

Abstract
Background/Aim: Dysregulation of mitochondrial pathways is implicated in several diseases, including cancer. Notably, mitochondrial respiration and mitochondrial biogenesis are favored in some invasive cancer cells, such as osteosarcoma. Hence, the aim of the current work was to investigate the effects of 2-methoxyestradiol (2-ME), a potent anticancer agent, on the mitochondrial biogenesis of osteosarcoma cells. Materials and Methods: Highly metastatic osteosarcoma 143B cells were treated with 2-ME separately or in combination with L-lactate, or with the solvent (non-treated control cells). Protein levels of α-syntrophin and peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1α) were determined by western blotting. Impact of 2-ME on mitochondrial mass, regulation of cytochrome c oxidase I (COXI) expression, and succinate dehydrogenase complex flavoprotein subunit A (SDHA) was determined by immunofluorescence analyses. Inhibition of sirtuin 3 (SIRT3) activity by 2-ME was investigated by fluorescence assay and also, using molecular docking and molecular dynamics simulations. Results: L-lactate induced mitochondrial biogenesis pathway via up-regulation of COXI. 2-ME inhibited mitochondrial biogenesis via regulation of PGC-1α, COXI, and SIRT3 in a concentration-dependent manner as a consequence of nuclear recruitment of neuronal nitric oxide synthase and nitric oxide generation. It was also proved that 2-ME inhibited SIRT3 activity by binding to both the canonical and allosteric inhibitor binding sites. Moreover, regardless of the mitochondrial biogenesis pathway, 2-ME affected the expression of SDHA. Conclusion: Herein, mitochondrial biogenesis pathway regulation and SDHA were presented as novel targets of 2-ME, and moreover, 2-ME was demonstrated as a potent inhibitor of SIRT3. L-lactate was confirmed to exert pro-carcinogenic effects on osteosarcoma cells via the induction of the mitochondrial biogenesis pathway. Thus, L-lactate level may be considered as a prognostic biomarker for osteosarcoma.
Keywords: 2-Methoxyestradiol, mitochondrial biogenesis, osteosarcoma, sirtuin 3, succinate dehydrogenase
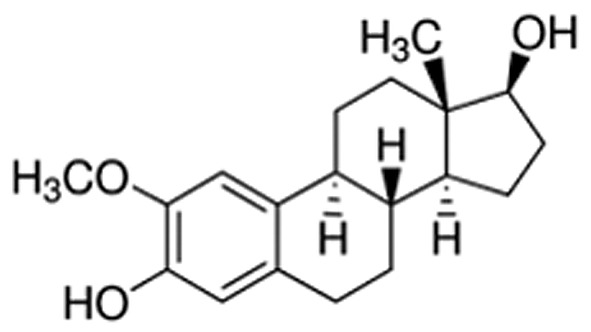
Mitochondria are highly specialized organelles that are crucial players in fundamental aspects of cell pathophysiology (1). Mitochondrial quality and content, crucial for proper cellular physiology and tissue formation, are under the control of mitochondrial biogenesis and mitophagy processes (2-5). Interestingly, increased mitochondrial biogenesis has been strictly associated with tumor proliferation and growth (6,7) and, therefore, comprises an important target for novel therapies against several malignancies, including osteosarcoma (4-11). One of the crucial regulators of mitochondrial biogenesis is the transcription coactivator peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PARGC1A, also known as PGC-1α) (11-13). Invasive cancer cells use PGC-1α to enhance oxidative phosphorylation, the oxygen consumption rate, and finally, mitochondrial biogenesis (11-13). Clinical analysis of human invasive breast cancers has revealed a strong correlation between PGC-1α expression in invasive cancer cells and the formation of distant metastases, while PGC-1α silencing has suspended their invasive potential and attenuated metastasis (11-13). Importantly, mitochondrial respiration and mitochondrial biogenesis remain under the strict control of nitric oxide (NO) (14-17). Furthermore, as indicated by Aquilano and coauthors, the nuclear recruitment of neuronal nitric oxide synthase (nNOS) and generation of NO in the nucleus are mandatory for the induction of the mitochondrial biogenesis pathway (14). Previously, we evidenced that generation of NO due to nuclear recruitment of nNOS constitutes an anticancer mechanism of 2-methoxyestradiol (2-ME) action (Figure 1) (18). 2-ME is a natural derivative of 17β-estradiol that possesses anticancer activities, as confirmed by in vitro and in vivo studies (18-29). Under the brand name Panzem, 2-ME is being evaluated in clinical trials for the treatment of numerous malignancies, such as breast and prostate cancers (19-22). Interestingly, previous studies have reported potential physiological anticancer activity of 2-ME and suggested to consider 2-ME not as a metabolite of 17β-estradiol but as a novel hormone (18,23,24,26). Nonetheless, potential hormonal properties of 2-ME need to be further elucidated by in vivo studies.
Figure 1. Chemical structure of 2-methoxyestradiol.
Therefore, the aim of the current study was to determine whether the anticancer potential of 2-ME is associated with the regulation of mitochondrial biogenesis as a consequence of NO generation due to nuclear nNOS localization. Our findings demonstrated that 2-ME significantly affects mitochondrial biogenesis especially at physiological relevant concentrations. It was also evidenced that regardless of the mitochondrial biogenesis pathway, 2-ME, at both physiologically- and pharmacologically-relevant concentrations, affects another important anticancer target, namely, mitochondrial inner membrane succinate dehydrogenase complex subunit A (SDHA).
Materials and Methods
Reagents. 2-Methoxyestradiol, the osteosarcoma 143B cell line, tissue culture media, antibiotic cocktail, heat inactivated fetal bovine serum (FBS), Charcoal stripped FBS, RIPA buffer, protease and phosphatase inhibitor cocktails were obtained from Sigma-Aldrich (Warsaw, Poland). The SIRT3 Direct Fluorescent Screening Assay Kit was obtained from Cayman Chemical (Warsaw, Poland). ProLong Gold Antifade Reagent, SlowFade Gold Antifade Reagent with DAPI, NucBlue™ Live Cell Stain, and MitoTracker® Green FM were purchased from Thermo Fisher Scientific (Waltham, MA, USA). The MitoBiogenesis™ ICC Kit and antibodies against α-syntrophin and PGC-1α were obtained from Abcam (Cambridge, UK). Antibody against β-Actin and secondary antibodies were obtained from Santa Cruz Biotechnology (Heidelberg, Germany). Cell Imaging TC coverglasses were purchased from Eppendorf (Warsaw, Poland) and CellView 4 Compartment Advanced TC plates were obtained from Greiner (Frickenhausen, Germany). Amersham ECL 4-12% gels were obtained from GE Healthcare Life Sciences (Warsaw, Poland).
Cell culture. 143B osteosarcoma cells were cultured at 37˚C in a humidified atmosphere and saturated with 5% CO2. Minimum Essential Medium Eagle was supplemented with 2 mM glutamine, 1% nonessential amino acids, and 10% heat-inactivated FBS (29). In order to avoid the effects of glucose and Warburg effect in osteosarcoma cells, low glucose, pyruvate and lactate-free Minimum Essential Medium Eagle was chosen for cell culture and treatment (29).
