

Abstract
Machine learning with maximization (support) of separating margin (vector), called support vector machine (SVM) learning, is a powerful classification tool that has been used for cancer genomic classification or subtyping. Today, as advancements in high-throughput technologies lead to production of large amounts of genomic and epigenomic data, the classification feature of SVMs is expanding its use in cancer genomics, leading to the discovery of new biomarkers, new drug targets, and a better understanding of cancer driver genes. Herein we reviewed the recent progress of SVMs in cancer genomic studies. We intend to comprehend the strength of the SVM learning and its future perspective in cancer genomic applications.
Keywords: Machine learning (ML), support vector machine (SVM), classifier, genomics, kernel function, gene expression, cancer classification, gene selection, biomarker discovery, drug discovery, driver gene, gene-gene interaction, review
Machine learning (ML) “learns” a model from past data in order to predict future data (1). The key process is the learning which is one of the artificial intelligences. Many different statistical, probabilistic, and optimization techniques can be implemented as the learning methods such as the logistic regression, artificial neural networks (ANN), K-nearest neighbor (KNN), decision trees (DT) and Naive Bayes. There are two main types of ML learning - supervised learning and unsupervised learning. The supervised learning builds a model by learning from known classes (labeled training data). In contrast, unsupervised learning methods learn the common features from unknown class data (unlabeled training data).
ML algorithms have been used for key feature training and recognition and for group classification. The strength of ML methods is it could detect hard-to-discern patterns from large, noisy or complex data sets. This capability is particularly well-suited to complex genomic data, especially in cancer studies. For example, ANN and DT have been used in cancer detection and diagnosis for nearly 20 years (2-3). The clinical implication of cancer heterogeneity and various cancer genomic data available motivate the applications of ML for cancer classification using genomic data.
SVM learning is one of many ML methods. Compared to the other ML methods SVM is very powerful at recognizing subtle patterns in complex datasets (4). SVM can be used to recognize handwriting, recognize fraudulent credit cards, identify a speaker, as well as detect face (5). Cancer is a genetic disease where the genomic feature patterns or feature function patterns may represent the cancer subtypes, the outcome prognosis, drug benefit prediction, tumorigenesis drivers, or a tumor-specific biological process. Therefore, the Artificial Intelligence of SVM can help us in recognizing these patterns in a variety of applications.
SVM Model
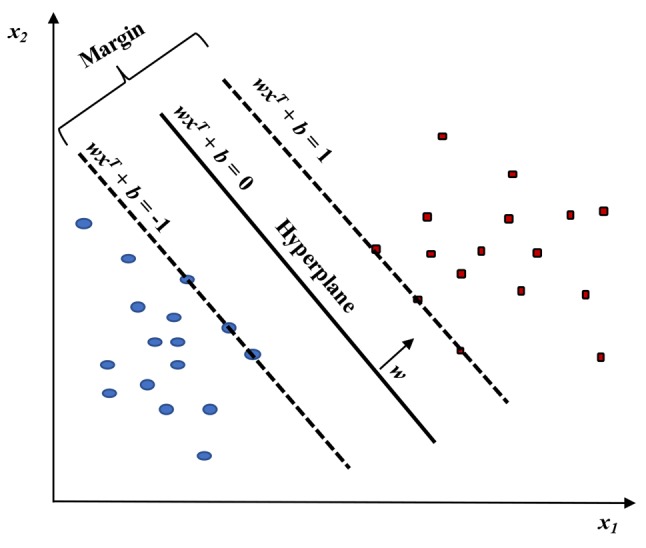
SVM is a powerful method for building a classifier. It aims to create a decision boundary between two classes that enables the prediction of labels from one or more feature vectors (6). This decision boundary, known as the hyperplane, is orientated in such a way that it is as far as possible from the closest data points from each of the classes. These closest points are called support vectors.
Given a labeled training dataset:
(x1, y1), ..., (xn, yn), xi ∈ Rd and yi ∈ (−1, +1)
where xi is a feature vector representation and yi the class label (negative or positive) of a training compound i.
The optimal hyperplane can then be defined as:
wxT + b=0
where w is the weight vector, x is the input feature vector, and b is the bias.
The w and b would satisfy the following inequalities for all elements of the training set:
wxiT + b ≥ +1 if yi=1
wxiT + b ≤ −1 if yi=–1
The objective of training an SVM model is to find the w and b so that the hyperplane separates the data and maximizes the margin 1 / || w ||2.
Vectors xi for which |yi| (wxiT + b)= 1 will be termed support vector (Figure 1).
Figure 1. Linear SVM model. Two classes (red versus blue) were classified.
The SVM algorithm was originally proposed to construct a linear classifier in 1963 by Vapnik (7). An alternative use for SVM is the kernel method, which enables us to model higher dimensional, non-linear models (8). In a non-linear problem, a kernel function could be used to add additional dimensions to the raw data and thus make it a linear problem in the resulting higher dimensional space (Figure 2). Briefly, a kernel function could help do certain calculations faster which would otherwise would need computations in high dimensional space.
Figure 2. Kernel function. Data that cannot be separated by linear SVM can be transformed and separated by a kernel function.

It is defined as:
K (x, y)=<f(x), f(y)>
Here K is the kernel function, x, y are n dimensional inputs. f is used to map the input from n dimensional to m dimensional space. <x, y> denotes the dot product. With kernel functions, we could calculate the scalar product between two data points in a higher dimensional space without explicitly calculating the mapping from the input space to the higher dimensional space. In many cases, computing the kernel is easy while going to the high dimensional space to compute the inner product of two feature vectors is hard. The feature vector for even simple kernels can blow up in size, and for kernels like the Radial Basis Function (RBF) kernel (KRBF(x, y) = exp (-γ||x - y||2), the corresponding feature vector is infinite dimensional. Yet, computing the kernel is almost trivial.
The choice of kernel function among other factors could greatly affect the performance of an SVM model. However, there is no way to figure out which kernel would do the best for a specific pattern recognition problem. The only way to choose the best kernel is through trials. We can start with a simple SVM and then experiment with a variety of ‘standard’ kernel functions. Depending on the nature of the problem, it is possible that one kernel is better than the other kernels. An optimal kernel function can be selected from a fixed set of kernels in a statistically rigorous fashion by using cross-validation.
