

SUMMARY
Tumors contain hostile inflammatory signals generated by aberrant proliferation, necrosis, and hypoxia. These signals are sensed and acted upon acutely by the Toll-like receptors (TLRs) to halt proliferation and activate an immune response. Despite the presence of TLR ligands within the microenvironment, tumors progress, and the mechanisms that permit this growth remain largely unknown. We report that self-renewing cancer stem cells (CSCs) in glioblastoma have low TLR4 expression that allows them to survive by disregarding inflammatory signals. Non-CSCs express high levels of TLR4 and respond to ligands. TLR4 signaling suppresses CSC properties by reducing retinoblastoma binding protein 5 (RBBP5), which is elevated in CSCs. RBBP5 activates core stem cell transcription factors, is necessary and sufficient for self-renewal, and is suppressed by TLR4 overexpression in CSCs. Our findings provide a mechanism through which CSCs persist in hostile environments because of an inability to respond to inflammatory signals.
In Brief
Alvarado et al. demonstrate that glioblastoma cancer stem cells express a lower level of the innate immune receptor TLR4 than surrounding cells, which allows them to avoid inhibitory innate immune signaling that would otherwise suppress self-renewal.
INTRODUCTION
In parallel to the stem cells that function during normal development and tissue homeostasis, a self-renewing, tumorigenic cancer stem cell (CSC) population has been identified in many advanced cancers (Visvader and Lindeman, 2012), including glioblastoma, the most common malignant primary brain tumor (Ostrom et al., 2015). CSCs drive tumor growth (Singh et al., 2004) and therapeutic resistance (Bao et al., 2006; Chen et al., 2012). CSCs are not governed solely by intrinsic programs but are also influenced by the tumor microenvironment, which is essential in maintaining the balance between self-renewal and differentiation (Lathia et al., 2015). Some of these external stimuli are byproducts of rapid and uncontrolled proliferation and include hypoxia, acidic stress, and necrosis, which are not favorable to cell survival; however, CSCs have adapted by developing mechanisms to self-renew despite these inhospitable conditions (Hjelmeland et al., 2011; Li et al., 2009). Although CSCs can persist under unfavorable conditions, the mechanisms that enable them to survive and even thrive in harsh environments have not been elucidated.
The innate immune system recognizes both damage- and pathogen-associated molecular patterns and has evolved as an early response mechanism to respond to potentially harmful insults (Bianchi, 2007). In the tumor microenvironment, the existence of damage-associated signaling could affect the phenotype and activation status of tumor cells as well as infiltrating immune cells. Reponses to these ligands are mediated by distinct classes of receptors that sense and initiate pro-inflammatory signaling cascades. Among these, the Toll-like receptor (TLR) family activates inflammatory response pathways and is essential for the recruitment of effector immune cells (O’Neill et al., 2013). Receptor diversity accommodates a wide spectrum of ligands and accounts for an equally diverse set of responses from members of the TLR family. In the context of cancer, TLRs have both pro- and anti-tumorigenic roles that are tumor and cell-type specific (Pradere et al., 2014). The anti-tumorigenic roles of TLRs are generally due to induction of anti-tumor immunity via activation of dendritic cells (Pradere et al., 2014). The pro-tumorigenic roles are mediated directly by tumor-associated macrophages, dendritic cells, and endothelial cells along with TLR-mediated cytokine production (Elinav et al., 2013; Hu et al., 2015; Vinnakota et al., 2013). In multiple tumor types, including hepatocellular carcinoma and colorectal, prostate, and bladder cancer, TLRs appear to function more in a pro-tumorigenic role (Cheah et al., 2015; Grimm et al., 2010; Zhao et al., 2015), and the precise outcome of TLR signaling in tumor cells, including CSCs, is an ongoing area of investigation.
There is accumulating evidence that TLRs may be important for CSC maintenance. In hepatocellular carcinoma, TLR4 directly regulates the core self-renewal gene NANOG to drive CSC maintenance (Chen et al., 2013). In GBM, it has recently been demonstrated that self-renewal is mediated by TLR9 (Herrmann et al., 2014). Similar pro-proliferative roles have been described for TLR4 in embryonic stem cells and mammary progenitor cells (Lee et al., 2009); however, anti-proliferative roles have also been described for TLR2, TLR3, and TLR4 in embryonic (Lathia et al., 2008; Okun et al., 2010) and adult neural progenitor cells (Rolls et al., 2007). These findings suggest that distinct TLRs can modulate the proliferation of non-immune cells in a positive or negative manner. As tumor cells reside within an inhospitable environment, we were interested in the adaptive mechanisms that CSCs possess to evade these damage-associated signals. The innate immune machinery, which is regulated by TLRs, is a critical regulator of damage signaling. We therefore interrogated GBM CSCs versus their non-stem cell (non-CSC) counterparts for the expression of TLRs and examined how these receptors affected self-renewal. We found that CSCs exhibit diminished levels of TLR4, allowing them to thrive in a hostile environment and avoid activation of inflammatory pathways. Functionally, TLR4 was able to suppress tumor growth by reducing the expression of retinoblastoma binding protein 5 (RBBP5), a transcription factor that we found to be elevated in CSCs and necessary and sufficient for their maintenance. Taken together, our findings demonstrate that CSCs have reduced innate immune signaling that, when activated, directly suppresses CSC maintenance.
