T, Mertes MM, Schoen CT, Tiburzy B, Herrmann A, Kohl J, Manz RA, Madaio MP, Berger M, Wardemann H, Ehlers M. T cell-independent B cell activation induces immunosuppressive sialylated IgG antibodies. J Clin Invest. 2013;123:3788–3796. doi: 10.1172/JCI65938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haas KM, Watanabe R, Matsushita T, Nakashima H, Ishiura N, Okochi H, Fujimoto M, Tedder TF. Protective and pathogenic roles for B cells during systemic autoimmunity in NZB/W F1 mice. J Immunol. 2010;184:4789–4800. doi: 10.4049/jimmunol.0902391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376:352–355. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- 45.Sato S, Steeber DA, Tedder TF. The CD19 signal transduction molecule is a response regulator of B-lymphocyte differentiation. Proc Natl Acad Sci USA. 1995;92:11558–11562. doi: 10.1073/pnas.92.25.11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haas KM, Poe JC, Tedder TF. CD21/35 promotes protective immunity to Streptococcus pneumoniae through a complement-independent but CD19-dependent pathway that regulates PD-1 expression. J Immunol. 2009;183:3661–3671. doi: 10.4049/jimmunol.0901218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanigaki K, Han H, Yamamoto N, Tashiro K, Ikegawa M, Kuroda K, Suzuki A, Nakano T, Honjo T. Notch-RBP-J signaling is involved in cell fate determination of marginal zone B cells. Nat Immunol. 2002;3:443–450. doi: 10.1038/ni793. [DOI] [PubMed] [Google Scholar]
- 48.Samanta DN, Palmetshofer A, Marinkovic D, Wirth T, Serfling E, Nitschke L. B cell hyperresponsiveness and expansion of mature follicular B cells but not of marginal zone B cells in NFATc2/c3 double-deficient mice. J Immunol. 2005;174:4797–4802. doi: 10.4049/jimmunol.174.8.4797. [DOI] [PubMed] [Google Scholar]
- 49.Haas KM. B-1 lymphocytes in mice and nonhuman primates. Ann N Y Acad Sci. 2015;1362:98–109. doi: 10.1111/nyas.12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parry SL, Hasbold J, Holman M, Klaus GG. Hypercross-linking surface IgM or IgD receptors on mature B cells induces apoptosis that is reversed by costimulation with IL-4 and anti-CD40. J Immunol. 1994;152:2821–2829. [PubMed] [Google Scholar]
- 51.Parry SL, Holman MJ, Hasbold J, Klaus GG. Plastic-immobilized anti-mu or anti-delta antibodies induce apoptosis in mature murine B lymphocytes. Eur J Immunol. 1994;24:974–979. doi: 10.1002/eji.1830240429. [DOI] [PubMed] [Google Scholar]
- 52.Mongini PK, Vilensky MA, Highet PF, Inman JK. Membrane IgM-stimulated human B lymphocytes succumb to activation-related apoptosis at a G1–>S transition: influence of ligand affinity and valency. Cell Immunol. 1998;188:137–50. doi: 10.1006/cimm.1998.1359. [DOI] [PubMed] [Google Scholar]
- 53.Mattson MP, Chan SL. Calcium orchestrates apoptosis. Nat Cell Biol. 2003;5:1041–1043. doi: 10.1038/ncb1203-1041. [DOI] [PubMed] [Google Scholar]
- 54.Demaurex N, Distelhorst C. Cell biology. Apoptosis–the calcium connection. Science. 2003;300:65–67. doi: 10.1126/science.1083628. [DOI] [PubMed] [Google Scholar]
- 55.Matsushita T, Horikawa M, Iwata Y, Tedder TF. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J Immunol. 2010;185:2240–52. doi: 10.4049/jimmunol.1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muller J, Obermeier I, Wohner M, Brandl C, Mrotzek S, Angermuller S, Maity PC, Reth M, Nitschke L. CD22 ligand-binding and signaling domains reciprocally regulate B-cell Ca2+ signaling. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:12402–7. doi: 10.1073/pnas.1304888110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Macauley MS, Pfrengle F, Rademacher C, Nycholat CM, Gale AJ, von Drygalski A, Paulson JC. Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. The Journal of clinical investigation. 2013;123:3074–83. doi: 10.1172/JCI69187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Keefe TL, Williams GT, Davies SL, Neuberger MS. Hyperresponsive B cells in CD22-deficient mice. Science. 1996;274:798–801. doi: 10.1126/science.274.5288.798. [DOI] [PubMed] [Google Scholar]