Cancer Classification and Subtyping
SVM as a classifier has been used in cancer classification since the high throughtput microarray gene expression data was avaliable in the early 2000’s. Golub et al. (9) first tried a linear SVM to classify two different types of leukemia using gene expression microarray data. In this study, 38 patients were used as training set. A simple learning algorithm called “weighted voting” was trained to recognize the distinction between two known (labeled) forms of leukemia. The Affymetrix Hgu6800 chips covered 7,129 gene features (gene expression probes) and each of the whole gene features was weighted in contributing to the two classes. The learned SVM model was used to test another independent data of 34 patients. This study has demonstrated the superior performance of SVM in classifying high-dimensional (gene features) and low sample size data. Subsequently, Vapnik et al. (10) improved upon the accuracy of the weighted voting method of the SVM, reducing the error rate from 6% (2 errors out of 34) to 0%. But in this study no feature selection was performed before the model development.
Moler et al. (11) applied SVM in a colon cancer tissue classification using selected features. They used a collection of 40 colon cancer tumors and 22 normal colon tissues. First, a feature selection metric, the naive Bayes relevance (NBR) score, was proposed, which was based on the probability of a class given the observed value of the feature, under a Gaussian model. SVMs were then trained using only the 50-200 top-ranked genes, which had the same or better generalization performance than the full repertoire of 1, 988 genes in discriminating non-tumor from tumor specimens. In addition, the performance of the SVM using various numbers of selected genes was compared to the performance of a naive Bayes classifier using the same genes. In each case, the SVM outperformed naive Bayes. Furey et al. (12) applied linear SVMs with feature selection to three cancer data sets. The first data set consisted of 31 tissues samples, including cancerous ovarian, normal ovarian and normal non-ovarian tissue. The other two sets were the leukemia (9) and colon cancer sets (11). In this study, A feature selection metric called the signal-to-noise ratio (9), which was closely related to the Fisher criterion score used in Fisher’s linear discriminant, was used to select genes for training the classifier. Overall, the SVM provided reasonably good performance across multiple data sets, although the experiments also demonstrated that several perceptron-based algorithms performed similarly. Segal et al. (13) proposed a genome-based SVM classification scheme for clear cell sarcoma, which displays characteristics of both soft tissue sarcoma and melanoma. Firstly, 256 genes were selected by student t-test. Subsequently, a linear SVM was trained to recognize the distinction between melanoma and soft tissue sarcoma using the selected genes. In a leave-one-out setting, the classifier correctly classified 75 out of 76 examples. In another study (14), SVM was applied to investigate the complex histopathology of adult soft tissue sarcomas. A data set including 51 samples that had been classified by pathologists into nine histologic subtypes was used. The SVM could successfully recognize the four subtypes for which molecular phenotypes are already known. Among the remaining samples, a combination of SVMs and hierarchical clustering could uncover a well-separated subset of the malignant fibrous histiocytoma subtype, which is a particularly controversial subtype.
Above SVMs are binary sample classifiers. Cancer is heterogeneous and multiclass classification is needed. For example, breast cancer consists of mainly four molecular subtypes (Lumina A, Lumina B, HER2-enriched and Basal). SVM can be extended for multiclass problems using the so-called one-vs-rest approach (15). For N class problems, SVMs will be trained independently between one specific class which is seen as the positive class and the other classes will form the negative cases. Li et al. (16) compared various state-of-the-art classification methods on numerous multiclass gene expression datasets and found that the multiclass classification problem was much more difficult than the binary one for the gene expression datasets due to the fact that the data are of high dimensionality and small sample size.
Besides mRNA expression features, DNA methylation was also applied in SVM modeling for cancer classification. Methylation is a molecular modification of DNA, where a methyl group is added to the nucleotide cytosine. Methylation patterns in the upstream regions of genes are considered a major factor in gene regulation. Twenty-five patients with two forms of leukemia were classified by methylation pattern that contained measurements from 81 positions along the DNA strand (17). In this study, various feature selection methods were employed prior to model training, including principle components analysis, the signal-to-noise ratio, the Fisher criterion score, the student t-test, and a method called backward elimination. Kim (18) proposed a weighted K-means support vector machine (wKM-SVM) method for two methylation profiles of breast and kidney cancer. Level 3 DNA methylation of beta values targeting on methylated and the un-methylated probes were downloaded from The Cancer Genome Atlas (TCGA) database (https://tcga- data.nci.nih.gov/tcga/). The breast cancer set consisted of methylation data of 10,121 genes for 316 tumor samples and 27 control cases. The kidney set included methylation data of 10,121 genes for 219 tumor samples and 199 control cases. They compared wKM-SVM with other different algorithms, including classification and regression tree (CART), KNN and random forest in recognizing tumor from normal. The wKM-SVM had the best performance. Yang et al. (19) used an extension of the random forest, Boruta, to select important DNA methylation features and establish an SVM classifier for liver cancer diagnosis. Alkuhlani et al. (20) developed a multistage approach to select the optimal CpG sites from three different DNA methylation cancer datasets (breast, colon and lung). Three different filter feature selection methods (Fisher Criterion, t-test and Area Under ROC Curve) were first combined to reduce the CpG sites. The final SVM Recursive Feature Elimination (SVM-RFE) resulted in classification accuracies of 96.02, 98.81 and 94.51% for the three cohorts, respectively. SVM was also applied in the identification and validation of the methylation biomarkers of non-small cell lung cancer (NSCLC) (21).
Other data types were also used in SVM modeling. An SVM algorithm has been used to classify or diagnose multiple cancers based on a protein chip that was fabricated with twelve monoclonal antibodies to quantify the tumor markers (22). Tyanova et al. (23) used proteomics data to train an SVM model to classify breast cancer subtypes. Copy number variations (24) and single nucleotide polymorphisms (SNPs) (25) were used to train SVM classifiers for bladder, uveal cancer and breast cancer respectively. Wu et al. (26) built three SVM classification models based on the identified pathways which effectively classified different breast cancer subtypes.