RESULTS
CSCs Have Reduced Response to Innate Immune Stimulation and Lower Expression of TLR4
Within a rapidly proliferating tumor, there is a concomitant generation of cellular debris. This process can initiate an immune response via TLRs, as the debris generated can stimulate TLR signaling. Although TLRs are known to mediate the activation of glioblastoma-associated immune cells, it remains unclear how tumor cells respond to TLR ligands. Work from the developing CNS suggests that TLR ligands may elicit signaling responses directly in non-immune cells, and therefore, these ligands may affect tumor cell behavior (Pradere et al., 2014). Moreover, recent work in CSCs demonstrates an elevation of TLR4 in hepatocellular carcinoma and of TLR9 in GBM (Chen et al., 2013; Herrmann et al., 2014). To systematically assess how CSCs respond to TLR ligands, we investigated the effect of several TLR ligands on the proliferation of both CSCs and non-CSCs isolated from patient-derived GBM xenografts. In all the specimens analyzed, ligands that stimulate TLR2 (Pam3C), TLR3 (polyinosinic:polycytidylic acid [PolyI:C]), and TLR9 (oligodeoxy-nucleotides [CpG]) showed no statistically significant effect on either cell population (Figures 1A and S1A). However, the TLR4 agonist lipopolysaccharide (LPS) showed a differential effect on tumor cells; CSCs proliferated normally, but the growth of non-CSCs was severely inhibited. To confirm that this effect was TLR4 specific, we used a signaling inhibitor (CLI-095) and observed that the decrease in proliferation induced by TLR4 stimulation was abrogated by the addition of CLI-095 (Figure S1B). In the brain, normal and tumor cells are rarely exposed to microbial components such as TLR ligands; however, there are endogenous proteins that can also serve as activators for this family of receptors. During necrosis and other injury- and stress-related cellular processes, proteins that are usually found inside cells can be released and act as damage-associated molecular patterns (DAMPs). These can be recognized by TLRs to activate inflammatory pathways and/or recruit immune cells from the periphery. We therefore stimulated cells with the endogenous TLR4 and TLR2 ligand high mobility group box 1 (HMGB1) (Figure 1A). As expected, only the non-CSC population was affected, showing a decrease in proliferation compared with the CSCs. Consistent with our findings, treating cells with CLI-095 blocked the decrease in proliferation induced by HMGB1 (Figure S1B). Of note, treatment of non-CSCs with CLI-095 alone increased cellular growth and led to higher expression of the stem cell markers SOX2, OCT4, and NANOG (Figures S1C and S1D, respectively), suggesting that TLR4 may suppress proliferation and stem cell marker expression. These observations are consistent with observations that TLR4 suppresses proliferation in adult neural stem cells and retinal progenitor cells (Rolls et al., 2007; Shechter et al., 2008).
Figure 1. CSCs and Non-CSCs Respond Differentially to Innate Immune Stimulation and Express Differential Levels of TLR4.

(A) CSCs (blue) and non-CSCs (black) were incubated with TLR ligands for 7 days, and proliferation relative to day 0 was assessed by CellTiter Glo assay and normalized to an untreated control. The following concentrations were used: LPS, 500 ng/mL; HMGB1, 1 μg/mL; CpG, 2 μM; Pam3C, 300 ng/mL; and Poly(I:C), 10 μg/mL.
(B) The mRNA expression level of all known human TLRs was evaluated by qRT-PCR in CSCs and non-CSCs; data were normalized to the non-CSC group.
(C and D) mRNA (C) and protein (D) levels of TLR4 were analyzed in CSCs and non-CSCs using qRT-PCR and western blot, respectively. Actin was used as an internal control for qRT-PCR and a loading control for western blotting.
Each experiment was performed at least three times. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 as assayed by one-way ANOVA. See also Figure S1.
We then evaluated the expression of human TLRs in CSCs and non-CSCs. Several significant differences were noted: TLR2 and TLR9 were highly expressed, while TLR3, TLR4, and TLR8 were expressed at lower levels in CSCs compared with non-CSCs (Figure 1B). We thus hypothesized that the reason CSCs were not affected by stimulation with TLR4 ligands (LPS and HMGB1) was their lower TLR4 expression. We confirmed these observations by analyzing additional specimens at both the mRNA (Figure 1C) and protein (Figure 1D) levels. Consistently, CSCs showed decreased levels of TLR4 expression compared with the non-CSC population. When we assessed TLR4 expression in GBM patient-derived tumorspheres, which contain both CSCs and non-CSCs, we observed a heterogeneous expression pattern (Figure S1E). These data confirm previous reports in which TLR4 was detected via immunohistochemistry in GBM specimens (Sarrazy et al., 2011; Tewari et al., 2012). To determine whether TLR4 was associated with differences in GBM patient prognosis, we assessed expression in the National Cancer Institute’s Rembrandt database and found that lower levels of TLR4 in patients correlated with a decrease in median survival (Figure S1F). Similar analyses performed for other TLRs (TLR3, TLR8, TLR9) and associated downstream signaling pathways did not demonstrate any differences in patient prognosis (Figure S1F and the MYD88, TRAF6, IRAK1, IRAK4, TICAM1, and TBK1 signaling molecules, data not shown). Moreover, when we compared GBM with astrocytoma, we found lower levels of TLR4 (Figure S1G), suggesting that there is a decrease in TLR4 in more malignant tumors. These data demonstrate that CSCs express lower levels of TLR4 compared with non-CSCs and may explain, in part, why CSCs are less responsive to TLR4-mediated damage signals compared with non-CSCs.
TLR4 Expression Negatively Correlates with CSC Features
To test the effect of TLR4 stimulation in vivo, we treated tumors established in immunocompromised mice with LPS or PBS and then evaluated tumor size (Figure 2A). We observed a dramatic decrease in tumor volume after treatment with a single LPS injection (150 and 300 μg) compared with PBS treatment (Figure 2B). These data are in line with a previously published report in which LPS decreased tumor growth in a syngeneic mouse model (Chicoine et al., 2001) and demonstrate that TLR4 stimulation alters tumor growth in the absence of the immune system. This confirms our in vitro data demonstrating that LPS treatment decreases the proliferation of TLR4-expressing tumor cells. Moreover, to assess the functional implications of TLR4 expression, we evaluated the stem cell characteristics of cells expressing the receptor. In unenriched GBM xenograft cells, 43% were TLR4 positive; however, after enrichment with the CSC marker CD133, fewer than 1% of cells were positive for TLR4 (Figure S2A). This confirms the expression results previously observed by RNA and protein analysis. We then assessed self-renewal in cells expressing different levels of TLR4 by limiting dilution analysis experiments. In multiple specimens, TLR4-low cells had higher stem cell frequencies than cells expressing high TLR4 levels (Figures 2C and S2B). Similarly, when we modulated stem cell conditions in culture by incubating CSCs in differentiation media (containing 10% serum), we observed a gradual increase in the expression of TLR4 concomitant with a decrease in the expression of the stem cell markers SOX2, OCT4, and NANOG and an increase in GFAP, a marker of astrocytic lineage commitment (Figure 2D and S2C). These data show that LPS reduces growth in vivo and that TLR4 is downregulated in CSCs and negatively correlates with self-renewal.
Figure 2. TLR4 Expression Negatively Correlates with Stemness.

(A and B) Experimental paradigm to assess the effect of LPS on tumor growth in vivo (A). Mice (n = 7 per arm) were injected with cells from xenograft T3832. After 14 days, tumors were injected with LPS (150 or 300 μg), and tumor volume (B) was measured using electronic calipers 3 days later. Tumor volume at day 3 was normalized to the volume of the tumor at day 0 when LPS was injected.