Cell treatment. In order to preserve lactate activity without affecting the pH of media, all the analyses were performed using sodium salt of L-lactate, indicated in the text as L-lactate. Treatments were performed in Minimum Essential Medium Eagle containing 1% charcoal-stripped fetal bovine serum which is used to elucidate the effects of hormones in a variety of in vitro systems (30). As previously described (29), 143B cells were treated with 2-ME alone and in combination with L-lactate. Initially, cells were pre-treated for 24 h with 10 mM L-lactate in order to induce mitochondrial biogenesis (6,7), followed by incubation with 10 mM L-lactate or 10 mM L-lactate and 2-ME (10 nM, 1 μM) for 8 h or 24 h, according to the experimental design. Cells treated separately with 10 nM or 1 μM of 2-ME for 8 h and 24 h were also used in the study. Incubation time of 8 h was chosen due to the observed highest level of nNOS induction and NO generation by 2-ME after this period of time (18,25). Control cells were treated with an equal volume of the solvent used to prepare 2-ME solution.
Live cell imaging. Osteosarcoma 143B cells were seeded onto CellView 4 Compartment Advanced TC plates at a density of 7×105 cells per well. After 24 h, cells were incubated with 2-ME and/or L-lactate according to the experimental design. Thirty minutes before the end of incubation, MitoTracker® Green FM (200 nM) was added to the culture medium (31). Afterwards, cells were rinsed with PBS and medium was replaced with RPMI 1640 without phenol red. Before imaging, ProLong™ Live Antifade and NucBlue™ Live ReadyProbes™ Reagent were added.
Immunofluorescence analyses. Osteosarcoma 143B cells were seeded onto Cell Imaging TC coverglasses at a density of 7×105 cells per well and cultured for 24 h. Thereafter, cells were incubated with 2-ME and/or L-lactate according to the experimental design and fixed with 4% paraformaldehyde for 15 min. Following antigen retrieval and washing with PBS, cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min and blocked in 10x blocking solution for 1 h at room temperature. Cells were then incubated with the antibody solution (1x antibody cocktail mouse anti-mitochondrial cytochrome c oxidase I (MtCOI or COXI) Alexa®488 and mouse anti-SDHA Alexa®594 antibody) overnight at 4˚C. Finally, SlowFade Gold Antifade Mountant containing DAPI was added.
Image acquisition and analysis. All samples were analyzed on a Zeiss Axio Observer Spinning Disc microscope equipped with a Plan-Apochromat 63x/1.4 Oil DIC objective and an Axiocam 503 mono CCD camera (Zeiss, Oberkochen, Germany). The following excitation lasers and emission filters were used: Blue channel: 405 nm diode laser, Green channel: 488 nm diode laser, 525/50 nm filter, and Red channel: 561 nm diode laser, 600/50 nm filter. Z-stacks were acquired for all samples applying an interval of 500 nm and maximum intensity projections were calculated. All image processing and analysis steps were performed with Zen blue 2.3 software (Zeiss, Oberkochen, Germany). To determine the mitochondrial mass, a region of interest was drawn around the whole mitochondrion in each cell and the intensity was measured (mean intensity of the green channel). The background was subtracted before analysis.
Screening of sirtuin 3 (SIRT3) deacetylase activity. Deacetylation activity of SIRT3 was determined using SIRT3 Direct fluorescence Screening Assay Kit. 2-ME at concentrations from 1 pM up to 500 μM was added to wells containing 25 μl Assay Buffer and 5 μl diluted SIRT3 peptide. Initial activity (100%) and background wells were prepared according to the assay protocol. The reaction was initiated by adding 15 μl Substrate Solution (SIRT3 peptide, NAD Solution, and diluted Assay Buffer). The plate was covered and incubated on a shaker for 45 min at 37˚C. Afterwards, 50 μl Stop/Developing solution was added to each well and the plate was incubated for 30 min at room temperature. The absorbance was then read at an excitation wavelength of 350-360 nm and an emission wavelength of 450-465 nm. Each experiment was performed at least three times. The activity of 2-ME was calculated according to the following formula: % Inhibition/Activation=[(Initial Activity- Sample Activity)/Initial Activity] ×100.
Western blot analysis. Osteosarcoma 143B cells were seeded at a density of 1×106 cells/dish, cultured in standard medium for 24 h. After incubation, cells were washed with ice-cold PBS and lysed in RIPA buffer containing protease and phosphatase inhibitor cocktail (0.5 ml per 1×106). Lysed cells were centrifuged for 20 min at 12000 rpm and supernatant was collected. Total protein (20 μg/sample) was resolved by polyacrylamide gel electrophoresis using Amersham ECL 4-12% gels. The membranes were then incubated with primary antibodies specific to α-syntrophin or PGC-1α (1:2000) overnight at 4˚C, or β-Actin (1:50000) for 30 min at room temperature. Analysis was performed as previously described (26). Chemiluminescence was detected using ImageQuant LAS 500 (GE Healthcare, Warsaw, Poland). The protein level was quantified by densitometry using Quantity One 4.5.2 software (Bio-Rad, Warsaw, Poland). The protein level of α-syntrophin and PGC-1α, as determined by chemiluminescent signal quantification, were normalized to loading control, β-Actin. Each experiment was performed at least three times.
System quality assessment and protein preparation. Crystal structure selection was based on the following criteria: high resolution of the crystal structure (below 3 Å) and electron density (ED) assessment. ED was evaluated using VHELIBS (32). The protein structure was obtained from the protein data bank (PDB) and Protein Preparation Wizard Maestro (Schrödinger) was used to add bond orders and hydrogen atoms to the crystal structure (33). Missing residues and atoms were fixed using Prime 4.0 (34,35). The protonation state of the protein and ligand was predicted using PropKa 3.1 (36,37). To validate the docking search algorithm, a “cognate docking” test was performed on the crystal. The cognate docking protocol provided a root mean square deviation (RMSD) below 2 Å, demonstrating good capability of correct placement of the ligand into the binding pocket.
Extra-precision (XP) docking. The docking grid was generated starting from the ligand present in the crystallized binding pocket. The docking protocol chosen was extra-precision one to minimize possible false poses (38). From the re-docking protocol, only the best poses of the ligand were retained for further studies.
Molecular mechanics generalized born surface area (MMGBSA). The MMGBSA calculation was performed on the best pose of the previous docking procedures. The VSGB 2.0 model (39) was used as the solvent and OPLS3 (40) as the force field for the molecular mechanics portion of the calculation.
Molecular dynamics. All molecular dynamics (MD) simulations were performed with DESMOND 4.2 using the OPLS3 force field (40). The protein-ligand systems were solved using TIP3P as the water model. Ions were added to neutralize charges. The equilibration steps were run at a temperature of 303.15 K and at 1.013 bar pressure using the NPT ensemble. A Nose-Hover thermostat and a Martyna-Tobia-Klein barostat were used. The integration time step was 2 fs. The SHAKE algorithm was adopted to maintain the hydrogen - heavy atom bonds rigid. Short-range Coulomb interactions were set with a cut-off radius of 9 Å and a smooth particle mesh Ewald was used for the long-range interactions. System stability was evaluated using the RMSD of the protein backbone set calculated against the initial frame.