New machine-learning methods have been developed to classify integrated multilayer heterogeneous genomic data (27). For example, for a given gene we might know the protein it encodes, the mRNA expression levels associated with the given gene for hundreds of patients, the occurrences of known or inferred transcription factor binding sites in the upstream region of that gene, and the identities of many of the proteins that interact with the given gene’s protein product. Each of these distinct data types provides one view of the molecular machinery of the cell. Thus, integrating the multi-layers of omics data could facilitate uncovering biological processes and capturing the interplay of multi-level genomic features. Several efforts have been made for multiple omics data integration in the context of SVM learning. Kim et al. (28) proposed a meta-analytic support vector machine (Meta-SVM) that can accommodate multiple omics data, making it possible to detect consensus genes associated with diseases across studies. The Meta-SVM method was applied to breast cancer expression profiles provided by TCGA including mRNA, copy number variation (CNV) and epigenetic DNA methylation. The three inter-omics features of breast cancer were aligned on identical protein coding regions. The results demonstrated that the Meta-SVM showed better performance in discovering the underlying true signals and in detecting gene sets enriched for cancer disease process validated as biologically significant.
Biomarker/Signature Discovery
Biomarkers discovery involves selecting biologically meaningful or associated gene expression, SNPs, DNA methylation, or micro-RNA from high-dimensional data and modeling scores based on the selected features to help cancer diagnosis, prognosis or treatment response (29). This process can be viewed as selecting features for classifications (cancer versus none cancer, good versus poor outcome classes, drug response versus no response classes). There are two main methods for selecting features: filter methods and wrapper methods. In filter methods, the features (i.e. genes) are selected by predetermined ranking criteria and then are fitted into cancer classifier algorithms (Figure 3). For example, genes can be ranked by correlation coefficients (9,12) and hypothesis testing statistics (30-33). The drawbacks with the gene-ranking methods are: (i) one has to specify the number of selected genes in advance and often subjectively and (ii) the selection is individual-based and hence ignores any significant gene-gene correlations that may occur in the data. Xu et al. (34) used differentially expressed genes (DEGs) and protein-protein interaction (PPI) network-based neighborhood scoring to select features and trained a SVM model of a 15-gene signature for prediction of colon cancer recurrence and prognosis. Hu et al. (35) built an SVM algorithm based on the structural risk minimization principle for the identification of thirty-eight markers involved in brain development from single-cell transcriptomic data. An SVM feature selection based on profiling of urinary RNA metabolites was applied to predict breast cancer (36). SVMs coupled with proteomics approaches were applied for detecting biomarkers predicting chemotherapy resistance in small cell lung cancer (37).
Figure 3. Feature selection methods. Two frameworks (Feature filter and wrapper) were presented.

In the wrapper methods, the gene selection and classifier modeling occur at the same time (38,39). Wrapper methods utilize the learning machine of interest as a black box to score subsets of variable according to their predictive power (Figure 3). Based on the inferences drawn from the previous modeling, gene features will be added to or removed from the current subset. These methods are usually computationally expensive. Some common examples of wrapper methods are forward feature selection, backward feature elimination, and recursive feature elimination.
One example is the SVM recursive feature elimination (SVM-RFE) proposed by Guyon et al. (39,40). The idea is that the orientation of the separating hyperplane modelled by the SVM can be used to select informative features, i.e. if the plane is orthogonal to a particular feature dimension, then that feature is informative, and vice versa. Thus, the SVM-RFE method could remove the least important features and select the most important features based on the weights of classifiers. Firstly, the SVM-RFE wrapper initializes the data set to contain all features. Then, it trains an SVM on the extended data set and applies a feature importance measure (i.e. criterion) to evaluate the importance of each feature. It ranks features in each iteration according to the criterion and constantly removes the lowest-ranked feature. Finally, the algorithm stops either when all features get confirmed or rejected.
The SVM-RFE algorithm has been tested on both the AML/ALL and the colon cancer data sets (40). In the leukemia dataset, SVM-RFE selected two genes which together yielded zero leave-one-out error. In the colon cancer dataset, SVM-RFE identified only 4 genes that yielded an accuracy of 98%. In addition, several other classification algorithms have been trained using the genes selected by SVM-RFE.
In biomarker discovery using SVM-RFE, the sampling variation may greatly influence subsequent biological validations. Abeel et al. (38) addressed this issue by introducing the ensemble concept into the original RFE method. In the ensemble SVM-RFE method, bootstrap was used to resample K times from the training data. SVM-RFE was then applied to each of the K resamples and thus K marker sets were obtained. In the final phase, the output of these separate marker selectors was aggregated and returned as the final (ensemble) result. This ensemble SVM-RFE method was tested in four microarray datasets: a Leukemia dataset with acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) tissues; a Colon Cancer dataset consisted of samples from 40 tumor and 22 normal colon tissues probed by an Affymetrix microarray chip measuring more than 6,500 genes; a lymphoma dataset from a study on diffuse large B-cell lymphoma; and a prostate dataset. The ensemble SVM-RFE was showing increases of up to almost 30% in robustness of the selected biomarkers, along with an improvement of about 15% in classification performance evaluated on the four microarray datasets compared to the original SVM-RFE. The stability improvement with ensemble methods is particularly noticeable for small signature sizes (a few dozens of genes), which is most relevant for the design of a diagnosis or prognosis model from a gene signature.
Chen et al. (41) developed a network-constrained support vector machine (netSVM) for identifying biologically network biomarkers using integration of gene expression data and protein-protein interaction data. The netSVM was tested in two breast cancer gene expression data sets to identify prognostic signatures for predicting cancer metastasis. The results showed that the network biomarkers identified by netSVM were highly enriched in biological pathways associated with breast cancer progression and helped improve the prediction performance when tested across different data sets. Specifically, many of the identified genes were related to apoptosis, cell cycle, and cell proliferation, which are hallmark signatures of breast cancer metastasis. Importantly, several novel hub genes, biologically important with many interactions in Protein-protein interaction (PPI) network but often showing little change in expression when compared to their downstream genes, were also identified as network biomarkers; the genes were enriched in signaling pathways such as the TGF-beta signaling pathway, MAPK signaling pathway, and JAK-STAT signaling pathway. These signaling pathways may provide new insight to the underlying mechanism of breast cancer metastasis.