(C) Bulk tumors derived from patient specimens were sorted for TLR4 surface expression using fluorescence-activated cell sorting (FACS). The 20% of cells with the highest expression and the 20% of cells with the lowest expression were plated in a limiting dilution manner, and the number of wells containing spheres was counted after 10 days to generate stem cell frequencies using the online algorithm detailed in the STAR Methods.
(D) CSCs were differentiated in DMEM containing 10% fetal bovine serum (FBS) for the indicated lengths of time, and protein levels of TLR4, SOX2, OCT4, NANOG, GFAP, and Actin were assessed by western blotting.
Experiments in (C) and (D) were performed at least three times. Data are represented as mean ± SEM. ***p < 0.001 as assayed by one-way ANOVA. See also Figure S2.
TLR4 Overexpression Reduces Proliferation and Inhibits CSC Maintenance
To evaluate the effect of modulating TLR4 expression on CSC function, we overexpressed TLR4 in CSCs using a transient expression vector and observed increases in TLR4 at both the mRNA (Figure 3A) and protein (Figure 3B) levels. We previously showed that TLR4 levels increase when CSCs are differentiated (Figure 2E), and we observed that forced TLR4 expression resulted in a decrease in pluripotency genes at the mRNA and protein levels (SOX2, NANOG, and OCT4; Figures 3B, S3A, and S3B). In addition, TLR4 overexpression decreased proliferation even in the absence of a TLR4 ligand (Figure 3C). When we added LPS, we observed a further decrease in the proliferation of cells overexpressing TLR4 (Figure 3D) but not in the control group (data not shown). This reduction was not dramatic and was likely due to the strong effect induced by TLR4 overexpression alone (data not shown). Along with the decrease in growth observed with TLR4 overexpression, cells overexpressing TLR4 displayed lower stem cell frequencies compared with cells expressing a control vector (Figure 3E). This effect (decreases from 1 in 35 and 1 in 27 in control cells to 1 in 120 and 1 in 71 in TLR4-overexpressing cells) supports the hypothesis that the activation of innate immune signaling can be detrimental for cancer cells. In vivo, the median survival of mice injected with CSCs overexpressing TLR4 was higher (44.5 days) than that of mice receiving control CSCs (37 days; Figure S3C). Although the difference in survival failed to reach statistical significance, this trend supports the in vitro studies showing that TLR4 overexpression decreased proliferation and self-renewal. The differences between the in vitro and in vivo results could be due to a variety of variables, including differences in the duration of in vitro and in vivo assessments, the selective pressure induced by the in vivo environment, or the presence of activating ligands in vivo. Together, these data suggest that signaling through the innate immune receptor TLR4 might feed into molecular pathways that regulate CSCs.
Figure 3. TLR4 Overexpression Decreases Proliferation and Reduces CSC Maintenance.

(A and B) Transient TLR4 overexpression in CSCs was measured at both the mRNA (A) and protein (B) levels using qRT-PCR and western blotting, respectively. Western blots were also stained for the pluripotency factors SOX2, NANOG, and OCT4. CSCs were nucleofected with pcDNA3-TLR4-YFP and used for experiments 3 days later. Actin was used as an internal control for qRT-PCR and a loading control for western blotting.
(C) Proliferation over 7 days was assessed in CSCs derived from the shown specimens using CellTiter Glo after overexpression of TLR4. Growth was normalized to cells containing an empty vector.
(D) CSCs overexpressing TLR4 were treated with LPS at 500 ng/mL, and proliferation was assessed for 7 days relative to cells containing a control vector.
(E) Limiting dilution analysis was used to estimate stem cell frequencies in CSCs after nucleofection with control or TLR4 overexpression vectors. Cells were plated in a limiting dilution manner, and the number of wells containing spheres was counted after 10 days to generate stem cell frequencies using the online algorithm detailed in the STAR Methods.
All experiments were performed at least three times. Data are represented as mean ± SEM. **p < 0.01 and ***p < 0.001 as assessed by one-way ANOVA. See also Figure S3.
TLR4 Overexpression Compromises CSC Characteristics by Repressing the Transcription Factor RBBP5
Given that TLR4 expression levels modulated the expression of key pluripotency transcription factors (SOX2, NANOG, and OCT4; Figures 3B and S3A), we hypothesized that TLR4 signaling might regulate the transcription of these key self-renewal genes. To gain mechanistic insight into this possibility, we first analyzed the promoter region of all three pluripotency genes using the University of California Santa Cruz (UCSC) Genome Browser and compiled a list of common transcription factors able to bind to all three regions. This list of factors was cross-referenced against an RNA sequencing (RNA-seq) data set (Suvà et al., 2014) that includes RNA expression in tumor propagating cells (TPCs) compared with differentiated glioblastoma cells (DGCs). We considered transcription factors that were greater than 1.5-fold differentially expressed and evaluated their expression in the context of TLR4 overexpression (Figure 4A). Notably, some of the differentially expressed genes in the RNA-seq data set did not appear to be affected by the levels of TLR4 (REST, SOX5, TEAD4, POU3F2, and MYC). JUN, a known downstream effector of TLR signaling, was downregulated when TLR4 was overexpressed; however, other subunits of this transcription factor complex (JUNB, JUND, and FOS; data not shown) did not show the same trend and were not evaluated in follow-up studies. The lack of a consistent change in the JUN complex was not surprising given the differential role of different activating protein (AP-1) subunits during inflammation and cancer (Hess et al., 2004), suggesting a more complex underlying biology. The most dramatic effect was seen for retinoblastoma binding protein 5 (RBBP5), in which overexpression of TLR4 resulted in a 60% decrease in RBBP5 expression (Figure 4A). In addition to its role binding retinoblastoma, as the name suggests, RBBP5 is part of a core complex that interacts with SET1 family members to tri-methylate lysine 4 on histone 3 (Ernst and Vakoc, 2012). This epigenetic mark is considered to be indicative of active transcription. When we analyzed the expression of RBBP5 in both CSCs and non-CSCs, we found that RBBP5 was more highly expressed in the CSC population (Figure 4B), as expected from our analysis of the RNA-seq data set. To directly assess the interaction between TLR4 and RBBP5, we overexpressed TLR4 in CSCs and found a decrease in the promoter activity of RBBP5 (Figure 4C). When we overexpressed TLR4 in CSCs, we also observed a decrease in the RBBP5 protein, with a concomitant increase in the differentiation marker GFAP (Figure 4D), suggesting that RBBP5 may be linked to the CSC state. To further assess this association, we differentiated CSCs, a process that we previously showed increased TLR4 levels (Figures 2D and S2C), and observed a decrease in RBBP5 (Figure 4E). These data identify RBBP5 as a downstream target of TLR4 and provide the first evidence for an association between TLR signaling and the CSC state via RBBP5.