Statistical analysis. The results represent the mean±SD from at least three independent experiments. All microscopic evaluations were conducted on randomized and coded slides. Differences between control samples versus 2-ME-treated samples were assessed by one-way analysis of variance (ANOVA) with post hoc testing using Dunnett’s multiple comparison test or t-test combined with the Wilcoxon test. A p-value less than 0.01 was considered to indicate statistical significance. Data were analyzed using GraphPad Prism software (GraphPad Software v.6).
Results
Effect of 2-ME on α-syntrophin and PGC-1α protein expression. α-syntrophin has been proved crucial for nuclear recruitment of nNOS and consequently, indirectly, for the induction of mitochondrial biogenesis pathway (14). Hence, the effect of 2-ME on intracellular α-syntrophin protein expression was determined in osteosarcoma cells. As demonstrated, 24 h treatment with 2-ME at a range of concentrations (10 pM, 100 pM, 1 nM, 10 nM, 100 nM, 1 μM) resulted in significant up-regulation of α-syntrophin levels by 2.0-, 3.8-, 2.1-, 4.6-, 7.4-, 5.6- fold, respectively (p<0.0001 compared to the control cells) (Figure 2A). Subsequently, the effect of 2-ME on PGC-1α expression in osteosarcoma cells was investigated. Osteoarcoma cells were treated 24 h with 2-ME at concentrations of 10 pM up to 10 μM. Only treatment with 2-ME at concentrations of 100 pM-100 nM showed a trend of slight decrease in PGC-1α protein levels (0.2-0.3 –fold, p<0.01 compared to the control cells) (Figure 2B).
Figure 2. 2-Methoxyestradiol (2-ME) affects α-syntrophin and peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1α) protein levels. Osteosarcoma 143B cells were incubated with 2-ME at concentrations ranging from 10 pM to 10 μM for 24 h. Non-treated cells were considered as control cells (c). Protein levels of α-syntrophin (A) and PGC-1α (B) were normalized to β-actin and presented as fold changes relative to control cells. The numbers below and above each lane represent the concentration of 2-ME and the fold change of protein levels, respectively.

2-ME reduces mitochondrial mass. The impact of representative physiologically (10 nM) and pharma-cologically (1 μM) relevant concentrations of 2-ME on the mitochondrial mass in osteosarcoma 143B cells was also tested. Importantly, cell treatment with 10 nM of 2-ME for 24 h resulted in statistically significant reduction in mitochondrial fluorescence per cell by 50±10.5% (p<0.00001 compared to the control cells) (Figure 3A and C). Pharmacological concentration of 2-ME also resulted in reduced mitochondrial fluorescence per cell by 17±8% (p<0.001 compared to the control cells) (Figure 3B,C).
Figure 3. Effect of 2-methoxyestradiol (2-ME) on mitochondrial mass. Osteosarcoma 143B cells were incubated with 10 nM (A) and 1 μM (B) of 2-ME for 24 h. Cells were stained with fluorescent dyes MitoTracker® Green FM (green) and Hoechst 33342 (blue) and imaged by a spinning disk microscopy. Representative maximum intensity projections of one hundred cells from three independent experiments are shown. The fluorescence density was presented in relative fluorescence units (RFUs). Quantification data of the mitochondrial mass (fluorescence intensity of labeled cells) are displayed in the chart (C). One hundred cells were analyzed and values represent the mean±SE of three independent experiments. Statistical significance level between various experimental pairs is indicated in the figure (**p<0.001, ****p<0.00001 versus control cells).

Regulation of COXI. To further strengthen our findings, the influence of 2-ME on COXI protein level, a typical marker of mitochondrial biogenesis encoded by mitochondrial DNA (14,41), was investigated. As demonstrated, incubation of osteosarcoma cells with 10 nM 2-ME for 8 h did not significantly affect COXI expression (Figure 4A, B), though treatment for 24 h significantly decreased COXI levels by 12±2.9% (p<0.01 compared to the control cells) (Figure 4A, C). On the contrary, pre-treatment of cells with 10 mM of L-lactate for 48 h, led to a statistically significant increase in the level of COXI by 28±10% (p<0.00001 compared to the control cells) (Figures 4,5). Further treatment with 10 nM of 2-ME for 8 h and 24 h significantly down-regulated the increased COXI levels by 21±5.9% and 29±5.5% (p<0.001, p<0.00001, respectively, compared to L-lactate-treated cells) (Figure 4A-C). Regarding the pharmacological concentration of 2-ME, cell treatment with 1 μM 2-ME alone did not change the level of COXI, compared to the control cells (Figure 5A-C). However, incubation of L-lactate pre-treated cells with 1 μM of 2-ME for 24 h significantly reversed the L-lactate-induced increased of COXI by 21±2.9% (p<0.0001 compared to L-lactate-treated cells) (Figure 5A,C).
Figure 4. The effect of 10 nM 2-methoxyestradiol (2-ME) on cytochrome c oxidase I (COXI) and succinate dehydrogenase complex flavoprotein subunit A (SDHA) protein expression. Osteosarcoma 143B cells, either treated (L-lactate for 48 h, 2-ME for 8 h or 24 h, or combination of L-lactate and 2-ME) or non-treated control (c), were stained for COXI (green) SDHA (red), and DAPI for nuclear staining (blue). Representative maximum intensity projections of spinning disc microscopy images from three independent experiments are shown (A). The quantification data of 2-ME effect on COXI (B, C) and SDHA (D, E) are presented as relative fluorescence units (RFUs) in charts. The total fluorescence of fifty cells was analyzed and values represent the mean±SE of three independent experiments. Statistical significance level between various experimental pairs is indicated in the figure (*p<0.01, **p<0.001, ***p<0.0001, ****p<0.00001).

Figure 5. The effect of 1 μM 2-methoxyestradiol (2-ME) on cytochrome c oxidase I (COXI) and succinate dehydrogenase complex flavoprotein subunit A (SDHA) protein expression. Osteosarcoma 143B cells either treated (with L-lactate for 48 h, 1 μM 2-ME 8 h or 24 h, or combination of L-lactate and 2-ME) or non-treated control (c), were stained for COXI (green), SDHA (red), and DAPI for nuclear staining (blue). Representative maximum intensity projections of spinning disc microscopy images from three independent experiments are shown (A). The quantification data of 2-ME effect on COXI (B, C) and SDHA (D, E) are presented as relative fluorescence units (RFUs) in charts. The total fluorescence of fifty cells was analyzed and values represent the mean±SE of three independent experiments. Statistical significance level between various experimental pairs is indicated in the figure (*p<0.01, **p<0.001, ***p<0.0001, ****p<0.00001).

Regulation of SDHA. In order to establish the role of 2-ME in the regulation of mitochondrial complex encoded by nuclear DNA, the effect of 2-ME and L-lactate, separately and in combination, on SDHA protein levels, important target for anticancer therapies, was also investigated. Treatment with 10 nM 2-ME for 8 h and 24 h significantly decreased the level of SDHA by 38±3% and 17±2.5% (p<0.00001, p<0.001, respectively, compared to the control cells) (Figure 4A, D and E). Moreover, incubation with 1 μM 2-ME for 8 and 24 h significantly decreased the expression of SDHA (21±4% p<0.001 and 37±2.8% p<0.00001, respectively, compared to the control cells) (Figure 5A, D and E). Notably, 10 mM L-lactate alone slightly enhanced the level of SDHA by 10±2.4% (p<0.01 versus control cells) (Figures 4,5). L-Lactate-induced SDHA up-regulation was significantly reversed after treatment with 1 μM of 2-ME for 24 h, as well as with 10 nM of 2-ME for 8 h and 24 h by 35.5±3.3% (p<0.00001), 22±5% (p<0.0001), 21±4% (p<0.00001), respectively, compared to L-lactate pre-treated cells (Figure 4A,D,E).