Drug Discovery for Cancer Therapy
Drugs for a variety of deadly cancers remain limited. The major challenges in cancer drug discovery include side effects of drugs, high toxicity and drug resistance towards current anticancer drugs. The traditional drug discovery process involves an iterative procedure of finding compounds that are active against a biological target, which is time-consuming when selecting from a large collection of compounds. Experimental techniques used for drug discovery are costly and time-consuming (42). Today SVM can aid this screening process using the maximum margin hyperplanes. This hyperplane separates the active from the inactive compounds and has the largest possible distance from any labeled compound.
Selecting anticancer drugs. Warmuth et al. (43) used active learning to develop an SVM model for selecting active compounds. Instead of separating the data into training set for deriving models and testing set for validating models, the learning set was incremented and a new, further improved model was learned with each round in active learning. Gupta et al. (44) applied an SVM model to prioritize anticancer drugs against a cancer using the genomic features of cancer cells. The drug profile of 24 anticancer drugs was tested against a large number of cell lines in order to understand the relation between drug resistance and altered genomic features of a cancer cell line. Bundela et al. (45) built an SVM-RBF model to identify potential therapeutic compounds for oral cancer from the large pool of compounds from the publicly available compound databases.
Matsumoto et al. (46) used SVM for virtual screening of radiation protection function and toxicity for radioprotectors targeting p53. Radiation therapy is one of the main approaches against cancer cells, although this therapy has adverse side effects, including p53-induced apoptosis of normal tissues and cells (47). It is considered that p53 would be a target for therapeutic and mitigative radioprotection to avoid the apoptotic fate. It was found that SVM is better than other machine learning in the case that the target protein is known and we search for a compound that bond to the target protein.
Identifying novel cancer drug targets. SVMs have been used for predicting druggability scores for targets (48). The model was trained using three to six global descriptors of protein binding sites accounting for size, compactness and physicochemical properties. With these descriptors, the druggability scores could be assigned to new targets (48). The SVM methodology was also used to prioritize docking poses. Li et al. (49) used a Support Vector Machine-based scoring function in regression mode (SVR) to assess target-ligand interactions. Knowledge-based pairwise potentials derived from complex crystal structures were used to develop the scoring function. In another study, a support vector regression (SVR) algorithm was derived from a set of descriptors and was applied to predict protein-ligand binding affinities (50).
The identification of drug target proteins (DTP) plays a critical role in biometrics. Wang et al. (51) designed a novel framework to retrieve DTPs from a collected protein dataset, which represents an overwhelming task of great significance. Previously reported methodologies for this task generally employ protein-protein interactive networks but neglect informative biochemical attributes. A novel framework was formulated utilizing biochemical attributes to address this problem. In the framework, a biased support vector machine (BSVM) was combined with the deep embedded representation extracted using a deep learning model, stacked auto-encoders (SAEs). In cases of non-drug target proteins (NDTPs) contaminated by DTPs, the framework is beneficial due to the efficient representation of the SAE and relief of the imbalance effect by the BSVM. The experimental results demonstrated the effectiveness of this framework, and the generalization capability was confirmed via comparisons to other models. This study is the first to exploit a deep learning model for IDTP. In summary, nearly 23% of the NDTPs were predicted as likely DTPs, which are awaiting further verification based on biomedical experiments.
Jeon et al. (52) utilized SVM to learn five genomic features from various types of high-throughput data for the genome-wide identification of cancer therapeutic targets. These features include gene essentiality, expression level, mutation, copy number and closeness in a PPI network. The SVM was trained by known cancer targets versus non-targets, and then used for novel target discovery.
Drug/nondrug classification. Singh et al. (53) developed a hybrid method of SVM on thousands of anticancer and non-anticancer molecules tested against 60 National Cancer Institute (NCI) cancer cell lines. This highly accurate hybrid method can be used for classification of anticancer and non-anticancer molecules. Also a non-linear machine learning techniques has been used to generate robust multiomic signatures that predict cancer cellular response to 17-AAG, AZD0530, AZD6244, Erlotinib, Lapatinib, Nultin-3, Paclitaxel, PD0325901, PD0332991, PF02341066, and PLX4720 using data from the CCLE, CGP, and NCI60 databases(54).
Anticancer drug sensitivity prediction. Computational models to predict the response of cancer cell lines to drug compounds facilitate cancer therapeutics development process. Hejase et al. (55) built an ensemble SVM model to predict the sensitivity of the breast cancer cell lines to previously untested drug compounds. The ensemble SVM model extracts features from different types of data (proteomic data, gene expression, RNA-seq, DNA methylation, and DNA copy number variation) rather than using different base algorithms on a single type of data. The ensemble model based on the different types of data enhanced and improved the accuracy of the overall model.
Predicting substrates of the cancer resistance. Human breast cancer resistance protein (BCRP) is an ATP-binding cassette (ABC) efflux transporter that confers multidrug resistance in cancers and also plays an important role in the absorption, distribution and elimination of drugs. Hazai et al. (56) developed SVM models to predict wild-type BCRP substrates based on a total of 263 known BCRP substrates and non-substrates collected from literature. The final SVM model had an overall prediction accuracy of approximately 73% for an independent external validation data set of 40 compounds.
Cancer Driver Gene Discovery
Cancer is initiated by somatic mutations, called cancer driver gene mutations. However, all various cancers cannot be explained by the handful number of driver genes currently reported. The continuing decline in the cost of genome sequencing, as well as the relative ease of interpreting the effects of mutations in many proteins via methods such as activity assays has led to a sustained drive to understand the effects of cancer derived mutations on cancer progression. The challenge of finding mechanistic links between mutations and cancer progression is made even more imperative by the fact that many cancer drugs target mutations that have specific effects, as well as the observation that many clinical trials fail due to patient cohorts that are not suitable for specific therapies (57). Sequencing efforts as well as the frequent failure of targeted therapies has led to an increasingly well-recognized principle that not all mutations confer selective advantage on cancer cells. These mutations are known as passenger mutations while mutations that confer some advantage are commonly referred to as driver mutations, because they can be seen as properties of the residues from the mutations. They showed that this classifier performs driving cancer progression (58).
