

Abstract
Multiple myeloma (MM) is a haematological malignancy of mature antibody‐secreting plasma cells. Currently, MM is incurable, but advances in drug treatments have increased patient lifespan. One of the characteristics of MM is the excessive production of monoclonal immunoglobulin (also referred to as paraprotein). This high level of protein production induces endoplasmic reticulum (ER) stress, and proteasomal degradation of the paraprotein is required to avoid ER stress‐induced cell death. Consequently, proteasomal inhibitors such as bortezomib have been particularly effective therapies. Unfortunately development of resistance to bortezomib is common. In this review, we address how control of endoplasmic reticulum stress is important in the development of MM and how the unfolded protein response and its associated stress response pathways are involved in the development of bortezomib resistance.
Keywords: bortezomib resistance, endoplasmic reticulum stress, multiple myeloma, proteasome inhibitors, unfolded protein response
The endoplasmic reticulum (ER) and ER stress
The endoplasmic reticulum is the powerhouse of protein synthesis within the cell and is responsible for modification and trafficking of many membranes and secreted proteins. The ER also has the ability to synthesise lipids and some carbohydrates. As a network of interconnected internal membranes, the ER is made up of the rough endoplasmic reticulum (RER) and the smooth endoplasmic reticulum (SER). Studded with ribosomes giving a rough physical appearance, the RER is a site of protein synthesis within eukaryotic cells. Lacking ribosomes, hence giving a smooth appearance, the SER is responsible for lipid and carbohydrate synthesis. While the ER is important in all eukaryotic cells, there is a particularly high level of dependence upon the ER in secretory cells such as plasma B cells, Paneth cells and pancreatic acinar cells. ER homoeostasis is essential for the correct functioning of these cells. ER stress occurs when there is an accumulation of unfolded and/or misfolded protein which exceeds the rate of protein refolding or degradation.1 Increased levels of unfolded and/or misfolded proteins are the result of a variety of contributing factors, such as unregulated gene transcription, expression of truncated/altered proteins encoded by mutated genes and the presence of damaged proteins caused by oxidative stress, chemotherapy and radiation. To cope with ER stress cells activate compensatory mechanisms, such as the unfolded protein response (UPR).2 In yeast models, the ER can expand in volume by up to fivefold in response to UPR‐inducing stimuli in an attempt to increase protein folding,3 and similar increases in ER size are seen in mammalian cells. The purpose of the UPR is to resolve the stress and restore ER homoeostasis. However, in instances where ER stress cannot be resolved, cells will normally succumb to apoptosis.
Regulation of the UPR
The mammalian UPR consists of signalling pathways that are regulated by three ER transmembrane proteins: IRE‐1 (inositol‐requiring enzyme‐1), PERK (double‐stranded RNA‐activated protein kinase [PKR]‐like ER kinase) and ATF6 (activating transcription factor 6) (Figure 1). In the absence of unfolded proteins, BiP, one of the 70 kDa heat‐shock proteins (HSP70s), is bound to these three ER membrane enzymes and keeps them in an inactive state. Exposed hydrophobic moieties on unfolded proteins bind preferentially to BiP, leading to the dissociation of BiP from the three ER proteins resulting in their activation.
Figure 1.
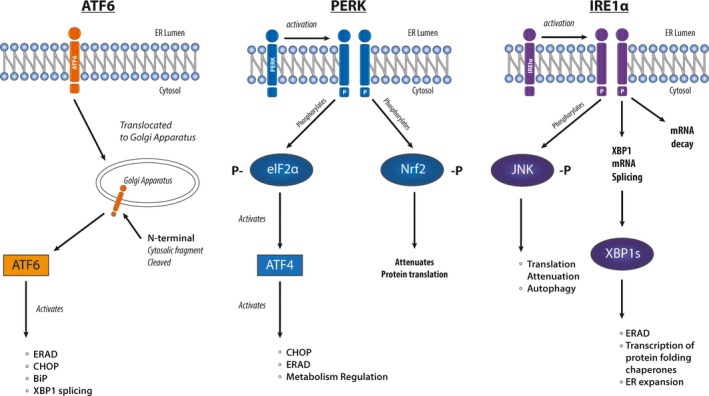
Transmembrane regulators of the unfolded protein response (UPR). During activation of the UPR, ATF6 translocates to the Golgi and is cleaved, before it activates a series of downstream targets, such as endoplasmic reticulum‐associated degradation (ERAD), CHOP, BiP and XBP1. PERK autophosphorylates, and in turn phosphorylates eIF2α and Nrf2, which targets downstream UPR targets. IRE1α is also activated by autophosphorylation, which phosphorylates JNK, while also activating XBP1. IRE1α removes the 26 base pair intron from unspliced XBP1 mRNA resulting in expression of the active isoform from the spliced mRNA.
In the absence of BiP, dimeric IRE‐1 (IRE‐1α and β) auto‐transphosphorylates and its endoribonuclease activity is induced. In an extranuclear splicing reaction, IRE‐1 cleaves a 26 base intron from XBP1 (X‐box Binding Protein) mRNA, producing an open reading frame shift that yields a longer polypeptide.4 The spliced XBP1 protein is a 54 kDa basic leucine zipper transcription factor, which consists of a DNA‐binding domain in the N‐terminus and a transactivation domain in the C‐terminus. Spliced XBP1 binds to promoter elements of multiple stress response genes inducing their expression. These genes include, DnaJ/Hsp40‐like genes, p58IPK, ERDj4, HEDJ, EDEM, protein disulphide isomerase‐P5, ribosome‐associated membrane protein 4 (RAMP4), ERDJ3 and BiP.5, 6 Their activation subsequently results in increased protein folding of unfolded and misfolded proteins, while also promoting the degradation of unfolded/misfolded proteins by endoplasmic reticulum‐associated degradation (ERAD). Unspliced XBP1 mRNA has a shorter reading frame and is translated to a protein that lacks the transactivation domain in the C‐terminus, but retains the DNA binding and dimerisation domains. Thus unspliced XBP1 protein acts as a dominant negative, thereby suppressing expression of target genes.4 This short version of XBP1 is unstable and degraded rapidly by the proteasome pathway.4
The second transmembrane component of the UPR, PERK, is a member of the eIF2α family of kinases. PERK phosphorylates (1) the α‐subunit of eukaryotic translation initiation factor 2 and (2) the bZIP transcription factor, Nrf2.7 Phosphorylated eIF2α interferes with the formation of the 43S translation initiation complex and thus attenuates protein translation.4 Specifically, phosphorylation of eIF2α induces the formation of stress granules (SG). SG are cytoplasmic foci where, during a stress response, some mRNA can be stored for later translation.8, 9 As such the level of translation is suppressed, relieving the burden on the ER. Nrf2 in unstressed cells associates with cytoskeletal anchor, Keap1. Phosphorylation of Nrf2 leads to its dissociation from Keap1 and translocation to the nucleus where it acts on antioxidant response elements (AREs). Via ARE binding, activated Nrf2 induces the transcription of ATF4, c‐Jun, Jun B and Jun D. AREs also control expression of genes that are involved in the phase II metabolism of xenobiotics, such as the A1 and A2 subunits of glutathione‐S‐transferase, NAD(P)H quinone oxidoreductase, glutamylcysteine synthetase, heme oxygenase 1 and UDP‐glucuronosyl transferase.10 Thus, there is a potential link between the UPR and cytotoxic drug detoxification.