SIRT3 activity in vitro. Subsequently, the in vitro inhibitory effects of 2-ME, at physiologically (1 pM-10 nM) and pharmacologically (100 nM-500 μM) relevant concentrations, on SIRT3 activity were determined. Importantly, as shown in Figure 6A, all concentrations of 2-ME significantly inhibited SIRT3 activity in vitro. The lowest percentage of SIRT3 activity inhibition (6%) was observed following treatment with 100 nM 2-ME (p<0.001) while treatment with 500 μM 2-ME resulted in the highest percent inhibition by 2-ME (32%) (p<0.00001) (Figure 6A).
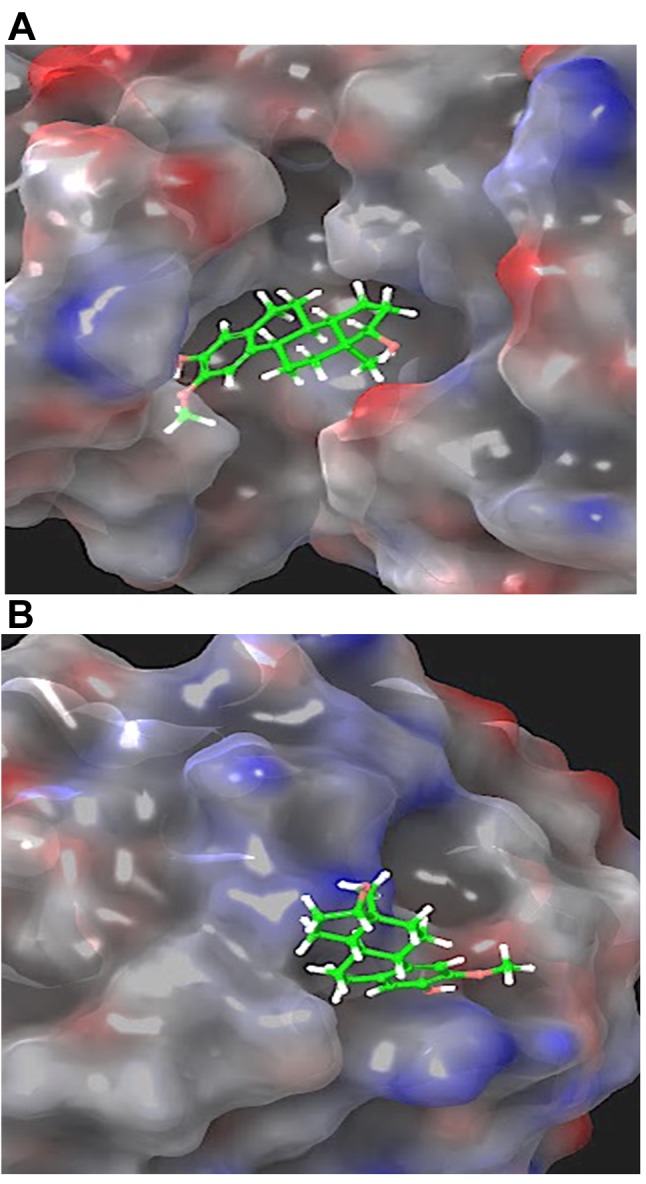
Figure 6. 2-Methoxyestradiol (2-ME) affects sirtuin 3 (SIRT3) activity in a concentration-dependent manner. The percentage of SIRT3 activity inhibition after in vitro treatment of osteosarcoma cells with a range of different 2-ME concentrations is presented in the chart (A). Values are presented as mean±SE of three independent experiments. Statistical significance level between each experimental group and control cells is indicated in the figure (*p<0.01, **p<0.001, ***p<0.0001, ****p<0.00001). In the 2D depiction of 2-ME within the canonical binding site of SIRT3 (B), H-bond and π-π interactions are highlighted in purple and green, respectively. In the 3D depiction of 2-ME within the canonical binding site of SIRT3 (C), H-bond and π-π interactions are highlighted in yellow and blue, respectively. Similarly, in the 2D depiction of 2-ME within the allosteric binding site of SIRT3 (D), H-bond and π-π interactions are highlighted in purple and green, respectively, and in the 3D depiction of 2-ME, H-bond interactions are highlighted in yellow (E).

Molecular docking. To enhance the reliability of our data, molecular modeling was performed to obtain information on the possible binding mode of 2-ME with SIRT3 using crystallographic coordinates from the PDB (ID 4C78). Figure 6 shows the 3D and 2D ligand receptor interactions demonstrated by molecular docking. Interestingly, the crucial interaction regions reported in the literature for known inhibitors (small molecules or peptides) were observed for 2-ME at both the canonical inhibitor and allosteric sites (42-46). The free energy (ΔG) value calculated for the canonical binding site was -33.64 kcal/mol. The major contribution is represented by H-bonds with Val 292 and Asp 231, π-π interactions with Phe 157 and Phe 180, and hydrophobic interactions with Ile230. Concerning the allosteric binding site, the calculated ΔG value was -20.80 kcal/mol. The binding was characterized by π-π interactions and H-bonds with Arg 139 and hydrophobic interactions with Met 311. The 2D and 3D interaction diagrams for the canonical and allosteric inhibitor binding sites are reposted in Figure 6B-E.
Molecular Dynamics (MD) Simulations. The RMSD plot obtained from the MD simulations is presented in Figure 7. Two different MD simulations were run to compare the host-guest stability when only one molecule of 2-ME was bound to SIRT3 in the canonical or allosteric inhibition site (Figure 7A-B). Moreover, a third MD simulation was run on sirtuin 3 protein presenting two 2-ME molecules in both the canonical and allosteric binding sites (Figure 8 and Figure 9). The 1:2 host/guest complex was as stable as the 1:2 complexes during 50 ns of MD simulation under the same conditions (Figures 8 and 9). The RMSD of the 2:1 complex was plotted and is shown in Figure 8A.
Figure 7. Root-mean-square deviation (RMSD) of 2-methoxyestradiol (2-ME) within the canonical inhibitor binding site of sirtuin 3 (SIRT3) (A). RMSD of 2-ME within the allosteric inhibitor binding site of SIRT3 (B).

Figure 8. Root mean square deviation (RMSD) of two 2-methoxyestradiol molecules within both the canonical and allosteric inhibitor binding sites of sirtuin 3 (A). 3D representation of the protein molecular surface of the 1:2 host-guest complex (B).

Figure 9. 3D representation of the protein molecular surface of the 1:2 host-guest complex. Detail of 2-methoxyestradiol (2-ME) molecule within the catalytic binding site (A) and the allosteric binding site of sirtuin 3 (B).
Discussion
Mitochondria are well-adapted endosymbiotic intracellular organelles that became efficient for energy production throughout the course of evolution (2,3,5,7). Beyond their conventional metabolic functions, they also play a crucial role in certain neurodegenerative diseases and cancer. Mitochondrial biogenesis due to its unquestionable role in regulation of cancer invasiveness has become a target for novel anticancer therapies (8,11).