SVM is one of the most widely used techniques to classify mutations specific to cancer. This essentially geometric method tries to find combinations of features that are common to mutations of different classes so that mutations of unknown class (i.e. driver or passenger) can be classified (59). SVM classifiers were also trained to predict whether mutations occur across the whole genome (60) as well as in a specific class of proteins (61). These methods based on cross-validation showed high accurate predictions, high receiver operating characteristic area under the curve (AUC), or high probability of distinguishing between examples of different classes.
Jordan et al. (59) developed an SVM method to predict the activation status of kinase domain mutations in cancer and the method showed it to be reliable with an accuracy of 78% when a balanced dataset was used. This method did not need to make any decisions in advance about which mutations are driver mutations, as many recent machine learning efforts have. It was also faster than molecular dynamics (MD) to predict the effect of kinase domain mutations which are ofted used to predict. Interestingly, the ability to affect salt bridge formation was demonstrated to be an important factor in determining whether a given mutation is likely to be a driver. Tan et al. (62) developed a novel missense-mutation-related feature extraction scheme for identifying driver mutations. A total of 126 features were investigated for each missense mutation, including (i) changes in the physiochemical properties of the residues from the mutations; (ii) substitution scoring matrix (SSM) features from published sources; (iii) protein sequence-specific (PSS) features, which extract various patterns of two consecutive amino acid residues or a six-letter exchange group in a protein sequence; and (iv) other annotated features derived from the UniProt KnowledgeBase, Swiss Prot variant page and COSMIC database. A classifier was derived based on the features using SVM. The classifier was then tested on a new data set using n-fold cross-validation. From the 126 candidate features, they were able to identify the top 70 features that were best able to discriminate between driver and passenger mutations. Most (61 of 70) of the top 70 features consisted of the SSM and PSS features rather than simple changes in the physiochemical better than the previous methods by comparing their ability (in terms of ROC and prediction precision) to identify 117 EGFR and 1029 TP53 missense mutations (58). Capriotti et al. (63) trained an SVM classifier on a set of 3163 cancer-causing variants and an equal number of neutral polymorphisms. The individual variants identified could be indicators of cancer risk. The method achieved 93% overall accuracy, a correlation coefficient of 0.86, and area under ROC curve of 0.98.
Bari et al. (64) built SVM models to uncover a new class of cancer-related genes that are neither mutated nor differentially expressed. This SVM-Assisted Network Inference (MALANI) algorithm assesses all genes regardless of expression or mutational status in the context of cancer etiology. 8807 expression arrays corresponding to 9 cancer types were used to build more than 2×108 SVM models for reconstructing a cancer network. Approximately 3% of ~19,000 not differentially expressed genes are the new class of cancer gene candidates.
Cancer Gene/Protein Interaction and Networks
Cancer is a complex disease of impacted biological processes with multiple genes or factors. Modeling the gene-gene interaction helps understand the underlying biological mechanisms. Traditional statistical tools are not appropriate for analyzing large-scale genetic data. However, it appears some of the computational limitations of detecting gene-gene interactions can be overcome using modern techniques, such as machine learning and data mining. The problem of detecting interactions among multiple genes can be considered as a combinatorial optimization problem: finding the best combination of gene features from a given dataset which can produce the highest prediction accuracy.
A few early studies (40,65,66) have shown that SVMs are promising predictors for the detection of gene-gene interactions. Later an applicable computational SVM framework for detecting gene-gene interactions was described (67). SVM and combinatorial optimization techniques (local search and genetic algorithm) were tailored to fit within this framework. Although the proposed approach is computationally expensive, the results indicate this is a promising tool for the identification and characterization of high order gene-gene and gene-environment interactions. On one hand, several advantages of this method, including the strong power for classification, less concern for overfitting, and the ability to handle unbalanced data and achieve more stable models, have been demonstrated. On the other hand, this method was computationally expensive.
In a study conducted by Guo et al. (68), SVM model derived from the primary sequences of proteins was used for predicting PPIs. The SVM model was developed with the aid of auto covariance (AC). AC was used to cover the information of interactions between amino acid residues a certain distance apart in the sequence. Thus, the neighboring effect was taken into consideration in this method. The AC and SVM combined method showed a very promising prediction result when performed on the yeast Saccharomyces cerevisiae PPI data. This method achieved an accuracy of 88.09% another independent data set of 11474 yeast PPIs. The superiority of this method over the existing sequence-based methods will make it useful for the study of protein networks. Chai et al. (69) built a new Net-SVM model which selected fewer but more relevant genes. This Net-SVM can be used to construct simple and informative PPI networks that are highly relevant to cancer.
Perspective
Cancer genomic data are high-dimensional, heterogeneous and noisy. The application of SVM learning in cancer genomics is a popular and successful undertaking (Table I). The appeal of SVM approach is due in part to the power of the SVM algorithm, and in part to the flexibility of the kernel approach to representing data. If the parameters C and r are appropriately chosen, SVMs can be robust, even when the training sample has some bias.
Table I. Summary of typical applications of SVM in cancer genomics.

*SVM-RFE: SVM recursive feature elimination; RBF-SVM: radial basis function SVM; netSVM: network-constrained SVM; Reg-SVM: regression SVM; CNV: copy number variation; SNP: single nucleotide polymorphism; COSMIC: the catalogue of somatic mutations in cancer; BCRP: human breast cancer resistance protein.
Although SVMs with non-linear kernels are extremely powerful classifiers, they do have some downsides as following: 1). Finding the best model requires testing of various combinations of kernels and model parameters; 2). It can be slow to train, particularly if the input dataset has a large number of features or examples; 3). Their inner workings can be difficult to understand because the underlying models are based on complex mathematical systems and the results are difficult to interpret. The success or failure of machine learning approaches on a given problem may vary strongly with the expertise of the user. Of special concern with supervised applications is that all steps involved in the classifier design (selection of input variables, model training, etc.) should be cross-validated to obtain an unbiased estimate for classifier accuracy. For instance, selecting the features using all available data and subsequently cross-validating the classifier training will produce an optimistically biased error estimate (70).