- 59.Nakashima H, Hamaguchi Y, Watanabe R, Ishiura N, Kuwano Y, Okochi H, Takahashi Y, Tamaki K, Sato S, Tedder TF, Fujimoto M. CD22 expression mediates the regulatory functions of peritoneal B-1a cells during the remission phase of contact hypersensitivity reactions. J Immunol. 2010;184:4637–4645. doi: 10.4049/jimmunol.0901719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pfrengle F, Macauley MS, Kawasaki N, Paulson JC. Copresentation of antigen and ligands of Siglec-G induces B cell tolerance independent of CD22. J Immunol. 2013;191:1724–31. doi: 10.4049/jimmunol.1300921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ding C, Ma Y, Chen X, Liu M, Cai Y, Hu X, Xiang D, Nath S, Zhang HG, Ye H, Powell D, Yan J. Integrin CD11b negatively regulates BCR signalling to maintain autoreactive B cell tolerance. Nat Communications. 2013;4:2813. doi: 10.1038/ncomms3813. [DOI] [PubMed] [Google Scholar]
- 62.Poe JC, Haas KM, Uchida J, Lee Y, Fujimoto M, Tedder TF. Severely-impaired B lymphocyte proliferation, survival and induction of the c-Myc:Cullin 1 ubiquitin ligase pathway resulting from CD22 deficiency on the C57BL/6 genetic background. J Immunol. 2004;172:2100–2110. doi: 10.4049/jimmunol.172.4.2100. [DOI] [PubMed] [Google Scholar]
- 63.Poe JC, Kountikov EI, Lykken JM, Natarajan A, Marchuk DA, Tedder TF. EndoU is a novel regulator of AICD during peripheral B cell selection. J Exp Med. 2014;211:57–69. doi: 10.1084/jem.20130648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buhl AM, Pleiman CM, Rickert RC, Cambier JC. Qualitative regulation of B cell antigen receptor signaling by CD19: Selective requirement for PI3-kinase activation, inositol-1,4,5-trisphosphate production and Ca2+ mobilization. J Exp Med. 1997;186:1897–1910. doi: 10.1084/jem.186.11.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Depoil D, Fleire S, Treanor BL, Weber M, Harwood NE, Marchbank KL, Tybulewicz VL, Batista FD. CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nat Immunol. 2008;9:63–72. doi: 10.1038/ni1547. [DOI] [PubMed] [Google Scholar]
- 66.Rickert RC. Regulation of B lymphocyte activation by complement C3 and the B cell coreceptor complex. Curr Opin Immunol. 2005;17:237–243. doi: 10.1016/j.coi.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 67.Haas KM, Tedder TF. Role of the CD19 and CD21/35 receptor complex in innate immunity, host defense and autoimmunity. Adv Exp Med Bio. 2005;560:125–139. doi: 10.1007/0-387-24180-9_16. [DOI] [PubMed] [Google Scholar]
- 68.Fujimoto M, Poe JC, Hasegawa M, Tedder TF. CD19 amplification of B lymphocyte Ca2+ responses: A role for Lyn sequestration in extinguishing negative regulation. J Biol Chem. 2001;276:44820–44827. doi: 10.1074/jbc.M107559200. [DOI] [PubMed] [Google Scholar]
- 69.Shih TA, Roederer M, Nussenzweig MC. Role of antigen receptor affinity in T cell-independent antibody responses in vivo. Nature immunology. 2002;3:399–406. doi: 10.1038/ni776. [DOI] [PubMed] [Google Scholar]
- 70.Sen G, Wu HJ, Bikah G, Venkataraman C, Robertson DA, Snow EC, Bondada S. Defective CD19-dependent signaling in B-1a and B-1b B lymphocyte subpopulations. Mol Immunol. 2002;39:57–68. doi: 10.1016/s0161-5890(02)00047-0. [DOI] [PubMed] [Google Scholar]
- 71.Dasu T, Sindhava V, Clarke SH, Bondada S. CD19 signaling is impaired in murine peritoneal and splenic B-1 B lymphocytes. Mol Immunol. 2009;46:2655–2665. doi: 10.1016/j.molimm.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dal Porto JM, Burke K, Cambier JC. Regulation of BCR signal transduction in B-1 cells requires the expression of the Src family kinase Lck. Immunity. 2004;21:443–453. doi: 10.1016/j.immuni.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 73.Yammani RD, Haas KM. Primate B-1 cells generate antigen-specific B cell responses to T cell-independent type 2 antigens. J Immunol. 2013;190:3100–3108. doi: 10.4049/jimmunol.1203058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dorner T, Shock A, Goldenberg DM, Lipsky PE. The mechanistic impact of CD22 engagement with epratuzumab on B cell function: Implications for the treatment of systemic lupus erythematosus. Autoimmun Rev. 2015;14:1079–86. doi: 10.1016/j.autrev.2015.07.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.