Figure 4. TLR4 Overexpression Compromises Stemness by Repressing the Transcription Factor RBBP5.

(A) The expression of the shown transcription factors was analyzed in CSCs overexpressing TLR4 using qRT-PCR and normalized to cells containing a control vector. CSCs were nucleofected with pcDNA3-TLR4-YFP and used for experiments 3 days later. Actin was used as an internal control.
(B) Protein levels of RBBP5 were analyzed in both CSCs and non-CSCs using western blot. Actin was used as a loading control.
(C) A reporter expressing luciferase under the control of the RBBP5 promoter region was used to measure RBBP5 expression after transient TLR4 overexpression in CSCs. CSCs were nucleofected with pcDNA3-TLR4-YFP and used for experiments 3 days later. Luciferase levels were normalized to those of control (NT) cells.
(D) Levels of RBBP5 and GFAP were determined by western blot in CSCs expressing both control and TLR4-overexpression vectors. Actin was used as a loading control.
(E) CSCs from the shown specimens were differentiated by incubating with DMEM with 10% FBS for the indicated times, and protein levels of RBBP5 were determined by western blot. Actin was used as a loading control.
All experiments were performed at least three times. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 as assayed by one-way ANOVA.
Targeting RBBP5 Mimics TLR4 Overexpression and Decreases CSC Characteristics
To evaluate whether RBBP5 directly regulates the CSC phenotype, we targeted it in CSCs using two independent short hairpin RNA (shRNA) lentiviral constructs. We confirmed downregulation of RBBP5 mRNA (Figure S4A) and protein (Figure 5A) in two specimens. Importantly, targeting RBBP5 also decreased SOX2, NANOG, and OCT4 mRNA (Figure S4A) and protein (Figure 5A). Both specimens analyzed showed a significant decrease in proliferation when RBBP5 was targeted with either shRNA, knockdown 1 (KD1) or knockdown 2 (KD2), compared with a non-targeting (NT) control (Figure 5B). A similar effect on self-renewal was observed when cells treated with these shRNA constructs were evaluated by limiting dilution analysis (Figures 5C and 5D). Stem cell frequencies decreased from 1 in 2 to 1 in 13 (KD1) and 1 in 12 (KD2) for specimen T387 and from 1 in 15 to 1 in 275 (KD1) and 1 in 133 (KD2) for specimen T4121. We further analyzed the effects of targeting RBBP5 in vivo in the T4121 specimen and observed a decrease in tumor initiation capacity compared with the NT control (Figure 5E). Mice injected intracranially with CSCs treated with RBBP5 shRNA failed to show neurological symptoms (KD1 and KD2) after 75 days, while NT control animals reached experimental endpoints within 55 days of injection (median survival 43 days). We then tested the effects of RBBP5 overexpression on both CSCs and non-CSCs using a lentiviral vector to stably transduce patient-derived cells (Figure S4B). As expected, CSCs overexpressing RBBP5 showed increased levels of proliferation (Figure 5F) as well as higher stem cell frequencies compared with control CSCs (Figure 5G). Importantly, RBBP5 overexpression in non-CSCs had similar effects on self-renewal (Figure 5G). These data highlight the importance of RBBP5 for the maintenance of CSCs and suggest that RBBP5 is not only necessary but also sufficient for the expression of pluripotency genes. Notably, knockdown of RBBP5 mimicked the effects of TLR4 overexpression, suggesting that RBBP5 suppression and the loss of the CSC state are downstream of TLR4 activation.
Figure 5. Targeting RBBP5 Mimics TLR4 Overexpression in CSCs.

(A) Validation of two shRNA constructs targeting RBBP5 at the protein level compared with non-targeting vector (NT). Western blots were also stained for the pluripotency factors SOX2, NANOG, and OCT4. CSCs were infected with lentivirus containing one of two different RBBP5 shRNA constructs, selected for stable expression, and assayed within five passages for all experiments. Actin was used as a loading control.
(B) The proliferation of CSCs stably expressing RBBP5 shRNA was measured and compared with an NT vector. The growth of the cells over 7 days as determined by CellTiter Glo was normalized to the growth of NT cells.
(C and D) Limiting dilution analysis of the effect of RBBP5 knockdown on two different specimens. Cells were plated in a limiting dilution manner, and the number of wells containing spheres was counted after 10 days to generate stem cell frequencies using the online algorithm detailed in the STAR Methods.
(E) Mice (n = 7 for KD, n = 6 for NT) were intracranially injected with 1000 T4121 NT or RBBP5-knockdown CSCs, and the time until endpoint was recorded. Kaplan-Meier survival plots are shown, and the log rank p value for significance between groups is shown next to the survival curve.
(F) Proliferation was analyzed after RBBP5 overexpression over 7 days in CSCs. Growth was assessed using CellTiter Glo and normalized to that of cells containing an empty vector. Cells were infected with lentivirus, selected for stable expression, and used within five passages for all experiments.
(G) Limiting dilution analyses of both CSCs and non-CSCs overexpressing RBBP5 compared with a control vector. Cells were plated in a limiting dilution manner, and the number of wells containing spheres was counted after 10 days to generate stem cell frequencies using the online algorithm detailed in the STAR Methods.
All in vitro experiments were performed at least three times. Data are represented as mean ± SEM. **p < 0.01 and ***p < 0.001 as assayed by one-way ANOVA. See also Figure S4.