The third ER transmembrane component is ATF6α (90 kDa), which, like XBP1, is a basic leucine zipper transcription factor.11 ATF6α is expressed constitutively in an inactive form. ER stress leads to dissociation of ATF6α from BiP resulting in the translocation of ATF6α to the Golgi apparatus. The subsequent proteolytic cleavage of its cytosolic bZIP domain allows for the release of ATF6α from the phospholipid bilayer. Once released, ATF6α enters the nucleus and activates ER stress response elements and ATF/cAMP response elements. BiP and CHOP are examples of genes that are activated by ATF6.10 The absence of ATF6α does not affect the activation of UPR genes and, therefore, ATF6 is not indispensable for the UPR to function. This might be due to other compensatory pathways, like XBP1 activation. Notably, ATF6 can induce XBP1, but ATF6 alone is not adequate for plasma cell differentiation and immunoglobulin production, which also requires the IRE‐1 induced splicing of XBP1 mRNA.11
Plasma cells
Plasma cells are long‐lived terminally differentiated B cells in the bone marrow that are responsible for the production of antigen‐specific immunoglobulin. The survival of plasma cells is dependent on the transcriptional activation of interferon regulatory factor 4 (IRF4), and activator Blimp1 is essential for immunoglobulin secretion in response to infection.12 In order to support this secretory function, Blimp1 induces the UPR, expansion of the ER and lysosomal trafficking.12 Blimp1 also activates multiple regulators of the UPR. More specifically, it activates ATF6 which induces XBP1 and activates IRE1 leading to splicing of cytoplasmic XBP1 and production of the active XBP1 transcription factor. XBP1 induces the transcription of stress response genes and chaperones which are important for the expression of the immunoglobulin heavy chain complex, activation of protein folding and targeting of proteins to the ER. Blimp1 regulates the cell size of plasma cells by upregulation of mammalian target of rapamycin (mTOR) complex 1 activity. The transcription profile of plasma cells leads to the activation of the UPR, which is essential in maintaining the secretory function of plasma cells.
Multiple myeloma (MM), genetic changes and paraprotein expression
Multiple myeloma is a plasma cell malignancy and is characterised by the proliferation of plasma cell clone/s and infiltration of the bone marrow by malignant plasma cells.13 MM is incurable, and the 5‐year relative survival during the period 2009–2013 in Australia was 48.9% (https://myeloma-cancer.canceraustralia.gov.au/statistics). Patients suffer from multiple systemic complications of the disease. The disruption to the bone marrow microenvironment and the normal functioning of the plasma cells eventually results in the development of anaemia, leukopenia, hypogammaglobulinemia and thrombocytopenia.13 Furthermore, 90% of patients with MM have osteolytic lesions which cause bone pain,14 increased risk of fractures15 and hypercalcaemia. Deposition of immunoglobulin light chains in the kidney tubules causes cast nephropathy and acute kidney injury. Excessive production of monoclonal immunoglobulin can also lead to hyperviscosity syndrome, which is characterised by bleeding, blurred vision, confusion, neurologic symptoms and thromboembolic disease.14
Hyperdiploidy and translocations of the IgH (immunoglobulin heavy chain) locus are the most frequently occurring chromosomal abnormalities identified in MM patients. A number of recurrent nonrandom abnormalities have been identified in MM within the VDJ recombination region, mediated by errors of IgH switch recombination. About 60% of chromosome translocations of the IgH locus (14q32 gene locus) lead to the juxtaposition of nonimmunoglobulin DNA sequences of gene loci from 11q13, 4p16.3, 16q23 or 6p21.16 Specifically, translocations activate oncogenes due to enhancer elements of the IgH gene locus acting on the oncogenes. Translocations of this locus are seen in up to 75% of MM cases, and almost all MM cell lines, resulting in the hypothesis that chromosome translocations may cause the initial transformation of a plasma cell into a malignant cell.16
An example of a prevalent chromosome translocation of the IgH locus is the translocation of the cyclin D1 gene at the 11q13 locus.17 In 15–20% of MM cases, the cyclin D1 protein is overexpressed as a result of translocation next to the powerful IgH 3′ enhancer.17, 18 Overexpression of cyclin D1 is restricted to MM, and mantle cell lymphoma as cyclin D1 is not expressed in normal B cells.18 The binding of cyclin D1 to cyclin‐dependent kinase inhibitor 1B (p27Kip1) blocks the inhibitory role of p27Kip1, thereby enabling cell cycle progression past the G1 phase.19 As a result, this promotes tumorigenesis in myeloma cells, which has also been linked to drug resistance in myeloma.18 Other major genetic mutations contributing to the development of MM are NRAS and KRAS activating mutations, present in ~50% of patients and C‐MYC overexpression, but this is predominantly increased in the later stages of the disease.20
Modulation of ER stress in MM
With the exception of a small subset of patients (1–5% of all MM patients),21 most patients diagnosed with MM characteristically produce excessive levels of immunoglobulin fragments (paraprotein). The continual overproduction of paraprotein subjects MM cells to continual ER stress2 making myeloma cells highly dependent upon the UPR for survival. But continual ER stress also drives utilisation of other stress pathways that enable cells to adapt to this extra burden. The dependence of MM cells on the UPR has made therapies that indirectly target the UPR effective in treating disease and improving the life expectancy of patients. Here, we will review a number of mechanisms utilised by MM to modulate ER stress, firstly in pathogenesis and then in response to treatment.