Previously, the inhibition of mitochondrial mass by 2-ME at the pharmacologically relevant concentration of 1 μM was suggested by Karbowski et al. (47). Herein, it is demonstrated for the first time that 2-ME may regulate mitochondrial biogenesis in osteosarcoma cells, especially at physiologically relevant concentrations. Aquilano et al. demonstrated that generation of NO in the nuclei of murine myocytes due to the nuclear localization of nNOS variant α (nNOSα) is critical for initiation of the mitochondrial biogenesis pathway (14). We have previously established that 2-ME exerts an anticancer effect at both physiologically and pharmacologically relevant concentrations by selective induction of nNOS, nuclear recruitment of nNOSα, and subsequent NO generation in the nuclei of osteosarcoma cells (18,25). As reported by Aquilano et al., α-syntrophin functions as the upstream mediator of nNOS translocation into nucleus and nNOS-dependent mitochondrial biogenesis (14). Indeed, in the current study, up-regulation of α-syntrophin was observed in osteosarcoma cells after cell treatment with both physiologically and pharmacologically relevant concentrations of 2-ME indicating its role in 2-ME-induced nuclear nNOS recruitment.
Notably, induction of α-syntrophin was accompanied by down-regulation of PGC-1α, a decrease in mitochondrial mass, and a decrease in the expression of COXI, though only after treatment with physiological concentrations of 2-ME. While, 2-ME, at pharmacologically relevant concentrations, caused only a slight decrease in mitochondrial mass without affecting PGC-1α and COXI expression. Up to date, no study has considered the effect of 2-ME on PGC-1α or any subunit of the mitochondrial cytochrome c oxidase (COX). Previously, the possibility for the hormone precursor of 2-ME, 17β-estradiol, to control reactive oxygen species generation and mitochondrial membrane potential via regulating COX activity has been suggested as a mechanism of estrogen-induced redox signaling by the mitochondria (41). This evidence, combined with the fact that sarcoma cells are characterized by high COX activity and mitochondrial DNA content (11) render mitochondrial biogenesis pathway a promising target for 2-ME-based osteosarcoma therapy. Expression of PGC-1α is strictly associated with increased cancer invasiveness (18,48,49), thus down-regulation of PGC-1α by low concentrations of 2-ME, observed in the current study, may be considered as a physiological anticancer mechanism of 2-ME. Distinct biological activity of physiologically- and pharmacologically-relevant 2-ME concentrations on the expression of PGC-1α and COXI may be explained by the bi-phasic activity of NO dependent on concentration, time of exposure, and the presence of other free radicals. Previously, our findings have revealed that treatment of osteosarcoma cells with 2-ME at physiological concentrations resulted in significantly lower nuclear NO levels than treatment with 2-ME at pharmacological concentrations (18). Differential effects depending on NO exposure time and concentration have previously been reported (17). In a long-term effect, a NO-dependent pathway has been shown to control mitochondrial biogenesis through up-regulating PGC-1α, the crucial regulator of mitochondrial biogenesis (15-17). In contrast, short-term treatment with NO donors leads to down-regulation of PGC-1α expression indicating that the modulation of PGC-1α by NO is time-dependent (15-17).
Noteworthy, PGC-1α may stimulate SIRT3, while not affecting other sirtuins (50,51). Thus, our research concerning regulation of mitochondrial biogenesis pathway via 2-ME was extended by analyzing the influence of 2-ME on SIRT3 activity. Human SIRT3 is a mitochondrial protein, class III histone deacetylase (50,51). Herein, for the first time the inhibitory activity of 2-ME, at both physiologically and pharmacologically relevant concentrations, towards SIRT3 was presented. To enhance the reliability of our data, molecular modeling was performed to obtain information on the possible binding mode of 2-ME with SIRT3 using crystallographic coordinates from the Protein Data Bank (PDB; ID 4C78) (52). The crystal structure, similar to human SIRT3 in complex with 4-bromo-Resveratrol and an inhibitor peptide, has been used by Nguyen et al. to demonstrate the possible role and inhibitory mechanism of 4-bromo-Resveratrol (45). The authors demonstrated that certain SIRT3 inhibitors bind to both the canonical inhibitor binding and allosteric sites. Thus, the binding of 2-ME to SIRT3 was investigated considering the possibility of a 2:1 stoichiometry between the ligand and protein, in order to mimic the possible concentration-dependent activity of 2-ME observed experimentally. Specifically, molecular docking of 2-ME was performed on the two possible binding sites, the canonical inhibitor site (NAD+ catalytic binding site in the C-terminus) and/or the allosteric site (N-terminus region). The best docking poses of 2-ME were selected and analysis of the molecular mechanics energies combined with the generalized Born and surface area continuum solvation (MMGBSA) was performed to evaluate the enthalpy and free energy contributions to the binding process (53-55). Finally, molecular dynamics simulations were performed to explore the stability of the ligand-protein complex and to evaluate the time evolution of the main host-guest interactions. Overall, the results of molecular modeling showed that 2-ME was clearly able to bind both the canonical and allosteric binding sites of SIRT3 protein, as evidenced by the negative ΔG values calculated for the binding interactions and, moreover, by the stability of the protein backbone observed during the MD simulations. Furthermore, the host-guest complex involving two ligand molecules and one protein shows analogous stability during 50 ns of MD simulation. These data provide information on the potential role and binding mode of 2-ME to SIRT3 and pave the way to additional experimental structural studies oriented to confirm that 2-ME binds to both the canonical and allosteric inhibitor binding sites, as observed for 4-bromo-Resveratrol (45). Interestingly, the 2:1 binding stoichiometry hypothesis is in excellent agreement with the concentration-dependent effect on the inhibition of SIRT3 activity observed experimentally. Sirtuins play an important role in many physiological and pathophysiological conditions, including metabolism, cell survival, aging, and cancer (50,51). SIRT3 inhibitors are currently investigated in potential therapy of numerous tumors including: head, neck tumors (56). Inhibition of SIRT3 alters also the sensitivity of cancer cells to apoptosis (57). Notably, silencing of SIRT3 expression has been associated with increased estrogen receptor β (ERβ) mitochondrial content, oxidative stress, and apoptosis. Indeed, in our recent study restoration of ERβ by 2-ME leading to osteosarcoma cell death was evidenced (58).
Importantly, the pro-cancerogenic effect of L-lactate due to mitochondrial biogenesis induction in highly metastatic osteosarcoma 143B cells was proved in the study. L-Lactate has previously been shown to fuel the cancer growth and proliferation via inducing metabolic reprogramming (6,7). Herein, we demonstrated that 2-ME, at both physiologically and pharmacologically relevant concentrations, is able to reverse pro-cancerogenic effect of L-lactate and is effective anticancer agent under metabolic reprogramming conditions. Hence, L-lactate level may be considered as a prognostic biomarker for cancer medicine.