On the basis of our observations that RBBP5 targeting compromised the expression of pluripotency genes at both the protein and mRNA levels, we wanted to further explore this mechanism at the transcriptional level using luciferase reporter vectors under the control of the promoter regions of the pluripotency genes SOX2, OCT4, and NANOG. In accordance with our previous results, knockdown of RBBP5 with two independent constructs decreased luciferase levels from all three gene promoters (Figure S4C). Similarly, we introduced the same constructs in non-CSCs overexpressing RBBP5, and as expected, the luciferase levels were higher for all three genes after RBBP5 introduction compared with an empty vector (Figure S4D). RBBP5 does not directly bind DNA but is a member of the WRAD complex that interacts with proteins that can modify the epigenetic state, such as mixed-lineage leukemia 1 (MLL1), which is essential for GBM CSCs (Gallo et al., 2013; Heddleston et al., 2012). To determine whether TLR4 and RBBP5 affect MLL1, we overexpressed TLR4 or knocked down RBBP5 in CSCs and assessed MLL1 levels. We found that either perturbation decreased MLL1 levels (Figures S4E and S4F). Because alterations in MLL1 should affect the epigenetic state of pluripotency genes, we assessed histone methylation at the promoters of these genes. We first analyzed the promoter regions of NANOG and OCT4 in both CSCs and non-CSCs. Our chromatin profiling of the NANOG and OCT4 loci showed higher enrichment of histone 3 lysine 4 tri-methylation (H3K4me3; 2- to 3-fold increased for NANOG and 4-fold increased for OCT4) (Figure S4G) but not histone 3 lysine 4 dimethylation (H3K4me2) or histone 3 lysine 4 mono-methylation (H3K4me1, data not shown) in CSCs compared with non-CSCs. Next, we assessed the effect of RBBP5 targeting on H3K4me3 in the promoter regions of OCT4 and NANOG in CSCs. Targeting RBBP5 with two independent shRNA constructs decreased the amount of H3K4me3 at both gene loci (Figure S4H) compared with a non-targeting control (by as much as 17-fold for NANOG and 15-fold for OCT4). To ensure that this effect was specific and not a result of a global chromatin change, we analyzed the same epigenetic modifications in an unrelated gene, solute carrier family 3 member 2 (SLC3A2) as a control and did not observe a significant change with RBBP5 knockdown (data not shown). Taken together, these data demonstrate that RBBP5 is necessary and sufficient for CSC phenotypes.
TLR4 Signals via TBK1 to Suppress RBBP5
To determine the signaling link between TLR4 and RBBP5, we used an Ingenuity Pathway Analysis using TLR4, SOX2, and RBBP5 as nodes to elucidate other members in this signaling pathway (data not shown). Interestingly, the network suggested a putative interaction between TLR4 and RBBP5 via tank-binding kinase 1 (TBK1), which is known to act downstream of TLR4 in a myeloid differentiation primary response gene 88 (MyD88)-independent manner. To confirm this interaction, we evaluated the phosphorylation status of TBK1 upon stimulation with TLR4 ligands. With each ligand, the activation of TLR4 signaling led to an increase in TBK1 phosphorylation compared with an untreated control (Figure 6A). To assess whether TBK1 is downstream of TLR4, we assessed proliferation in non-CSCs upon TLR4 stimulation in the context of TBK1 inhibition via a TBK1 signaling inhibitor, BX795. Treatment of non-CSCs with BX795 abrogated the decrease in proliferation induced by treatment with both LPS and HMGB1 (Figure 6B). To assess how RBBP5 is linked to TBK1, we determined the expression of RBBP5 mRNA in conditions with LPS stimulation and TBK1 inhibition and found that the LPS-induced reduction in RBPP5 levels in non-CSCs was rescued by TBK1 inhibition (Figure 6C). To examine this pathway in CSCs, we overexpressed TLR4 and then analyzed the phosphorylation of TBK1. Forced expression of TLR4 increased the phosphorylation of TBK1 and IRAK1 (Figure 6D), indicating that TLR4 expression activates this signaling pathway downstream of the receptor. To evaluate the impact of the TLR4-TBK1 signaling axis on RBBP5 and pluripotency transcription factor expression, we assessed CSCs with or without forced expression of TLR4 and treated with the TBK1 inhibitor. As expected, TLR4 overexpression decreased the levels of RBBP5 and the stem cell genes SOX2, OCT4, and NANOG (Figure S5). However, treatment with BX795 in TLR4-overexpressing cells rescued the mRNA levels of all the aforementioned genes to levels comparable to those of the control group. It is important to note that treatment of control cells with BX795 also led to an increase in the expression of RBBP5 and stem cell genes, raising the possibility that other signaling pathways could be involved in regulating CSC maintenance. The relationship between TBK1 and RBBP5 was also observed when TBK1 protein was reduced by RNAi in non-CSCs, which increased RBBP5 levels (data not shown). These data further confirm the effect of TLR4 in regulating pluripotency transcription factors via RBBP5 and provide evidence that TBK1 mediates the effects of this signaling cascade (Figure 6E).
Figure 6. TBK1 Acts as a Signaling Intermediate between TLR4 and RBBP5.

(A) Non-CSCs were treated with LPS (500 ng/mL) or HMGB1 (1 μg/mL) for 1 day, and the phosphorylation of TBK1 was analyzed by western blot with a p-TBK1-specific antibody. Total protein was used as a loading control.
(B) Proliferation of non-CSCs treated with LPS (500 ng/mL) or HMGB1 (1 μg/mL) in the presence or absence of TBK1 inhibitor (BX795; 100 nM) for 1 day and normalized to untreated controls.
(C) Non-CSCs were treated with LPS (500 ng/mL) with and without the addition of TBK1 inhibitor (BX795; 100 nM) for 1 day, and the mRNA levels of RBBP5 were measured and normalized to the control group. Actin was used as an internal control.
(D) TLR4 was transiently overexpressed in CSCs using nucleofection of pcDNA3-TLR4-YFP, and phosphorylation and levels of TBK1 and IRAK1 were analyzed by western blotting and compared with control vector. Total protein was used as a loading control.
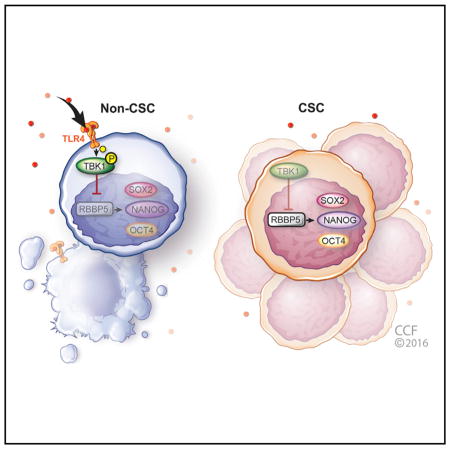
(E) Reduced TLR4 expression in cancer stem cells enables them to persist in hostile environments. Working model showing the signaling axis where TLR4 activation leads to the phosphorylation of TBK1, which suppresses RBBP5. RBBP5 in turn regulates stem cell genes and is critical for CSC maintenance. In non-CSCs (left), TLR4 expression allows for the activation of TBK1 in the presence of appropriate TLR4 ligands, which consequently inhibits RBBP5, decreasing stem cell gene expression. In contrast (right), the lack of TLR4 expression in CSCs suppresses the inhibition of RBBP5 and promotes the activation of self-renewal programs with consequences on proliferation, growth, and tumor initiation.