Multiple myeloma is dependent on the activation of the UPR. Genes involved in protein homoeostasis, RNA processing, protein translation and the UPR were identified as frequently mutated in MM in a whole genome sequencing study.22 Gene expression profiling of MM patients reveals common upregulation of genes involved in intracellular protein transport.23 Transgenic mice with sustained spliced XBP‐1 expression develop monoclonal gammopathy and MM.24 This shows that activation of UPR is a hallmark of MM. As described above, the UPR is regulated through three ER transmembrane proteins PERK, ATF6 and IRE1α. ER stress activates all three arms of the UPR in myeloma cells,11, 25 and there is increased expression of XBP1 and ATF6 in MM cells from patients.26 Increased UPR mediated signalling results in induction of additional downstream stress pathways and a cascade of events resulting in decreased transcriptional and translational rates, but upregulation of transcription of molecular chaperones responsible for protein folding and degradation.2 Moreover, the UPR can coordinate and activate autophagy and the ubiquitin‐proteasome pathway and HSP as strategies to reduce ER stress.27 Consequently, UPR activation is associated with increased survival of myeloma cells.28
ATF6 and PERK in MM development/initiation are considerably under studied relative to XBP1. While studied in the progression of myeloma in the later stages of the disease, the roles of ATF6 and PERK in the initiation/early stages of the disease are yet to be explored. XBP1 on the other hand has been well characterised in MM development/initiation using mouse modelling. Overexpression of the spliced isoform of XBP1 in transgenic mice has been found to drive monoclonal gammopathy and MM development, resulting in the expansion of the B‐cell compartment, spikes in immunoglobulin secretion of IgG and IgM types, bone lytic lesions, increases of up to 5–30% in the number of plasma cells in the bone marrow and 1119 differentially expressed genes.24 Differential expression of Cyclin D1, Cyclin D2, MAF and MAFB, all of which are altered in MM patients, were also identified in the transgenic mice.24 Furthermore, MM development could be a result of chronic B‐cell hyperproliferation and IL‐6 activation driven by overexpression of XBP1.24
The ubiquitin‐proteasome system (UPS) is an additional degradation pathway that is also utilised to reduce ER stress in MM (Figure 2). This system of intracellular proteolysis is highly substrate specific, tightly controlled and distinct from the nonspecific lysosomal degradation process. UPS is utilised and activated by the UPR through a process known as ERAD.29 There are two main steps in the UPS, the first being the transport of misfolded or damaged proteins into the cytoplasm and their covalent tagging with ubiquitin moieties. The second is the degradation of the tagged proteins by the 26S proteasome and recycling of ubiquitin. The 26S proteasome is a multicomponent protease, consisting of a 20S cylindrical core and two 19S subunits. Misfolded or damaged proteins, or proteins otherwise destined for degradation, are recognised by the ubiquitin ligases that mediate the sequential binding of ubiquitin moieties to the proteins to form a covalently bound polyubiquitin chain.30 The polyubiquitin chain is recognised by the 26S proteasome, and the 19S subunits bind to the polyubiquitin chains, unfold the protein and insert it into the channel of the 20S catalytic core for proteolysis.30 Ubiquitin is subsequently recycled back into the UPS so that it can be reused.31 Myeloma cells are heavily dependent on this degradation system to reduce the load of paraprotein and demand on the ER and protein folding. This has made inhibition of the proteasome one of the most effective current treatments for MM.
Figure 2.
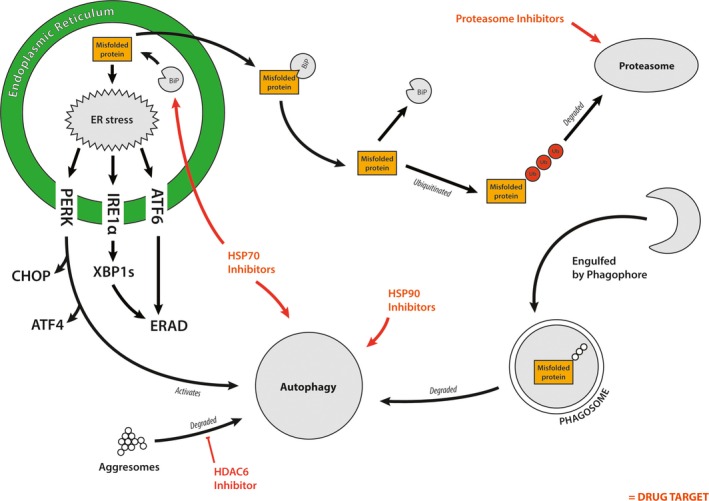
Molecular mechanisms responsible for modulating endoplasmic reticulum (ER) stress. Accumulating misfolded proteins trigger ER stress, which in turn activates the UPR. In response to this, PERK, ATF6 and IRE1α activate CHOP, ATF4, endoplasmic reticulum‐associated degradation and XBP1 to alleviate ER stress. The BiP protein traffics misfolded proteins into the cytoplasm where they are ubiquitinated. Once ubiquitinated, misfolded proteins can be degraded by either autophagy or the proteasome. The proteasome is inhibited (red) in multiple myeloma as a way of treating the disease. Other mechanisms are also relied upon to assist in alleviating ER stress such as autophagy and proteasomal degradation of proteins. These stress pathways are also potential targets for treating MM (red).
Autophagy is an evolutionarily conserved cellular mechanism important for maintaining cellular homoeostasis. It is the process of degradation of cellular organelles and proteins, to maintain cellular biosynthetic capacity during nutrient deprivation or metabolic stress.32 Autophagy is important for adapting to nutrient starvation, intracellular protein and organelle clearance, development, microbial destruction, apoptosis, tumor suppression and antigen presentation.33, 34 Cytoplasmic components such as proteins and cytoplasmic organelles like mitochondria are degraded by autophagy by first being engulfed by an isolation membrane.35 The isolation membrane, surrounds the cytoplasmic debris to form an autophagosome, a double membrane‐enclosed structure.34 Once formed, the autophagosome fuses with endosomes and eventually fusion of lysosome outer membranes and autophagosomes occurs.35 Lysosomal hydrolase enzymes degrade the engulfed contents along with the inner membrane of the autophagosome.35 The recycled macromolecules are then released into the cytosol. A major function of this pathway is to eliminate damaged cellular organelles or unused/damaged proteins.33, 35
Studies have found that the UPR can trigger autophagic activation in response to the accumulation of unfolded proteins.36 Autophagy activation in response to ER stress can be initiated by PERK‐dependent eIF2α phosphorylation, which leads to the upregulation of autophagy‐related protein 12 (Atg12).37 Basal autophagy levels are increased in myeloma cells in response to ER stressors, and autophagy activation is important for maintaining myeloma cell viability.36 Autophagy is upregulated in response to proteasome inhibition using therapeutic agent bortezomib (Bz).27, 38 Combinational therapy using Bz and bafilomycin A1 enhances the cytotoxic effect of these drugs on MM cells as the combination enhances ER stress levels by preventing paraprotein degradation, but also by inhibiting the compensatory mechanism of autophagy.39 Numerous studies have shown that autophagy inhibition significantly enhances cytotoxicity of proteasome inhibition.39, 40, 41 However, contradicting studies show combined autophagy and proteasome inhibition can be antagonistic.36, 39, 42 This is likely due to individual inhibitors targeting different parts of the autophagic pathway, which in turn may have downstream effects on other stress pathways, causing their upregulation and a limited response to treatment. Studies addressing this hypothesis are ongoing.