Another important issue of the present study is the effect of 2-ME on the expression of mitochondrial succinate dehydrogenase (SDH). Mitochondrial SDH, also known as Complex II or succinate: ubiquinone oxidoreductase (SQR), contains four nuclear encoded subunits: SDH A-D (59). SDH has the unique characteristic of being the component that links the Krebs cycle and the electron transport chain. SDH complex has attracted attention after the reveal of its role in human disease. It plays an important role both in cell survival and death depending on the physiological or experimental conditions. Inhibition of SDH during ischemia/reperfusion attenuates reactive oxygen species-induced damage, while has also been shown to induce apoptosis (60). One mechanism that has been described for SDH-induced apoptosis involves its disassembly in the low pH environment of distressed cells that results in excessive production of reactive oxygen species from the SDHA (60). SDH inhibitors exhibit potent and selective anti-proliferative activity in multiple cancer cell lines, acting to inhibit mitochondrial electron transport (1,61). Thus, SDH is considered a target for novel anticancer therapies (62). On the other hand, mutations in the structural subunits of the SDH are implicated in a variety of human diseases. Great number of tumors including head and neck paragangliomas, pheochromocytomas, gastrointestinal stromal tumors (GIST), thyroid cancer, renal tumors, and even neuroblastoma could arise from SDH mutations (63-66). Herein, it was evidenced that 2-ME, at both physiologically and pharmacologically relevant concentrations, decreased the expression of SDHA in osteosarcoma cell death model. Estrogen has been previously shown to cause multiple effects at the level of SDH, including the inhibition of electron transfer, maintenance of mitochondrial membrane potential and an increase in SDH activity (41), whereas no study has reported the effects of 2-ME. However, inhibition of mitochondrial respiration in both intact cells and submitochondrial particles due to the inhibition of mitochondrial Complex I by 2-ME was proposed as one mechanisms of its anticancer action (67).
Despite the dual activity of SDH in cancer formation and/or anticancer effects, mutations in SDHA gene are also associated with Leigh syndrome, neuronal cell death and/or some form of progressive neurodegenerative disease (59,63). The association of SDHA deficiency with neurodegeneration may explain the plausible neurotoxic effects of 2-ME previously observed by us in hippocampal experimental model (24,25,68). Importantly, 2-ME, at both pharmacologically and physiologically relevant concentrations, may down-regulate the expression of SDHA and reverse the effect of L-lactate. It has been also demonstrated that L-lactate slightly increased the expression of mitochondrial SDH, which may represent one of the mechanisms of L-lactate that result in increased mitochondrial phosphorylation, metabolism, and ultimately enhanced tumor cell growth and metastatic potential. We also hypothesize that 2-ME may also modulate SDHA activity via its impact on SIRT3 activity, since it is the major deacetylase that controls the SDHA acetylation level (56,60). Reversible acetylation in mouse SDHA has been shown to attenuate its catalytic activity. Regulation of SDH subunits and electron transport from succinate via 2-ME may be of great interest regarding the anticancer and plausible neurotoxic potential of 2-ME and needs to be further elucidated.
Conclusion
The current study presented new insights into the anticancer mechanism of action of 2-ME. Using an osteosarcoma experimental model, it was demonstrated that 2-ME inhibits mitochondrial biogenesis, especially at low physiological concentrations, targeting PGC-1α, COXI, and SIRT3 as a consequence of the nuclear recruitment of nNOS. 2-ME was also evidenced to be a potent inhibitor of SIRT3 through binding to both the canonical inhibitor binding and allosteric sites. Moreover, it was established that, regardless of mitochondrial biogenesis, 2-ME, at both physiologically- and pharmacologically-relevant concentrations, down-regulates the expression of SDHA, and plansibly may regulate its activity via SIRT3. Potential physiological activity of 2-ME needs to be further elucidated. Our findings also confirmed pro-cancerogenic effects of L-lactate in osteosarcoma cells due to the induction of mitochondrial biogenesis pathway, suggesting that L-lactate level may serve as a prognostic biomarker for cancer medicine. (Figure 10)
Figure 10. New molecular mechanism of action of 2-methoxyestradiol (2-ME). 2-ME via nuclear hijacking of neuronal nitric oxide synthase (nNOS) and nitric oxide generation leads to DNA damage, inhibition of mitochondrial biogenesis, downregulation of succinate dehydrogenase complex flavoprotein subunit A, and finally, results in anticancer and plausible neurotoxic effects. NMDA, N-methyl-D-aspartate; ERβ, estrogen receptor beta.

Conflicts of Interest
The Authors declare no conflict of interest.
Acknowledgements
This research and manuscript publication were funded by Iuventus Plus programme of the Polish Ministry of Science and Higher Education No IP 2015 022074.
References
- 1.Wang H, Huwaimel B, Verma K, Miller J, Germain TM, Kinarivala N, Pappas D, Brookes P, Trippier PC. Synthesis and antineoplastic evaluation of mitochondrial complex II (Succinate dehydrogenase) inhibitors derived from atpenin A5. ChemMedChem. 2017;12(13):1033–1044. doi: 10.1002/cmdc.201700196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palikaras K, Daskalaki I, Markaki M, Tavernarakis N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol Ther. 2017;178:157–174. doi: 10.1016/j.pharmthera.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Palikaras K, Tavernarakis N. Mitochondrial homeostasis: the interplay between mitophagy and mitochondrial biogenesis. Exp Gerontol. 2014;56:182–188. doi: 10.1016/j.exger.2014.01.021. [DOI] [PubMed] [Google Scholar]
- 4.Kim HK, Noh YH, Nilius B, Ko KS, Rhee BD, Kim N, Han J. Current and upcoming mitochondrial targets for cancer therapy. Semin Cancer Biol. 2017;47:154–167. doi: 10.1016/j.semcancer.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 5.Grimm A, Eckert A. Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem. 2017;143(4):418–431. doi: 10.1111/jnc.14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonuccelli G, Avnet S, Grisendi G, Salerno M, Granchi D, Dominici M, Kusuzaki K, Baldini N. Role of mesenchymal stem cells in osteosarcoma and metabolic reprogramming of tumor cells. Oncotarget. 2014;5(17):7575–7588. doi: 10.18632/oncotarget.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, Flomenberg N, Howell A, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Ketones and lactate "fuel" tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010;9(17):3506–3514. doi: 10.4161/cc.9.17.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim B, Suh DH, Song YS. Anticancer strategy targeting mitochondrial biogenesis in ovarian cancer. J Cancer Sci Ther. 2014;6:422–428. [Google Scholar]