All experiments were performed at least three times. Data are represented as mean ± SEM. *p < 0.05 and **p < 0.01 as assayed by one-way ANOVA. See also Figure S5.
DISCUSSION
TLR activation limits proliferation in developing neural systems, yet these pathways have been shown to be essential for proliferation in many tumors including hepatocellular carcinoma and glioblastoma. This functional shift is surprising considering the number of developmental programs activated in CSCs. Our findings demonstrate that TLR4 is anti-proliferative for CSCs in GBM and provide a mechanism by which CSCs can persist in a hostile microenvironment by losing the ability to sense damage signals. Our findings also add to the increasing understanding of non-immune cell functions for TLRs in the brain that regulate proliferation, survival, and self-renewal in the context of development, neurodegeneration, and neoplasia. The brain has a distinct set of immune regulatory mechanisms, and diverse TLR expression in neurons, astrocytes, oligodendrocytes, and microglia contributes to normal brain function and homeostasis (Jack et al., 2005; Okun et al., 2009; van Noort and Bsibsi, 2009). It appears that TLRs also regulate GBM growth and progression. TLR9 functions in a pro-tumorigenic manner to drive self-renewal (Herrmann et al., 2014), and a versican-TLR2-MT1-MMP signaling axis has been linked to tumor invasion and expansion (Hu et al., 2015). However, TLR2 activation on dendritic cells leads to tumor regression, and this is mediated via HMGB1, which is generated by stressors such as GBM standard-of-care irradiation and chemotherapy (temozolomide) (Curtin et al., 2009). Along with more comprehensive studies to appreciate the cell-type-specific role of individual TLRs during tumor growth, a more detailed analysis of endogenous ligands is required. It is surprising how little information is available as to the precise DAMPs present within the tumor microenvironment and how therapies may alter DAMP signature. This is essential information that may help explain the alterations in the immune system within GBM and reveal TLR-dependent interventions for follow-up studies. Moreover, this information may be useful in activating the immune system for complementary immunotherapy approaches, as exposure to TLR ligands is currently one approach being assessed (Deng et al., 2014).
Traditionally, TLR activation in non-tumorigenic cells has been associated with inflammation and the progression of tumor growth (Elinav et al., 2013). Recent evidence has also linked TLR activation in tumor cells to tumorigenesis in different oncogenic contexts such as hepatocellular carcinoma and colorectal and prostate and bladder tumors. TLR4 in particular has been associated with driving hepatocellular carcinoma progression (Dapito et al., 2012) and interacts with the CSC signaling network via NANOG and STAT3 (Chen et al., 2013; Uthaya Kumar et al., 2016). In this report, we describe the opposite role for TLR4 in GBM, where the receptor suppresses tumor growth by directly attenuating self-renewal circuitry (Figure 6E). Functionally, self-renewal was suppressed by TLR4 overexpression as assessed by limiting dilution assays that integrate proliferation, survival, and self-renewal. In this context, CSCs give rise to non-CSCs in heterogeneous tumorspheres, but the majority of divisions are symmetric in that a CSC gives rise to daughter cells that remain CSCs (Lathia et al., 2011). Overexpression of TLR4 altered the dynamics of tumorsphere formation, which was consistent with decreases in proliferation and pluripotency gene expression. The tumor-suppressive functions we observed for TLR4 in GBM likely reflect the differences in the specialized immune co-adaptors and co-stimulatory molecules within the brain compared with those in the liver. From a signaling perspective, there are also differences in how TLR4 links to the self-renewal signaling network. We observed a repression of NANOG expression upon TLR4 activation, while work in hepatocellular carcinoma observed an alcohol-induced activation of NANOG with coordinate activation of AKT and TGFβ (Chen et al., 2013), both of which are major drivers of CSC maintenance in GBM (Anido et al., 2010; Bleau et al., 2009; Eyler et al., 2008). This divergence in signaling response could be due to ligand specificity or downstream pathway activation, because TBK1 is a component of the MyD88-independent response and the activation of TLR4 may occur via a MyD88-dependent mechanism in hepatocellular carcinoma. Further elucidation of these differences in signaling pathways is an immediate priority, as it may clarify the pro- and anti-tumorigenic roles of TLR4. For example, it has recently been shown that TLR4 is necessary for epidermal growth factor receptor (EGFR) signaling in mammary epithelial cells via MyD88 (De et al., 2015). Given the importance of EGFR in GBM progression (Brennan et al., 2013) and CSC self-renewal (Mazzoleni et al., 2010), the activation of MyD88-dependent or -independent pathways may provide diametrically opposite results. Furthermore, clarifying this difference will be vital information as TLR modulation is being explored as an adjuvant immunotherapy for brain tumors (Deng et al., 2014).
On the basis of our findings, there may be therapeutic opportunities to target this new TLR4-TBK1-RBBP5 signaling axis. Although TLR4 suppresses self-renewal, it remains a poor choice for therapeutic intervention, as overexpressing a receptor is challenging to achieve in vivo. TBK1 faces the same issue, as modulation of this axis would require an overexpression strategy. Our data suggest that RBBP5 is a critical member of this pathway, and our pre-clinical genetic studies indicate that its reduction attenuates stem cell gene expression, self-renewal, and tumor initiation. RBBP5 could be targeted by the development of specific small molecules or specific micro-RNAs that suppress its expression. Recently, another member of the retinoblastoma binding protein family has been demonstrated to be important for chemosensitivity in GBM (Kitange et al., 2016), suggesting that targeting this family may have therapeutic benefit. Although these approaches would require substantial development, they may provide next-generation anti-CSC targeting strategies. Taken together, our findings reveal that a reduced ability to sense damage signals and activate innate immune signaling via TLR4 provides CSCs a survival advantage in hostile environments. Leveraging this mechanism may sensitize CSCs to damage signals in the tumor microenvironment and provide a unique therapeutic opportunity to compromise CSC maintenance.