Heat‐shock proteins are a diverse family of proteins important in processes including protein folding, protein scaffolding and trafficking, and maintaining protein conformation during normal or stressed conditions.43, 44 HSPs were first discovered in response to heat‐induced stress; however, research has now shown that HSPs are also induced under a range of different stress conditions, including DNA damage, infection, exposure to toxins, starvation and hypoxia.43 A number of HSPs play important roles in the UPR and MM (Figure 2). Heat‐shock protein 90 kDa (HSP90) is an important stabiliser protein for the kinase activity of PERK and IRE1α proteins during the UPR.45 As described above, BiP, another HSP, though of a different class of HSPs (HSP70s), is important for initiating the UPR by dissociating from PERK, ATF6 and IRE1α. The binding of the BiP protein to PERK, ATF6 and IRE1α suppresses the conformational change of these three proteins, which is required for their activation.6 There are increased expression levels of BiP and HSP90 in MM, where they control the UPR in response to accumulating paraprotein.45, 46 BiP is also an important component of the protein translocation complex responsible for trafficking proteins out of the ER into the cytoplasm for proteasome degradation.47 HSP72 and HSP73 promote tumor cell survival in MM by contributing to the chaperone function of HSP90.48 Inhibition of these proteins induces apoptosis, further highlighting the important role of HSP70 proteins in the biology of MM and ER stress management.44, 48 Their relative upregulation in MM and known role in modulating ER stress has led to the development of a number of therapies targeting these proteins (see below for more detail). Furthermore, the glucose‐regulated protein 94 (GRP94) (also known as HSP90B1) protein plays a significant role in ER protein quality control, via protein folding capabilities, and also serves an equally important role in orchestrating ERAD.49 Within MM, GRP94 is associated with advanced stages of the disease with proposed diagnostic and prognostic potential.50
As such the UPR response regulates multiple pathways and plays roles in both MM survival and potentially in MM death in response to prolonged activation of the UPR. Apoptotic activation by PERK is mediated by downstream ATF4/ATF3 that activates C/EBP homologous protein (CHOP), leading to growth arrest and DNA damage‐inducible protein (GADD34) activation.51, 52 Pro‐apoptotic signalling can also be initiated during ER stress by IRE1α by directly interacting with the pro‐apoptotic regulators BAX and BAK of the BCL‐2 family.9, 53 It is the UPRs role to monitor misfolded/unfolded protein levels, and orchestrate their folding by chaperone folding proteins, or alternatively their removal via degradation pathways, such as the UPS. In instances where ER stress levels surpass the UPRs folding ability, the pathway will trigger apoptosis.
Current treatments for MM patients
There are a variety of drugs used for the treatment of MM, which fall into six major categories (Table 1 and Figure 2). These classes are DNA‐damaging agents, Glucocorticoids, Immunomodulatory Drugs (IMiDs), Proteasome Inhibitors (PIs), Histone deacetylase inhibitors (HDACi) and Monoclonal Antibodies. However, of the six major classes, only PIs target ER stress mechanisms. DNA‐damaging agents frequently used include melphalan and Cyclophosphamide. Glucocorticosteroid is a backbone of most treatment strategies. IMiDs have immunostimulatory effects and suppress MM survival factors MYC and IRF4. Monoclonal antibody, Daratumumab directly targets CD38, a surface molecule of myeloma cells. The inhibition of the proteasome exploits the dependence of myeloma cells on the UPR by preventing the degradation of pro‐apoptotic proteins and increasing protein load within the cell. The disruption of intracellular homoeostasis leads to proteotoxicity as a result of accumulating paraprotein.54
Table 1.
Current treatments for multiple myeloma patients and the class of action
| Class of drug | DNA damaging | Glucocorticoids | IMiDs | Monoclonal antibodies | PIs | HDACi |
|---|---|---|---|---|---|---|
| Drugs |
Melphalan Cyclophosphamide Lipo‐doxorubicin Bendamustine |
Dexamethasone Prednisone |
Thalidomide Lenalidomide Pomalidomide |
Elotuzumab (Targets SLAMF7) Daratumumab (Targets CD138) |
Bortezomib Carfilzomib Ixazomib |
Panobinostat |
HDACi, histone deacetylase inhibitors; IMiDs, immunomodulatory drugs; PIs, proteasome inhibitors.
Bortezomib (Bz) was the first proteasome inhibitor to be approved for clinical use and has improved the survival of MM patients since its introduction.54 Bz selectively and reversibly inhibits the chymotryptic protease activity of the 26S proteasome through the binding of the boronic acid peptide to the active threonine site within the 26S proteasome.55 The effectiveness of Bz is further enhanced by its combination with other therapies used to treat MM within the clinic. However, like many other chemotherapy drugs, resistance develops in the majority of patients. This has led to the development of new PIs, such as carfilzomib and ixazomib to improve the efficacy of PIs and overcome resistance (Table 1). Carfilzomib is a novel proteasome inhibitor that has a more potent effect than Bz, largely attributed to its irreversible inhibitory action on the chymotrypsin‐like activity of the proteasome.56 Promisingly, some Bz‐resistant MM patients and Bz‐resistant cell lines respond to Carfilzomib treatment.57
HDAC inhibitors have multiple cellular effects. Panobinostat, a pan‐histone deacetylase inhibitor, is approved by the FDA for the treatment of relapsed and refractory MM. One of its mechanisms of action is the inhibition of aggresome formation which leads to increased ER stress and synergism with bortezomib.58
The following drugs are potential modulators of the UPR and are undergoing preclinical and clinical testing. HSP inhibitors may be effective against myeloma cells.25 The role HSPs play in the UPR, ERAD and autophagic pathways, has prompted the development of new chemotherapies targeting these proteins. Inhibition of HSP90 has cytotoxic effects within a number of cancers.59 Inhibition of HSP90 in MM induces cell death as a result of elevated ER stress and triggering UPR activated cell death.25 Further, inhibition of HSP70 in combination with HSP90 inhibitors increases the cytotoxicity compared to HSP90 inhibitors alone in MM cell lines.60 The hypothesised reason behind this is that inhibiting HSP70 counters the compensatory upregulation of HSP70 as a result of HSP90 inhibition. The combination of Bz with the HSP90 inhibitor, KW‐2478, has also exhibited synergistic antitumor activity in vitro and in vivo.61 The tumor burden was also seen to decrease within the bone marrow of an orthotopic myeloma model as a result of the combination treatment.61 Phase I clinical trials of KW‐2478 in patients with relapsed/refractory B‐cell malignancies have shown promising results.59
Research is starting to explore autophagy inhibition as an alternative method of treating MM. Chloroquine is an autophagy inhibitor. Hydrochloroquine could increase the effectiveness of Carfilzomib in MM cell lines and patient cells but had less effect when combined with Bz.62 However, chloroquine is being tested in combination with Bz in early phase clinical trials with promising safety data,63 but the efficacy in MM patients is not known yet.