The cancer genomic and epigenomic data are exponentially increased as the new generation of sequencing technologies advances. The challenge in analyzing these large complex data motivates us to use artificial intelligent approaches. Developing new kernel functions will aid to discover new targets and new target drugs for various cancers, especially for those deadly and heterogeneous cancers, such as triple-negative breast cancers (TNBCs), soft tissue sarcomas (STS), etc.
Acknowledgements
This study has been supported by University of Manitoba Faculty of Science Interdisciplinary/New Directions Research Collaboration Initiation Grant, and partially by the Canadian Breast Cancer Foundation, the Research Institute of Oncology and Hematology Summer student research fund, and CancerCare Manitoba Foundation (CCMF).
References
- 1.Cruz JA, Wishart DS. Applications of machine learning in cancer prediction and prognosis. Cancer Inform. 2006;2:59–77. [PMC free article] [PubMed] [Google Scholar]
- 2.Cicchetti D. Neural networks and diagnosis in the clinical laboratory: state of the art. Clin Chem. 1992;38(1):9–10. [PubMed] [Google Scholar]
- 3.Simes RJ. Treatment selection for cancer patients: application of statistical decision theory to the treatment of advanced ovarian cancer. J Chronic Dis. 1985;38(2):171–186. doi: 10.1016/0021-9681(85)90090-6. [DOI] [PubMed] [Google Scholar]
- 4.Aruna S, Rajagopalan SP. A novel SVM based CSSFFS feature selection algorithm for detecting breast cancer. Int J Comput Appl. 2011;31(8):14–20. [Google Scholar]
- 5.Noble W. Support vector machine applications in computational biology. In: Kernel methods in computational biology. Schölkopf B, Tsuda K and Vert JP (eds.) Cambridge, MA, MIT Press. 2004;In:71–92. [Google Scholar]
- 6.Noble WS. What is a support vector machine. Nat biotechnol. 2006;24(12):1565–1557. doi: 10.1038/nbt1206-1565. [DOI] [PubMed] [Google Scholar]
- 7.Vapnik V. Pattern recognition using generalized portrait method. Autom Remote Control. 1963;24:774–780. [Google Scholar]
- 8.Aizerman MA, Braverman EM, Rozoner LI. Theoretical foundations of the potential function method in pattern recognition learning. Autom Remote Control. 1964;25:821–837. [Google Scholar]
- 9.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286(5439):531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 10.Vapnik V, Mukherjee S. Support VectorMachine for Multivariate Density Estimation Advances in Neural Information Processing Systems. Leen T, Solla S and Muller KR (eds.) Cambridge, MA, MIT Press. 2000:659–665. [Google Scholar]
- 11.Moler E, Chow M, Mian I. Analysis of molecular profile data using generative and discriminative methods. Physiol Genomics. 2000;4(2):109–126. doi: 10.1152/physiolgenomics.2000.4.2.109. [DOI] [PubMed] [Google Scholar]
- 12.Furey TS, Cristianini N, Duffy N, Bednarski DW, Schummer M, Haussler D. Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics. 2000;16(10):906–914. doi: 10.1093/bioinformatics/16.10.906. [DOI] [PubMed] [Google Scholar]
- 13.Segal NH, Pavlidis P, Noble WS, Antonescu CR, Viale A, Wesley UV, Busam K, Gallardo H, DeSantis D, Brennan MF. Classification of clear cell sarcoma as melanoma of soft parts by genomic profiling. J Clin Oncol. 2003;21:1775–1781. doi: 10.1200/JCO.2003.10.108. [DOI] [PubMed] [Google Scholar]
- 14.Segal NH, Pavlidis P, Antonescu CR, Maki RG, Noble WS, DeSantis D, Woodruff JM, Lewis JJ, Brennan MF, Houghton AN. Classification and subtype prediction of adult soft tissue sarcoma by functional genomics. Am J Pathol. 2003;163(2):691–700. doi: 10.1016/S0002-9440(10)63696-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang Y. Deep learning using linear support vector machines. arXiv preprint 1306.0239. 2013 [Google Scholar]
- 16.Li T, Zhang C, Ogihara M. A comparative study of feature selection and multiclass classification methods for tissue classification based on gene expression. Bioinformatics. 2004;20(15):2429–2437. doi: 10.1093/bioinformatics/bth267. [DOI] [PubMed] [Google Scholar]
- 17.Model F, Adorjan P, Olek A, Piepenbrock C. Feature selection for DNA methylation based cancer classification. Bioinformatics. 2001;17(suppl 1):S157–164. doi: 10.1093/bioinformatics/17.suppl_1.s157. [DOI] [PubMed] [Google Scholar]
- 18.Kim S. Weighted K-means support vector machine for cancer prediction. Springerplus. 2016;5(1):1162. doi: 10.1186/s40064-016-2677-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Z, Jin M, Zhang Z, Lu J, Hao K. Classification based on feature extraction for hepatocellular carcinoma diagnosis using high-throughput dna methylation sequencing data. Procedia Comput Sci. 2017;107:412–417. [Google Scholar]
- 20.Alkuhlani A, Nassef M, Farag I. Multistage feature selection approach for high-dimensional cancer data. Soft Comput. 2017;21:6895–6906. [Google Scholar]
- 21.Guo S, Yan F, Xu J, Bao Y, Zhu J, Wang X, Wu J, Li Y, Pu W, Liu Y, Jiang Z, Ma Y, Chen X, Xiong M, Jin L, Wang J. Identification and validation of the methylation biomarkers of non-small cell lung cancer (NSCLC) Clin Epigenetics. 2015;7:3. doi: 10.1186/s13148-014-0035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Z, Fu X, Zhang L, Yang X, Liu F, Hu G. A protein chip system for parallel analysis of multi-tumor markers and its application in cancer detection. Anticancer Res. 2004;24:1159–1165. [PubMed] [Google Scholar]