- 9.Tengan CH, Moraes CT. NO control of mitochondrial function in normal and transformed cells. Biochim Biophys Acta. 2017;1858(8):573–581. doi: 10.1016/j.bbabio.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez-Outschoorn UE, Pavlides S, Sotgia F, Lisanti MP. Mitochondrial biogenesis drives tumor cell proliferation. Am J Pathol. 2014;178(5):1949–1952. doi: 10.1016/j.ajpath.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bera S, Ray M. The transcriptional cascade associated with creatine kinase down-regulation and mitochondrial biogenesis in mice sarcoma. Cell Mol Biol Lett. 2009;14(3):481–496. doi: 10.2478/s11658-009-0014-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo C, Widlund HR, Puigserver P. PGC-1 coactivators: shepherding the mitochondrial biogenesis of tumors. Trends Cancer. 2016;2(10):619–631. doi: 10.1016/j.trecan.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, Asara JM, Kalluri R. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014;16(10):992–1003. doi: 10.1038/ncb3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aquilano K, Baldelli S, Ciriolo MR. Nuclear recruitment of neuronal nitric-oxide synthase by α-syntrophin is crucial for the induction of mitochondrial biogenesis. J Biol Chem. 2013;289(1):365–378. doi: 10.1074/jbc.M113.506733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borniquel S, Valle I, Cadenas S, Lamas S, Monsalve M. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1alpha. FASEB J. 2006;20(11):1889–1891. doi: 10.1096/fj.05-5189fje. [DOI] [PubMed] [Google Scholar]
- 16.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299(5608):896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 17.Raharijaona M, Le Pennec S, Poirier J, Mirebeau-Prunier D, Rouxel C, Jacques C, Fontaine JF, Malthiery Y, Houlgatte R, Savagner F. PGC-1-related coactivator modulates mitochondrial-nuclear crosstalk through endogenous nitric oxide in a cellular model of oncocytic thyroid tumours. PLoS One. 2009;4(11):e7964. doi: 10.1371/journal.pone.0007964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorska M, Kuban-Jankowska A, Zmijewski M, Marino Gammazza A, Cappello F, Wnuk M, Gorzynik M, Rzeszutek I, Daca A, Lewinska A, Wozniak M. DNA strand breaks induced by nuclear hijacking of neuronal NOS as an anti-cancer effect of 2-methoxyestradiol. Oncotarget. 2015;6(17):15449–15463. doi: 10.18632/oncotarget.3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruce J Y, Eickhoff J, Pili R, Logan T, Carducci M, Arnott J, Liu G. A phase II study of 2-methoxyestradiol nanocrystal colloidal dispersion alone and in combination with sunitinib malate in patients with metastatic renal cell carcinoma progressing on sunitinibmalate. Invest New Drugs. 2010;30(2):794–802. doi: 10.1007/s10637-010-9618-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrison MR, Hahn NM, Pili R, Oh W K, Hammers H, Sweeney C, Liu G. A phase II study of 2-methoxyestradiol (2ME2) NanoCrystalA (R) dispersion (NCD) in patients with taxane- refractory, metastatic castrate-resistant prostate cancer (CRPC) Invest New Drugs. 2011;29(6):1465–1474. doi: 10.1007/s10637-010-9455-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kulke MH, Chan JA, Meyerhardt JA, Zhu AX, Abrams TA, Blaszkowsky LS, Regan E, Sidor C, Fuchs CS. A prospective phase II study of 2-methoxyestradiol administered in combination with bevacizumab in patients with metastatic carcinoid tumors. Cancer Chemother Pharmacol. 2011;68(2):293–300. doi: 10.1007/s00280-010-1478-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matei D, Schilder J, Sutton G, Perkins S, Breen T, Quon C, Sidor C. Activity of 2 methoxyestradiol (Panzem (R) NCD) in advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis: A Hoosier Oncology Group trial. Gynecol Oncol. 2009;115(1):90–96. doi: 10.1016/j.ygyno.2009.05.042. [DOI] [PubMed] [Google Scholar]
- 23.Vijayanathan V, Venkiteswaran S, Nair SK, Verma A, Thomas TJ, Zhu BT, Thomas T. Physiologic levels of 2-methoxyestradiol interfere with nongenomic signaling of 17 beta estradiol in human breast cancer cells. Clin Cancer Res. 2006;12(7Pt1):2038–2048. doi: 10.1158/1078-0432.CCR-05-2172. [DOI] [PubMed] [Google Scholar]
- 24.Gorska M, Kuban-Jankowska A, Slawek J, Wozniak M. New insight into 2-methoxyestradiol- a possible physiological link between neurodegeneration and cancer cell death. Curr Med Chem. 2016;23(15):1513–1527. doi: 10.2174/0929867323666160316123443. [DOI] [PubMed] [Google Scholar]
- 25.Gorska M, Kuban-Jankowska A, Zmijewski MA, Gorzynik M, Szkatula M, Wozniak M. Neuronal nitric oxide synthase induction in the antitumorigenic and neurotoxic effects of 2-methoxyestradiol. Molecules. 2014;19(9):13267–13281. doi: 10.3390/molecules190913267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorska-Ponikowska M, Perricone U, Kuban-Jankowska A, Lo Bosco G, Barone G. 2-methoxyestradiol impacts on amino acids-mediated metabolic reprogramming in osteosarcoma cells by interaction with NMDA receptor. J Cell Physiol. 2017;232(11):3030–3049. doi: 10.1002/jcp.25888. [DOI] [PubMed] [Google Scholar]
- 27.Fujii H, Honoki K, Tsujiuchi T, Kido A, Yoshitani K, Takakura Y. Growth inhibition and induction of apoptosis by 2-methoxyestradiol in rat osteosarcoma and malignant fibrous histiocytoma cell lines. In Vivo. 2008;22(1):21–25. [PubMed] [Google Scholar]
- 28.Maran A, Shogren KL, Benedikt M, Sarkar G, Turner RT, Yaszemski MJ. 2-methoxyestradiol-induced cell death in osteosarcoma cells is preceded by cel cycle arrest. J Cell Biochem . 2008;104(5):1937–1945. doi: 10.1002/jcb.21758. Erratum in: J Cell Biochem 105(4): 1146, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gorska-Ponikowska M, Kuban-Jankowska A, Daca A, Nussberger S. 2 methoxyestradiol reverses the pro-carcinogenic effect of L-lactate in osteosarcoma 143B Cells. Cancer Genomics Proteomics. 2017;14(6):483–493. doi: 10.21873/cgp.20058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bagati A, Koch Z, Bofinger D, Goli H, Weiss LS, Dau R, Thomas M, Zucker SN. A modified in vitro invasion assay to determine the potential role of hormones, cytokines and/or growth factors in mediating cancer cell invasion. J Vis Exp. 2015;98 doi: 10.3791/51480. doi: 10.3791/51480. [E-pub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agnello M, Morici G, Rinaldi AM. A method for measuring mitochondrial mass and activity. Cytotechnology. 2008;56(3):145–149. doi: 10.1007/s10616-008-9143-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cereto-Massagu A, Ojeda MJ, Joosten RP, Valls C, Mulero M, Salvado MJ, Arola-Arnal A, Arola L, Garcia-Vallvé S, Pujadas G. The good, the bad and the dubious: VHELIBS, a validation helper for ligands and binding sites. J Cheminform. 2013;5(1):36. doi: 10.1186/1758-2946-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 34.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determi,ning protein side-chain conformations. J Mol Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 35.Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55(2):351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 36.Olsson MH, SØndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 37.Søndergaard CR, Olsson MHM, Rostkowski M, Jensen J H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J Chem Theory Comput. 