Mechanisms modulating ER stress in acquired bortezomib resistance
As described above, resistance to bortezomib (Bz) eventually develops in the majority of MM patients. The underlying causes of resistance are yet to be determined; however, a number of alterations within the cell are likely to be contributing (Figure 2).
Proteasome mutations
Acquired Bz resistance had been linked to a point mutation within the gene encoding the proteasome b5 subunit (PSMB5), leading to a conformational change within the bortezomib‐binding pocket.64 Cells with the mutated PSMB5 subunit displayed greater resistance to apoptosis induced by Bz, while cells with the wild‐type PSMB5 subunit were more susceptible to Bz‐induced apoptosis.64 Resistance could be further attributed to upregulation of the mutant β5 subunit in various Bz‐resistant cells.65 However, in two independent studies, this mutation was not identified in patients with Bz refractory or relapsed myeloma.66, 67 Instead, resistant myeloma cells had higher numbers of active proteasomes than sensitive cells.42
Reduced dependence on the UPR
Bz resistance can be associated with decreased UPR activity. Within Bz‐resistant clones, the expression of ATF6 and XBP1 (spliced and total XBP1 mRNA levels) are decreased substantially.68, 69, 70 ATF6 and XBP1 expressions are considered a predictor of Bz sensitivity, as the expression of both genes is seen to mirror sensitivity to Bz.68, 70 Further, Bz‐resistant cells have smaller ER lumen widths and overall ER size.70 Expansion of the ER plays an integral part in the UPR, with ER expansion directly controlled by XBP1. The UPR is also dependant on increased ER biogenesis and phosphatidylcholine synthesis, which is enhanced by ATF6.71 This response is critical during the UPR to alleviate ER stress caused by accumulation of unfolded and misfolded proteins. Therefore, the reduced ER size in Bz‐resistant cells is likely due to reduced XBP1 and ATF6 expression.
The decrease in XBP1 expression in Bz‐resistant patients has been associated with a single nucleotide polymorphism (SNP) occurring in the splice site of the XBP1 gene, preventing XBP1 splicing and activation.72 Under ER‐induced stress, cells transfected with wild‐type XBP1 are able to undergo splicing and activate the UPR pathway; however, cells with mutated XBP1 were incapable of splicing the mRNA,72 thus preventing activation of the UPR via the IRE1α‐XBP1 pathway. This in turn, reduced/compromised the UPR in myeloma cells, reducing the sensitivity of myeloma cells to Bz, as sensitivity is related to the dependence of the myeloma cells on the UPR. However, the occurrence of this mutation within patients is also low. In one study, only two of 38 MM patients analysed possessed a mutation within the splice site of XBP1.22 One patient diagnosed with progressive disease, possessed an XBP1‐P326R mutation and this patient failed cyclophosphamide, Bz and dexamethasone treatment.22, 69 The second patient had a XBP1‐L167I mutation, though died prior to undergoing treatment.69 Both mutations were reported to have lethal consequences, though there is limited evidence to suggest that they caused Bz resistance.
Autophagy
Autophagy has begun to emerge as an additional factor that may confer resistance to proteasome inhibition. Within resistant cells, Sequestosome‐1 (SQSTM1)‐dependent autophagy provides a protection mechanism to proteasome inhibitor‐induced protein aggregation and cell death.73 Inhibition of SQSTM1 within resistant cells sensitised cells to proteasome inhibition.73 Additional studies have supported this hypothesis by identifying increased expression of high mobility group box 1 (HMGB1), an important regulator of autophagy, within Bz‐resistant cells.74 Within the same study, Bz administered in combination with the autophagy inhibitor lycorine not only resensitised myeloma cells to Bz, but enhanced its effect in in vitro and in vivo models.74 It is doubtful that HMGB1 alone is responsible for driving Bz resistance, as regulation of autophagy is a highly involved and complex mechanisms may require multiple changes. So, while these findings do provide initial evidence that autophagy is a potential mechanism for Bz resistance, further investigation is required.
Deubiquitinating enzymes (DUBs) and ubiquitin ligase inhibitors
Deubiquitinating enzymes are a class of enzymes responsible for the removal ubiquitin labelling from cellular components marked for degradation. Their role and importance have been illustrated in MM, particularly Bz‐resistant myeloma. The DUBs, USP14 and UCHL5, promote MM survival and possibly contribute to Bz resistance by reducing ubiquitinated protein levels in myeloma cells.75 Plasma cells isolated from MM patients exhibited high expression levels of both USP14 and UCHL5, while there is no detectable expression of these proteins in normal plasma cells.75 Inhibiting the deubiquitinating activity with a novel 19S regulatory particle inhibitor, b‐AP15, in combination with knockdown of USP14 and UCHL5 resulted in a reduction in cell viability and inhibition of proliferation.76 Bz‐resistant cells were also shown to become more sensitive to Bz. Other studies have also found that b‐AP15 inhibition of USP14 and UCHL5 triggers apoptosis in MM cell lines in a time‐dependent and dose‐dependent manner.77 Similar cytotoxic effects, as those seen with b‐AP15 treatment, occur within myeloma cells when treated with an alternative DUB inhibitor, copper pyrithione.77 Targeting E1 ubiquitin‐activating enzyme and E3 ubiquitin ligases has synergistic activity with bortezomib on cell lines.78, 79 An inhibitor of USP7, P5091, can interfere with ubiquitin binding and overcome bortezomib resistance in vitro and in vivo.80 NIMA‐related kinase 2 (NEK2) overexpression is associated with resistance to multiple drugs and poor prognosis in MM, and NEK2 inhibitor has been shown to decrease proteasome activity.81 Further investigation into its role in Bortezomib resistance is required.
In summary, there are a number of stress mechanisms that play an important role in mediating Bz resistance. Abnormalities in the functioning of the UPR in Bz‐resistant MM result in a number of stress mechanisms that compensate for the UPR in alleviating ER stress levels. Bz resistance is complex and involves a number of cellular mechanisms. It is difficult to determine the precise cause of Bz resistance; however, mechanisms described in this section contribute to managing ER stress levels in Bz‐resistant MM cells. It is highly likely that there are additional mechanisms in MM which allow Bz‐resistant cells to adapt to ER stress. For example, nonsense‐mediated decay (NMD) is the mechanism by which cells identify and degrade mRNA transcripts with premature termination codons. NMD is known to regulate the UPR by both reducing the load of truncated and potentially misfolded proteins as well as directly regulating the RNA levels of key UPR proteins including ATF4.82, 83 How nonsense‐mediated decay is regulated during MM development or during acquired drug resistance is currently unknown and needs further investigation. Understanding this and other mechanisms for controlling ER stress will allow for development of better treatment options for relapsed/refractory MM patients.