STAR★METHODS
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TLR4 | Santa Cruz Biotechnology | Cat# sc-293072; RRID: AB_10611320 |
| Actin | Santa Cruz Biotechnology | Cat# sc-7210; RRID: AB_2223518 |
| SOX2 | R&D Systems | Cat# mAb2018; RRID: AB_358009 |
| RBBP5 | Cell Signaling | Cat# 13171 |
| GFAP | Invitrogen | Cat# NB300-141 |
| TBK1 | Cell Signaling | Cat# 3504; RRID: AB_2255663 |
| phospho-TBK1 | Cell Signaling | Cat# 5483 |
| IRAK | Abcam | Cat# ab238; RRID: AB_303024 |
| phospho-IRAK | Abcam | Cat# ab139739 |
| MLL | Bethyl Laboratories | Cat# A300-374A; RRID: AB_345243 |
| OCT4 | Cell Signaling | Cat# 2750; RRID: AB_823583 |
| NANOG | Cell Signaling | Cat# 4903 |
| TLR4-PE | Santa Cruz Biotechnology | Cat# sc-13593 PE; RRID: AB_628366 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CpG | InvivoGen | Cat# tlrl-2216 |
| Pam3C | InvivoGen | Cat# tlrl-pms |
| PolyI:C | InvivoGen | Cat# tlrl-pic |
| CLI-095 | InvivoGen | Cat# tlrl-cli95 |
| BX795 | InvivoGen | Cat# tlrl-bx7 |
| LPS | Sigma | Cat# L2880 |
| HMGB1 | GenScript | Cat# Z02803 |
| Geltrex | Life Technologies | Cat# A1413201 |
| Lipofectamine 2000 | Life Technologies | Cat# 11668019 |
| Critical Commercial Assays | ||
| Cell Titer Glo | Promega | Cat# G7572 |
| RIPA lysis buffer | Santa Cruz Biotechnology | Cat# sc-24948 |
| BCA Protein Assay | Pierce Biotechnology | Cat# 23225 |
| ECL Plus Western Blotting Substrate | Pierce Biotechnology | Cat # 32132 |
| Papain Dissociation Kit | Worthington Biochemical | Cat# LK003150 |
| Secrete-Pair Gaussia Luciferase | Genecopoeia | Cat# SPGA-G100 |
| Mouse Neural Stem Cell Nucleofector Kit | Lonza | Cat# VPG-1004 |
| qSCRIPT cDNA Supermix | Quanta Biosciences | Cat# 95048-100 |
| SYBR-Green Mastermix | SA Biosciences | Cat# 330523 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | JAX: 005557 |
| Recombinant DNA | ||
| psPAX2 | Addgene | Plasmid #12260 |
| pMD2.G | Addgene | Plasmid #12259 |
| RBBP5 shRNA vector1 | Sigma-Aldrich Mission | TRCN0000353567 |
| RBPP5 shRNA vector2 | Sigma-Aldrich Mission | TRCN0000369176 |
| pLKO.1 non-targeting vector | Sigma-Aldrich Mission | SHC002 |
| RBBP5 vector | Applied Biological Materials | LV283511 |
| TLR4 vector | Applied Biological Materials | LV335816 |
| Control vector | Applied Biological Materials | LV590 |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| pcDNA3-YFP | Addgene | Plasmid #13033 |
| pcDNA3-TLR4-YFP | Addgene | Plasmid #13018 |
| Sequence-Based Reagents | ||
| qRT-PCR primers (Table S1) | This paper | N/A |
| ChIP primer, NANOG Reverse (−220 to −242): GGCT CTATCACCTTAGACCCACC | This paper | N/A |
| ChIP primer, NANOG Forward (−288 to −311): GAGAC TGGTAGACGGGATTAACTG | This paper | N/A |
| ChIP primer, OCT4 Reverse (−162 to −183): TCCAG GACCTCAGTGCAGGTC | This paper | N/A |
| ChIP primer, OCT4 Forward (−234 to −254): CTGCAC TCCAGTCTGGGCAAC | This paper | N/A |
| Software and Algorithms | ||
| Extreme limiting-dilution analysis | Hu and Smyth, 2009 | http://bioinf.wehi.edu.au/software/elda/ |
| GlioVis | Bowman et al., 2016 | http://gliovis.bioinfo.cnio.es |
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for reagents should be directed to the Lead Contact, Dr. Justin D. Lathia, at lathiaj@ccf.org.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
CSC derivation and xenograft maintenance
Established GBM xenografts representing the classical (T4121), proneural (T3832), and mesenchymal (T387) subtypes were previously reported (Bao et al., 2006; Schonberg et al., 2015) and were obtained via a material transfer agreement from Duke University, where they were originally established under an IRB-approved protocol that faciliated the genetration of xenografts in a de-identified manner from excess tissue taken from consented patients. For experimental studies, GBM cells were dissociated from established xenografts under Cleveland Clinic-approved Institutional Animal Care and Use Committee protocols. Xenografts were passaged in immune-deficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (obtained from The Jackson Laboratory, Bar Harbor, ME, USA) for maintenance of tumor heterogeneity. Six-week-old female mice were unilaterally injected subcutaneously in the flank with freshly dissociated human GBM cells, and animals were sacrificed by CO2 asphyxiation and secondary cervical dislocation when tumor volume exceeded 5% of the animal’s body weight.
In vivo patient-derived GBM cell injections
For in vivo tumor formation, live CSCs containing NT vector or RBBP5 shRNA or empty or TLR4 overexpression vector were transplanted into the striate nucleus of NSG mice at 1000 cells per mouse (n = 7 for KD, n = 6 for NT, and n = 8 for empty and TLR4 overexpression vectors) in accordance with Cleveland Clinic-approved Institutional Animal Care and Use Committee protocols. Six-week-old female mice were randomly assigned to one of the groups. Five mice were housed per cage, with a 12 hr light/dark cycle, and were provided food and water ad libitim. Mice were monitored daily and sacrificed upon the development of neurological signs, which include but were not limited to lethargy, ataxia, and seizures, along with associated weight loss and reduction in grip strength. Animals were sacrificed by CO2 asphyxiation and secondary cervical dislocation.