- 23.Tyanova S, Albrechtsen R, Kronqvist P, Cox J, Mann M, Geiger T. Proteomic maps of breast cancer subtypes. Nat Commun. 2016;7:10259. doi: 10.1038/ncomms10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rapaport F, Barillot E, Vert JP. Classification of arrayCGH data using fused SVM. Bioinformatics. 2008;24(13):i375–i382. doi: 10.1093/bioinformatics/btn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vura S, Wang X, Guda C. Classification of breast cancer patients using somatic mutation profiles and machine learning approaches. BMC Syst Biol. 2016;10(suppl 3):62. doi: 10.1186/s12918-016-0306-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu T, Wang Y, Jiang R, Lu X, Tian J. A pathways-based prediction model for classifying breast cancer subtypes. Oncotarget. 2017;8(35):58809–58822. doi: 10.18632/oncotarget.18544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin E, Lane HY. Machine learning and systems genomics approaches for multi-omics data. Biomark Res. 2017;5(1):2. doi: 10.1186/s40364-017-0082-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim S, Jhong JH, Lee J, Koo JY. Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 2017;10(1):2. doi: 10.1186/s13040-017-0126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yiu AJ, Yiu CY. Biomarkers in colorectal cancer. Anticancer Res. 2016;36(3):1093–1102. [PubMed] [Google Scholar]
- 30.He W. A spline function approach for detecting differentially expressed genes in microarray data analysis. Bioinformatics. 2004;20(17):2954–2963. doi: 10.1093/bioinformatics/bth339. [DOI] [PubMed] [Google Scholar]
- 31.Thomas JG, Olson JM, Tapscott SJ, Zhao LP. An efficient and robust statistical modeling approach to discover differentially expressed genes using genomic expression profiles. Genome Res. 2001;11(7):1227–1236. doi: 10.1101/gr.165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan W. A comparative review of statistical methods for discovering differentially expressed genes in replicated microarray experiments. Bioinformatics. 2002;18(4):546–554. doi: 10.1093/bioinformatics/18.4.546. [DOI] [PubMed] [Google Scholar]
- 33.Troyanskaya OG, Garber ME, Brown PO, Botstein D, Altman RB. Nonparametric methods for identifying differentially expressed genes in microarray data. Bioinformatics. 2002;18(11):1454–1461. doi: 10.1093/bioinformatics/18.11.1454. [DOI] [PubMed] [Google Scholar]
- 34.Xu G, Zhang M, Zhu H, Xu J. A 15-gene signature for prediction of colon cancer recurrence and prognosis based on SVM. Gene. 2017;604:33–40. doi: 10.1016/j.gene.2016.12.016. [DOI] [PubMed] [Google Scholar]
- 35.Hu Y, Hase T, Li HP, Prabhakar S, Kitano H, Ng SK, Ghosh S, Wee LJ. A machine learning approach for the identification of key markers involved in brain development from single-cell transcriptomic data. BMC Genomics . 2016;17(suppl 13):1025. doi: 10.1186/s12864-016-3317-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henneges C, Bullinger D, Fux R, Friese N, Seeger H, Neubauer H, Laufer S, Gleiter CH, Schwab M, Zell A, Kammerer B. Prediction of breast cancer by profiling of urinary RNA metabolites using Support Vector Machine-based feature selection. BMC Cancer. 2009;9:104. doi: 10.1186/1471-2407-9-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han M, Dai J, Zhang Y, Lin Q, Jiang M, Xu X, Liu Q, Jia J. Support vector machines coupled with proteomics approaches for detecting biomarkers predicting chemotherapy resistance in small cell lung cancer. Oncol Rep. 2012;28(6):2233–2238. doi: 10.3892/or.2012.2037. [DOI] [PubMed] [Google Scholar]
- 38.Abeel T, Helleputte T, Van de Peer Y, Dupont P, Saeys Y. Robust biomarker identification for cancer diagnosis with ensemble feature selection methods. Bioinformatics. 2009;26(3):392–398. doi: 10.1093/bioinformatics/btp630. [DOI] [PubMed] [Google Scholar]
- 39.Guyon I, Elisseeff A. An introduction to variable and feature selection. J Mach Learn Res. 2003;3:1157–1182. [Google Scholar]
- 40.Guyon I, Weston J, Barnhill S, Vapnik V. Gene selection for cancer classification using support vector machines. Mach Learn. 2002;46(1):389–422. [Google Scholar]
- 41.Chen L, Xuan J, Riggins RB, Clarke R, Wang Y. Identifying cancer biomarkers by network-constrained support vector machines. BMC Syst Biol. 2011;5(1):161. doi: 10.1186/1752-0509-5-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stagos D, Karaberis E, Kouretas D. Assessment of antioxidant/anticarcinogenic activity of plant extracts by a combination of molecular methods. In Vivo. 2005;19(4):741–747. [PubMed] [Google Scholar]
- 43.Warmuth MK, Liao J, Rätsch G, Mathieson M, Putta S, Lemmen C. Active learning with support vector machines in the drug discovery process. J Chem Inf Comput Sci. 2003;43(2):667–673. doi: 10.1021/ci025620t. [DOI] [PubMed] [Google Scholar]
- 44.Gupta S, Chaudhary K, Kumar R, Gautam G, Nanda JS, Dhanda SK, Brahmachari SK, Raghava GPS. Prioritization of anticancer drugs against a cancer using genomic features of cancer cells: A step towards personalized medicine. Sci Rep. 2016;6:23857. doi: 10.1038/srep23857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bundela S, Sharma A, Bisen PS. Potential compounds for oral cancer treatment: resveratrol, nimbolide, lovastatin, bortezomib, vorinostat, berberine, pterostilbene, deguelin, andrographolide, and colchicine. PLoS One. 2015;10(11):e0141719. doi: 10.1371/journal.pone.0141719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsumoto A, Aoki S, Ohwada H. Comparison of random forest and SVM for raw data in drug discovery: prediction of radiation protection and toxicity case study. Int J Mach Learn Comput. 2016;6(2):145–148. [Google Scholar]
- 47.Morita A, Ariyasu S, Wang B, Asanuma T, Onoda T, Sawa A, Tanaka K, Takahashi I, Togami S, Nenoi M. AS-2, a novel inhibitor of p53-dependent apoptosis, prevents apoptotic mitochondrial dysfunction in a transcription-independent manner and protects mice from a lethal dose of ionizing radiation. Biochem Biophys Res Commun. 2014;450(4):1498–1504. doi: 10.1016/j.bbrc.2014.07.037. [DOI] [PubMed] [Google Scholar]