Future treatments for proteasome inhibitor‐resistant MM patients
Future treatment of proteasome inhibitor‐resistant MM patients may include drugs described above – autophagy inhibitors, HSP inhibitors, DUB inhibitors and ubiquitin ligase inhibitors. However, these are likely to be used in combination with other classes of drugs to maximise response and minimise the emergence of resistance. Novel classes of drugs include Venetoclax (BCL‐2 inhibitor), chimeric antigen receptor (CAR)‐T cells, anti‐BMCA, MCL inhibitor and Nuclear Export inhibitor.84 These treatments may not modulate ER stress directly but have promising efficacy in PI‐resistant MM patients. Genomics of individual patients may inform actionable targets that contribute to resistance and progressive disease.
Currently, there are no drugs that specifically target resistance to proteasome inhibitors or resensitise MM to proteasome inhibitors. The available approved drugs that can be used to treat proteasome inhibitor‐resistant MM patients are limited. They include immunomodulatory drugs and monoclonal antibodies, which have a completely different mechanism of action to proteasome inhibitors. Panobinostat has synergistic effect with Bortezomib in MM in vitro and in vivo. This is due to the dual inhibition of the aggresomal and proteasomal protein degradation pathways. Panobinostat is approved by the FDA for the treatment of patients with MM after two or more prior lines of therapies (Table 1). Panobinostat in combination with Bortezomib and dexamethasone is associated with increased response rate and prolonged progression‐free survival compared with Bortezomib and dexamethasone.58 Potential targets that can resensitise cells to proteasome inhibitors include inhibition of autophagy, DUBs, HSP, the NMD pathways and induction of ER stress (using Nelfinavir).
ER stress in other haematological disorders
While the bulk of research into ER stress and its regulation in disease development and drug resistance has focussed on MM, these processes are also important in other haematological disorders. Proteasomal inhibition is also effective in the treatment of mantle cell lymphoma (MCL) and Waldenstrom macroglobulinemia (WM). This could be due to their dependence on the ER and UPR for immunoglobulin secretory function and B‐cell maturation. Similarly to the situation in MM, in MCL, the chromosomal translocation of Cyclin D1 with the Ig heavy chain gene results in the continual overexpression of Cyclin D1.85 The increased expression of Cyclin D1 not only places stress on the ER, but also promotes unregulated cellular proliferation.86 This in turn places extra load on the cells ER, contributing to further ER stress. WM is a low‐grade lymphoma also associated with excessive IgM secretion, which is also heavily dependent upon the ER and UPR.87
Summary
In conclusion, the management of ER stress in B‐cell malignancies, in particular MM, is not solely dependent on a single stress pathway to ameliorate ER stress. Instead, there is a multifaceted network of stress mechanisms involved that are predominantly orchestrated by the UPR. Due to the dependence of many B‐cell malignancies on the UPR, inhibitors such as Bz are effective in treating the disease. However, the emergence of Bz resistance has made the treatment of MM increasingly difficult. Abnormalities associated with the functioning of the UPR and regulated stress pathways are potential causes of Bz resistance in MM. Upregulation of alternative stress mechanisms, such as DUBs and autophagy, compensates for the role of the UPR in managing ER stress and preventing cell death. Novel therapies are starting to target these newly identified mechanisms, though it is currently unclear how effective they will be in the treatment of Bz resistance and whether combinations of drugs targeting multiple stress mechanisms will be required. As such, research is required to better understand the involvement of these stress mechanisms in MM and related malignancies, and also how each mechanism is linked and regulated in Bz resistance. Furthermore, other pathways which regulate the UPR, such as nonsense‐mediated decay, may be promising drug targets, but a better understanding of their role in MM and Bz resistance is required. By understanding the regulatory network for ER stress mechanisms, novel therapeutic approaches can be developed for relapsed/refractory MM patients.
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Silvia Ling, Email: Silvia.Ling@health.nsw.gov.au.
Tara Laurine Roberts, Email: Tara.Roberts@westernsydney.edu.au.
References
- 1. Xu C, Bailly‐Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 2005; 115: 2656–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nikesitch N, Ling SC. Molecular mechanisms in multiple myeloma drug resistance. J Clin Pathol 2016; 69: 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 2006; 4: e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem 2005; 74: 739–789. [DOI] [PubMed] [Google Scholar]
- 5. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 2012; 13: 89–102. [DOI] [PubMed] [Google Scholar]
- 6. Gardner BM, Pincus D, Gotthardt K et al Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol 2013; 5: a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rozpedek W, Pytel D, Mucha B et al The role of the PERK/eIF2alpha/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med 2016; 16: 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kedersha N, Anderson P. Stress granules: sites of MRNA triage that regulate MRNA stability and translatability. Biochem Soc Trans 2002; 30: 963–969. [DOI] [PubMed] [Google Scholar]
- 9. Brown JA, Roberts TL, Richards R et al A novel role for hSMG‐1 in stress granule formation. Mol Cell Biol 2011; 31: 4417–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bertolotti A, Zhang Y, Hendershot LM et al Dynamic interaction of BiP and ER stress transducers in the unfolded‐protein response. Nat Cell Biol 2000; 2: 326–332. [DOI] [PubMed] [Google Scholar]
- 11. Shoulders MD, Ryno LM, Genereux JC et al Stress‐independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep 2013; 3: 1279–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shapiro‐Shelef M, Lin KI, McHeyzer‐Williams LJ et al Blimp‐1 is required for the formation of immunoglobulin secreting plasma cells and pre‐plasma memory B cells. Immunity 2003; 19: 607–620. [DOI] [PubMed] [Google Scholar]
- 13. Kyle RA, Gertz MA, Witzig TE et al Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc 2003; 78: 21–33. [DOI] [PubMed] [Google Scholar]
- 14. Roodman GD. Pathogenesis of myeloma bone disease. J Cell Biochem 2010; 109: 283–291. [DOI] [PubMed] [Google Scholar]
- 15. Terpos E, Moulopoulos LA, Dimopoulos MA. Advances in imaging and the management of myeloma bone disease. J Clin Oncol 2011; 29: 1907–1915. [DOI] [PubMed] [Google Scholar]