2011;7:2284–2295. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 38.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 39.Li J, Abel R, Zhu K, Cao Y, Zhao S, Friesner RA. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins Struct Funct Bioinforma. 2011;79(10):2794–2812. doi: 10.1002/prot.23106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J Chem Theory Comput. 2016;12(1):281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- 41.Felty Q, Roy D. Estrogen, mitochondria, and growth of cancer and non-cancer cells. J Carcinog. 2005;4:1. doi: 10.1186/1477-3163-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen B, Wang J, Huang Y, Zheng W. Human SIRT3 tripeptidic inhibitors containing Nε-thioacetyl-lysine. Bioorg Med Chem Lett. 2015;25:3481–3487. doi: 10.1016/j.bmcl.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 43.Chen Y, Fu LL, Wen X, Wang XY, Liu J, Cheng Y, Huang J. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014;5:e1047. doi: 10.1038/cddis.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Disch JS, Evindar G, Chiu CH, Blum CA, Dai H, Jin L, Schuman E, Lind KE, Belyanskaya SL, Deng J, Coppo F, Aquilani L, Graybill TL, Cuozzo JW, Lavu S, Mao C, Vlasuk GP, Perni RB. Discovery of thieno(3,2-d )pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3. J Med Chem. 2013;56:3666–3679. doi: 10.1021/jm400204k. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen GTT, Gertz M, Steegborn C. Crystal structures of Sirt3 complexes with 4’-bromo-resveratrol reveal binding sites and inhibition mechanism. Chem Biol. 2013;20:1375–1385. doi: 10.1016/j.chembiol.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 46.Patel K, Sherrill J, Mrksich M, Scholle MD. Discovery of SIRT3 inhibitors using SAMDI mass spectrometry. J Biomol Screen. 2015;20(7):842–848. doi: 10.1177/1087057115588512. [DOI] [PubMed] [Google Scholar]
- 47.Karbowski M, Spodnik JH, Teranishi M, Wozniak M, Nishizawa Y, Usukura J, Wakabayashi T. Opposite effects of microtubule-stabilizing and microtubule-destabilizing drugs on biogenesis of mitochondria in mammalian cells. J Cell Sci. 2001;114:281–291. doi: 10.1242/jcs.114.2.281. [DOI] [PubMed] [Google Scholar]
- 48.Villena JA. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015;282(4):647–672. doi: 10.1111/febs.13175. [DOI] [PubMed] [Google Scholar]
- 49.Jones AW, Yao Z, Vicencio JM, Karkucinska-Wieckowska A, Szabadkai G. PGC-1 family coactivators and cell fate: roles in cancer, neurodegeneration, cardiovascular disease and retrograde mitochondria-nucleus signalling. Mitochondrion. 2012;12(1):86–99. doi: 10.1016/j.mito.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 50.Parihar P, Solanki I, Mansuri ML, Parihar MS. Mitochondrial Sirtuins: emerging roles in metabolic regulations, energy homeostasis and diseases. Exp Gerontol. 2015;61:130–141. doi: 10.1016/j.exger.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 51.Brenmoehl J, Hoeflich A. Dual control of mitochondrial biogenesis by Sirtuin 1 and Sirtuin 3. Mitochondrion. 2013;13(6):755–761. doi: 10.1016/j.mito.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 52.Berman HM. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaillard T, Panel N, Simonson T. Protein side chain conformation predictions with an MMGBSA energy function. Proteins. 2016;84:803–819. doi: 10.1002/prot.25030. [DOI] [PubMed] [Google Scholar]
- 54.Ylilauri M, Pentikäinen OT. MMGBSA as a tool to understand the binding affinities of filamin-peptide interactions. J Chem Inf Model. 2013;53:2626–2633. doi: 10.1021/ci4002475. [DOI] [PubMed] [Google Scholar]
- 55.Suenaga A, Okimoto N, Hirano Y, Fukui K. An efficient computational method for calculating ligand binding affinities. PLoS One. 2012;8:e42846. doi: 10.1371/journal.pone.0042846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alhazzazi TY, Kamarajan P, Xu Y, Ai T, Chen L, Verdin E, Kapila YL. A novel Sirtuin-3 inhibitor, LC-0296, inhibits cell survival and proliferation,and promotes apoptosis of head and neck cancer cells. Anticancer Res. 2016;36(1):49–60. [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang L, Ren X, Cheng Y, Huber-Keener K, Liu X, Zhang Y, Yuan YS, Yang JW, Liu CG, Yang JM. Identification of Sirtuin 3, a mitochondrial protein deacetylase, as a new contributor to tamoxifen resistance in breast cancer cells. Biochem Pharmacol. 2013;86(6):726–733. doi: 10.1016/j.bcp.2013.06.032. [DOI] [PubMed] [Google Scholar]
- 58.Gorska M, Wyszkowska RM, Kuban-Jankowska A, Wozniak M. Impact of Apparent Antagonism of Estrogen Receptor β by Fulvestrant on Anticancer Activity of 2-Methoxyestradiol. Anticancer Res. 2016;36(5):2217–2226. [PubMed] [Google Scholar]
- 59.Kluckova K, Bezawork-Geleta A, Rohlena J, Dong L, Neuzil J. Mitochondrial Complex II, a novel target for anti-cancer agents. Biochim Biophys Acta. 2013;1827(5):552–564. doi: 10.1016/j.bbabio.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 60.Pfleger J, He M, Abdellatif M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015;6(7):e1835. doi: 10.1038/cddis.2015.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo L, Shestov AA, Worth AJ, Nath K, Nelson DS, Leeper DB, Glickson JD, Blair IA. Inhibition of mitochondrial Complex II by the anticancer agent lonidamine. J Biol Chem. 2016;291(1):42–57. doi: 10.1074/jbc.M115.697516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bezawork-Geleta A, Rohlena J, Dong L, Pacak K, Neuzil J. Mitochondrial Complex II: At the crossroads. Trends Biochem Sci. 2017;42(4):312–325. doi: 10.1016/j.tibs.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rutter J, Winge DR, Schiffman JD. Succinate dehydrogenase - Assembly,regulation and role in human disease. Mitochondrion. 2010;10(4):393–401. doi: 10.1016/j.mito.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou W, Liotta LA, Petricoin EF. The Warburg effect and mass spectrometry-based proteomic analysis. Cancer Genomics Proteomics. 2017;14(4):211–218. doi: 10.21873/cgp.20032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Braun S, Riemann K, Kupka S, Leistenschneider P, Sotlar K, Schmid H, Blin N. Active succinate dehydrogenase (SDH) and lack of SDHD mutations in sporadic paragangliomas. Anticancer Res. 2005;25(4):2809–2814. [PubMed] [Google Scholar]
- 66.Santi R, Rapizzi E, Canu L, Ercolino T, Baroni G, Fucci R, Costa G, Mannelli M, Nesi G. Potential pitfalls of SDH immunohistochemical detection in paragangliomas and phaeochromocytomas harbouring germline SDHx gene mutation. Anticancer Res. 2017;37(2):805–812. doi: 10.21873/anticanres.11381. [DOI] [PubMed] [Google Scholar]
- 67.Hagen T, D’Amico G, Quintero M, Palacios-Callender M, Hollis V, Lam F, Moncada S. Inhibition of mitochondrial respiration by the anticancer agent 2-methoxyestradiol. Biochem Biophys Res Commun. 2004;322(3):923–929. doi: 10.1016/j.bbrc.2004.07.204. [DOI] [PubMed] [Google Scholar]
- 68.Gorska M, Zmijewski MA, Kuban-Jankowska A, Wnuk M, Rzeszutek I, Wozniak M. Neuronal nitric oxide synthase-mediated genotoxicity of 2-methoxyestradiol in hippocampal HT22 cell line. Mol Neurobiol. 2016;53(7):5030–5040. doi: 10.1007/s12035-015-9434-5. [DOI] [PubMed] [Google Scholar]