- 48.Volkamer A, Kuhn D, Grombacher T, Rippmann F, Rarey M. Combining global and local measures for structure-based druggability predictions. J Chem Inf Model. 2012;52(2):360–372. doi: 10.1021/ci200454v. [DOI] [PubMed] [Google Scholar]
- 49.Li L, Wang B, Meroueh SO. Support vector regression scoring of receptor-ligand complexes for rank-ordering and virtual screening of chemical libraries. J Chem Inf Model. 2011;51(9):2132–2138. doi: 10.1021/ci200078f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li GB, Yang LL, Wang WJ, Li LL, Yang SY. ID-Score: a new empirical scoring function based on a comprehensive set of descriptors related to protein-ligand interactions. J Chem Inf Model. 2013;53(3):592–600. doi: 10.1021/ci300493w. [DOI] [PubMed] [Google Scholar]
- 51.Wang Q, Feng Y, Huang J, Wang T, Cheng G. A novel framework for the identification of drug target proteins: Combining stacked auto-encoders with a biased support vector machine. PloS One. 2017;12(4):e0176486. doi: 10.1371/journal.pone.0176486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jeon J, Nim S, Teyra J, Datti A, Wrana JL, Sidhu SS, Moffat J, Kim PM. A systematic approach to identify novel cancer drug targets using machine learning, inhibitor design and high-throughput screening. Genome Med. 2014;6(7):57. doi: 10.1186/s13073-014-0057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singh H, Kumar R, Singh S, Chaudhary K, Gautam A, Raghava GP. Prediction of anticancer molecules using hybrid model developed on molecules screened against NCI-60 cancer cell lines. BMC Cancer. 2016;16(1):77. doi: 10.1186/s12885-016-2082-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stetson LC, Pearl T, Chen Y, Barnholtz-Sloan JS. Computational identification of multi-omic correlates of anticancer therapeutic response. BMC Genomics. 2014;15(7):S2. doi: 10.1186/1471-2164-15-S7-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hejase HA, Chan C. Improving Drug Sensitivity Prediction Using Different Types of Data. CPT Pharmacometrics Syst Pharmacol. 2015;4:98–105. doi: 10.1002/psp4.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hazai E, Hazai I, Ragueneau-Majlessi I, Chung SP, Bikadi Z, Mao Q. Predicting substrates of the human breast cancer resistance protein using a support vector machine method. BMC Bioinformatics. 2013;14:130. doi: 10.1186/1471-2105-14-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Normanno N, Rachiglio AM, Roma C, Fenizia F, Esposito C, Pasquale R, La Porta ML, Iannaccone A, Micheli F, Santangelo M. Molecular diagnostics and personalized medicine in oncology: challenges and opportunities. J Cell Biochem. 2013;114(3):514–524. doi: 10.1002/jcb.24401. [DOI] [PubMed] [Google Scholar]
- 58.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jordan EJ, Radhakrishnan R. Machine learning predictions of cancer driver mutations. Proceedings of the 2014 6th International Advanced Research Workshop on In Silico Oncology and Cancer Investigation. 2014 doi: 10.1109/iarwisoci.2014.7034632. doi: 10.1109/IARWISOCI.2014.7034632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Capriotti E, Altman RB. A new disease-specific machine learning approach for the prediction of cancer-causing missense variants. Genomics. 2011;98(4):310–317. doi: 10.1016/j.ygeno.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Izarzugaza JM, del Pozo A, Vazquez M, Valencia A. Prioritization of pathogenic mutations in the protein kinase superfamily. BMC Genomics. 2012;13(4):S3. doi: 10.1186/1471-2164-13-S4-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tan H, Bao J, Zhou X. A novel missense-mutation-related feature extraction scheme for ‘driver’mutation identification. Bioinformatics. 2012;28(22):2948–2955. doi: 10.1093/bioinformatics/bts558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Capriotti E, Altman RB. A new disease-specific machine learning approach for the prediction of cancer-causing missense variants. Genomics. 2011;98(4):310–317. doi: 10.1016/j.ygeno.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bari MG, Ung CY, Zhang C, Zhu S, Li H. Machine Learning-assisted network inference approach to identify a new class of genes that coordinate the functionality of cancer networks. Sci Rep. 2017;7:6993. doi: 10.1038/s41598-017-07481-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Listgarten J, Damaraju S, Poulin B, Cook L, Dufour J, Driga A, Mackey J, Wishart D, Greiner R, Zanke B. Predictive models for breast cancer susceptibility from multiple single nucleotide polymorphisms. Clin Cancer Res. 2004;10(8):2725–2737. doi: 10.1158/1078-0432.ccr-1115-03. [DOI] [PubMed] [Google Scholar]
- 66.Schwender H, Zucknick M, Ickstadt K, Bolt HM, The GENICA network A pilot study on the application of statistical classification procedures to molecular epidemiological data. Toxicol Lett. 2004;151(1):291–299. doi: 10.1016/j.toxlet.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 67.Chen SH, Sun J, Dimitrov L, Turner AR, Adams TS, Meyers DA, Chang BL, Zheng SL, Grönberg H, Xu J. A support vector machine approach for detecting gene–gene interaction. Genet Epidemiol. 2008;32(2):152–67. doi: 10.1002/gepi.20272. [DOI] [PubMed] [Google Scholar]
- 68.Guo Y, Yu L, Wen Z, Li M. Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res. 2008;36(9):3025–3030. doi: 10.1093/nar/gkn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chai H, Huang HH, Jiang HK, Liang Y, Xia LY. Protein-protein interaction network construction for cancer using a new L1/2-penalized Net-SVM model. Genet Mol Res. 2016;15(3):gmr.15038794. doi: 10.4238/gmr.15038794. [DOI] [PubMed] [Google Scholar]
- 70.Tarca AL, Carey VJ, Chen XW, Romero R, Drăghici S. Machine learning and its applications to biology. PLoS Comput Biol. 2007;3(6):e116. doi: 10.1371/journal.pcbi.0030116. [DOI] [PMC free article] [PubMed] [Google Scholar]