- 16. Seidl S, Kaufmann H, Drach J. New insights into the pathophysiology of multiple myeloma. Lancet Oncol 2003; 4: 557–564. [DOI] [PubMed] [Google Scholar]
- 17. Bergsagel PL, Kuehl WM. Chromosome translocations in multiple myeloma. Oncogene 2001; 20: 5611–5622. [DOI] [PubMed] [Google Scholar]
- 18. Bustany S, Bourgeais J, Tchakarska G et al Cyclin D1 unbalances the redox status controlling cell adhesion, migration, and drug resistance in myeloma cells. Oncotarget 2016; 7: 45214–45224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Casimiro MC, Crosariol M, Loro E et al Cyclins and cell cycle control in cancer and disease. Genes Cancer 2012; 3: 649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chng WJ, Huang GF, Chung TH et al Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia 2011; 25: 1026–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Prasad R, Yadav RR, Singh A et al Case report. Non‐secretory multiple myeloma presenting with diffuse sclerosis of affected bones interspersed with osteolytic lesions. Br J Radiol 2009; 82: e29–e31. [DOI] [PubMed] [Google Scholar]
- 22. Chapman MA, Lawrence MS, Keats JJ et al Initial genome sequencing and analysis of multiple myeloma. Nature 2011; 471: 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fabris S, Ronchetti D, Agnelli L et al Transcriptional features of multiple myeloma patients with chromosome 1q gain. Leukemia 2007; 21: 1113–1116. [DOI] [PubMed] [Google Scholar]
- 24. Carrasco DR, Sukhdeo K, Protopopova M et al The differentiation and stress response factor XBP‐1 drives multiple myeloma pathogenesis. Cancer Cell 2007; 11: 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davenport EL, Moore HE, Dunlop AS et al Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood 2007; 110: 2641–2649. [DOI] [PubMed] [Google Scholar]
- 26. Patterson J, Palombella VJ, Fritz C et al IPI‐504, a novel and soluble HSP‐90 inhibitor, blocks the unfolded protein response in multiple myeloma cells. Cancer Chemother Pharmacol 2008; 61: 923–932. [DOI] [PubMed] [Google Scholar]
- 27. Zhu K, Dunner K Jr, McConkey DJ. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010; 29: 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nakamura M, Gotoh T, Okuno Y et al Activation of the endoplasmic reticulum stress pathway is associated with survival of myeloma cells. Leuk Lymphoma 2006; 47: 531–539. [DOI] [PubMed] [Google Scholar]
- 29. Carvalho AS, Rodriguez MS, Matthiesen R. Review and literature mining on proteostasis factors and cancer. Methods Mol Biol 2016; 1449: 71–84. [DOI] [PubMed] [Google Scholar]
- 30. Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin‐proteasome pathway in normal and disease states. J Am Soc Nephrol 2006; 17: 1807–1819. [DOI] [PubMed] [Google Scholar]
- 31. Amm I, Sommer T, Wolf DH. Protein quality control and elimination of protein waste: the role of the ubiquitin‐proteasome system. Biochim Biophys Acta 2014; 1843: 182–196. [DOI] [PubMed] [Google Scholar]
- 32. Yang ZJ, Chee CE, Huang S et al The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011; 10: 1533–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mizushima N. Autophagy: process and function. Genes Dev 2007; 21: 2861–2873. [DOI] [PubMed] [Google Scholar]
- 34. Lapierre LR, Kumsta C, Sandri M et al Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 2015; 11: 867–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct 2002; 27: 421–429. [DOI] [PubMed] [Google Scholar]
- 36. Hoang B, Benavides A, Shi Y et al Effect of autophagy on multiple myeloma cell viability. Mol Cancer Ther 2009; 8: 1974–1984. [DOI] [PubMed] [Google Scholar]
- 37. Verfaillie T, Salazar M, Velasco G et al Linking ER stress to autophagy: potential implications for cancer therapy. Int J Cell Biol 2010; 2010: 930509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Milani M, Rzymski T, Mellor HR et al The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with bortezomib. Cancer Res 2009; 69: 4415–4423. [DOI] [PubMed] [Google Scholar]
- 39. Kawaguchi T, Miyazawa K, Moriya S et al Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: crosstalk among proteasome, autophagy‐lysosome and ER stress. Int J Oncol 2011; 38: 643–654. [DOI] [PubMed] [Google Scholar]
- 40. Escalante AM, McGrath RT, Karolak MR et al Preventing the autophagic survival response by inhibition of calpain enhances the cytotoxic activity of bortezomib in vitro and in vivo . Cancer Chemother Pharmacol 2013; 71: 1567–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Auner HW, Cenci S. Recent advances and future directions in targeting the secretory apparatus in multiple myeloma. Br J Haematol 2015; 168: 14–25. [DOI] [PubMed] [Google Scholar]
- 42. Oliva L, Cenci S. Autophagy in plasma cell pathophysiology. Front Immunol 2014; 5: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Feder ME, Hofmann GE. Heat‐shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Annu Rev Physiol 1999; 61: 243–282. [DOI] [PubMed] [Google Scholar]
- 44. Zuiderweg ER, Hightower LE, Gestwicki JE. The remarkable multivalency of the Hsp70 chaperones. Cell Stress Chaperones 2017; 22: 173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marcu MG, Doyle M, Bertolotti A et al Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1alpha. Mol Cell Biol 2002; 22: 8506–8513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Duus J, Bahar HI, Venkataraman G et al Analysis of expression of heat shock protein‐90 (HSP90) and the effects of HSP90 inhibitor (17‐AAG) in multiple myeloma. Leuk Lymphoma 2006; 47: 1369–1378. [DOI] [PubMed] [Google Scholar]
- 47. Fernandez‐Fernandez MR, Gragera M, Ochoa‐Ibarrola L et al Hsp70–a master regulator in protein degradation. FEBS Lett 2017; 591: 2648–2660. [DOI] [PubMed] [Google Scholar]
- 48. Chatterjee M, Andrulis M, Stuhmer T et al The PI3k/Akt signaling pathway regulates the expression of Hsp70, which critically contributes to Hsp90‐chaperone function and tumor cell survival in multiple myeloma. Haematologica 2013; 98: 1132–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Eletto D, Dersh D, Argon Y. GRP94 in ER quality control and stress responses. Semin Cell Dev Biol 2010; 21: 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chhabra S, Jain S, Wallace C et al High expression of endoplasmic reticulum chaperone grp94 is a novel molecular hallmark of malignant plasma cells in multiple myeloma. J Hematol Oncol 2015; 8: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harding HP, Novoa I, Zhang Y et al Regulated translation initiation controls stress‐induced gene expression in mammalian cells. Mol Cell 2000; 6: 1099–1108. [DOI] [PubMed] [Google Scholar]
- 52. Marciniak SJ, Yun CY, Oyadomari S et al Chop induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 2004; 18: 3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hetz C, Bernasconi P, Fisher J et al Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 2006; 312: 572–576. [DOI] [PubMed] [Google Scholar]
- 54. Chen D, Frezza M, Schmitt S et al Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets 2011; 11: 239–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Adams J, Palombella VJ, Sausville EA et al Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res 1999; 59: 2615–2622. [PubMed] [Google Scholar]
- 56. Ruschak AM, Slassi M, Kay LE et al Novel proteasome inhibitors to overcome bortezomib resistance. J Natl Cancer Inst 2011; 103: 1007–1017. [DOI] [PubMed] [Google Scholar]
- 57. Kuhn DJ, Chen Q, Voorhees PM et al Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin‐proteasome pathway, against preclinical models of multiple myeloma. Blood 2007; 110: 3281–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. San‐Miguel JF, Hungria VT, Yoon SS et al Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double‐blind phase 3 trial. Lancet Oncol 2014; 15: 1195–1206. [DOI] [PubMed] [Google Scholar]
- 59. Yong K, Cavet J, Johnson P et al Phase I study of KW‐2478, a novel Hsp90 inhibitor, in patients with B‐cell malignancies. Br J Cancer 2016; 114: 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Davenport EL, Zeisig A, Aronson LI et al Targeting heat shock protein 72 enhances Hsp90 inhibitor‐induced apoptosis in myeloma. Leukemia 2010; 24: 1804–1807. [DOI] [PubMed] [Google Scholar]
- 61. Ishii T, Seike T, Nakashima T et al Anti‐tumor activity against multiple myeloma by combination of KW‐2478, an Hsp90 inhibitor, with bortezomib. Blood Cancer J 2012; 2: e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Baranowska K, Misund K, Starheim KK et al Hydroxychloroquine potentiates carfilzomib toxicity towards myeloma cells. Oncotarget 2016; 7: 70845–70856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Montanari F, Lu M, Marcus S et al A phase II trial of chloroquine in combination with bortezomib and cyclophosphamide in patients with relapsed and refractory multiple myeloma. Blood 2014; 124: 5775. [Google Scholar]
- 64. Ri M, Iida S, Nakashima T et al Bortezomib‐resistant myeloma cell lines: a role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia 2010; 24: 1506–1512. [DOI] [PubMed] [Google Scholar]
- 65. Franke NE, Niewerth D, Assaraf YG et al Impaired bortezomib binding to mutant Beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012; 26: 757–768. [DOI] [PubMed] [Google Scholar]
- 66. Politou M, Karadimitris A, Terpos E et al No evidence of mutations of the PSMB5 (Beta‐5 subunit of proteasome) in a case of myeloma with clinical resistance to bortezomib. Leuk Res 2006; 30: 240–241. [DOI] [PubMed] [Google Scholar]
- 67. Shuqing L, Jianmin Y, Chongmei H et al Upregulated expression of the PSMB5 gene may contribute to drug resistance in patient with multiple myeloma when treated with bortezomib‐based regimen. Exp Hematol 2011; 39: 1117–1118. [DOI] [PubMed] [Google Scholar]
- 68. Ling SC, Lau EK, Al‐Shabeeb A et al Response of myeloma to the proteasome inhibitor bortezomib is correlated with the unfolded protein response regulator XBP‐1. Haematologica 2012; 97: 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Leung‐Hagesteijn C, Erdmann N, Cheung G et al Xbp1s‐negative tumor B cells and pre‐plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell 2013; 24: 289–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nikesitch N, Tao C, Lai K et al Predicting the response of multiple myeloma to the proteasome inhibitor bortezomib by evaluation of the unfolded protein response. Blood Cancer J 2016; 6: e432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Aragon IV, Barrington RA, Jackowski S et al The specialized unfolded protein response of B lymphocytes: ATF6alpha‐independent development of antibody‐secreting B cells. Mol Immunol 2012; 51: 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hong SY, Hagen T. Multiple myeloma Leu167ile (C.499c>a) mutation prevents XBP1 mRNA splicing. Br J Haematol 2013; 161: 898–901. [DOI] [PubMed] [Google Scholar]
- 73. Milan E, Perini T, Resnati M et al A plastic SQSTM1/p62‐dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy 2015; 11: 1161–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Roy M, Liang L, Xiao X et al Lycorine downregulates HMGB1 to inhibit autophagy and enhances bortezomib activity in multiple myeloma. Theranostics 2016; 6: 2209–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tian Z, D'Arcy P, Wang X et al A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014; 123: 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Feng X, Holmlund T, Zheng C et al Proapoptotic effects of the novel proteasome inhibitor b‐AP15 on multiple myeloma cells and natural killer cells. Exp Hematol 2014; 42: 172–182. [DOI] [PubMed] [Google Scholar]
- 77. Liu N, Liu C, Li X et al A novel proteasome inhibitor suppresses tumor growth via targeting both 19S proteasome deubiquitinases and 20S proteolytic peptidases. Sci Rep 2014; 4: 5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhuang J, Lee H, Kuiatse I et al The anti‐tumor effect of the ubiquitin‐activating enzyme (UAE) inhibitor TAK‐243 on pre‐clinical models of multiple myeloma. Blood 2016; 128: 3296. [Google Scholar]
- 79. Gu Y, Kaufman JL, Bernal L et al MLN4924, an NAE inhibitor, suppresses AKT and mTOR signaling via upregulation of REDD1 in human myeloma cells. Blood 2014; 123: 3269–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chauhan D, Tian Z, Nicholson B et al A small molecule inhibitor of ubiquitin‐specific protease‐7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012; 22: 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhou W, Yang Y, Xia J et al NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell 2013; 23: 48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Roberts TL, Ho U, Luff J et al Smg1 haploinsufficiency predisposes to tumor formation and inflammation. Proc Natl Acad Sci USA 2013; 110: E285–E294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Karam R, Lou CH, Kroeger H et al The unfolded protein response is shaped by the NMD pathway. EMBO Rep 2015; 16: 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rasche L, Weinhold N, Morgan GJ et al Immunologic approaches for the treatment of multiple myeloma. Cancer Treat Rev 2017; 55: 190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer 2007; 7: 750–762. [DOI] [PubMed] [Google Scholar]
- 86. Kampmeyer C, Nielsen SV, Clausen L et al Blocking protein quality control to counter hereditary cancers. Genes Chromosom Cancer 2017; 56: 823–831. [DOI] [PubMed] [Google Scholar]
- 87. Leleu X, Xu L, Jia X et al Endoplasmic reticulum stress is a target for therapy in Waldenstrom Macroglobulinemia. Blood 2009; 113: 626–634. [DOI] [PubMed] [Google Scholar]