For LPS treatment of tumor-bearing mice, a total of 5×105 cells from xenograft T3832 were injected subcutaneously unilaterally into the flank of 6-week-old female mice (n = 7) in accordance with Cleveland Clinic-approved Institutional Animal Care and Use Committee protocols. After 14 days, mice received either PBS, 150 μg LPS, or 300 μg LPS via intratumoral injection. These doses were based on a previous report in a syngeneic mouse model (Chicoine et al., 2001). Tumors were measured at day 0 and at day 3 using electronic calipers, and tumor volume was calculated using the formula: tumor volume (mm3) = width2 × length × 0.52, with the assumption that the tumors were hemi-elliptical.
Tumor dissociation and GBM cell culture
Xenografted tumors were dissected and mechanically dissociated using papain dissociation kits (Worthington Biochemical Corporation, Lakewood, NJ, USA), and cells were cultured overnight in neurobasal medium (Life Technologies, Carlsbad, CA, USA) supplemented with B27 (Life Technologies), 1% penicillin/streptomycin (Life Technologies), 1 mM sodium pyruvate, 2 mM L-glutamine, 20 ng/mL EGF (R&D Systems, Minneapolis, MN, USA,), and 20 ng/mL FGF-2 (R&D Systems) in a humidified incubator with 5% CO2. CSCs were enriched using the CD133 Magnetic Bead Kit for Hematopoietic Cells (CD133/2; Miltenyi Biotech, San Diego, CA, USA) and cultured in supplemented neurobasal medium. This enrichment method reliably enriches CSCs that have increased self-renewal compared with their non-CSC counterparts (Bao et al., 2006; Schonberg et al., 2015). Non-stem cells (non-CSCs) were obtained in parallel and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Sigma, St. Louis, MO, USA) and 1% penicillin/streptomycin to preserve differentiation status. Both populations of tumor cells were cultured in the appropriate complete medium until the day they were used; only low (< 5) passage cells were used for experiments to prevent cellular drift. Differentiation of CSCs was induced by exposure to differentiation medium and confirmed via immunoblotting for a reduction in CSC markers (SOX2, NANOG, OCT4) and the acquisition of differentiation markers (GFAP). Differentiation was further validated by GFAP staining, which resulted in an increase in the number of GFAP-positive cells and overall staining intensity (data not shown).
METHOD DETAILS
In vitro functional analysis: tumorsphere formation and cell proliferation
Cell proliferation experiments were conducted by plating cells of interest at a density of 1000 cells/well in a 96-well plate in triplicate. Cell number was measured every other day and normalized to the initial reading at day 0 using the CellTiter-Glo assay kit (Promega, Madison, WI, USA). The experiments shown represent growth at day 7 relative to day 0, and similar effects were observed at days 3 and 5. For self-renewal experiments, cells were sorted using a flow cytometer (BD FACSAria II) into 96-well plates at a density of 1, 5, 10, and 20 live cells per well (24 wells for each density). Cells were maintained for 10 days before sphere formation was evaluated. Spheres larger than 10 cells in diameter were considered for analysis. Reported numbers represent either number of cells per well or stem cell frequency as calculated using the Walter and Eliza Hall Institute Bioinformatics Division ELDA analyzer (http://bioinf.wehi.edu.au/software/elda/) (Hu and Smyth, 2009). All tumorsphere and proliferation experiments were performed at least three times.
Toll-like receptor signaling modulation
The following TLR ligands and inhibitors were obtained from InvivoGen (San Diego, CA, USA): CpG (2 μM), Pam3C (300 ng/mL), PolyI:C (10 μg/mL), CLI-095 (1 μM), and BX795 (100 nM). LPS (500 ng/mL) was obtained from Sigma. HMGB1 (1 μg/mL) was obtained from GenScript (Piscataway, NJ, USA). Concentrations used in the experiments presented are specified in parentheses. Cells were treated for 7 days before growth was assessed or 1 day before harvesting for mRNA or protein. All signaling modulation experiments were performed at least three times.
Immunofluorescence analysis
Tumorspheres from GBM patient xenografts were fixed in 4% paraformaldehyde for 30 min at room temperature, followed by overnight cryoprotection with 20% sucrose in PBS at 4°C prior to sectioning at a thickness of 7 microns. Sections were then post-fixed with ice-cold methanol prior to staining with an anti-TLR4 antibody (Santa Cruz Biotechnology, Dallas, TX, USA; 1:75). Images were obtained using a Leica SP5 confocal microscope, and images were processed in Adobe Photoshop CS6 (Adobe, San Jose, CA). All immunofluorescence experiments were performed at least three times.
Immunoblotting analysis
Cell populations were lysed using RIPA lysis buffer (containing PMSF, protease inhibitor cocktail, and sodium orthovanadate; Santa Cruz Biotechnology, Dallas, TX, USA), and protein concentrations were calculated using a BCA protein assay (Pierce Biotechnology, Rockford, IL, USA). After denaturation with Laemmli buffer (BioRad Laboratories, Hercules, CA, USA), 10 μg of total protein was loaded on 10% polyacrylamide SDS-PAGE gels, transferred to polyvinyl difluoride (PVDF) membranes (Millipore, Billerica, MA, USA), and probed using the following antibodies: TLR4 (Santa Cruz Biotechnology, 1:1000), SOX2 (R&D Systems, 1:1000), RBBP5 (Cell Signaling, Danvers, MA, USA, 1:2000), GFAP (Invitrogen, 1:1000), TBK1 (Cell Signaling, 1:500), phospho-TBK1 (Cell Signaling, 1:500), IRAK1 (Abcam, 1:1000), phospho-IRAK1 (Abcam, 1:1000), MLL1 (Bethyl Laboratories, 1:1000), OCT4 (Cell Signaling, 1:1000), and NANOG (Cell Signaling, 1:1000); β-Actin (Santa Cruz Biotechnology, 1:2000) was used as a loading control. Species-specific horseradish peroxidase (HRP)-conjugated secondary antibodies were used for detection (Invitrogen, 1:5000). Membranes were developed using ECL-2 reagent (Pierce Biotechnology). All western blots were performed at least three times.
qRT-PCR
RNA from cells of interest was extracted using TRIzol (Life Technologies), and cDNA was synthesized using qSCRIPT cDNA Super-mix (Quanta Biosciences). qPCR reactions were performed using an ABI 7900HT system using SYBR-Green Mastermix (SA Biosciences, Valencia, CA, USA). For qPCR analysis, the threshold cycle (CT) values for each gene were normalized to expression levels of β-Actin. Dissociation curves were evaluated for primer fidelity, and only threshold cycles below 35 cycles were reported. The primers (Integrated DNA Technologies) used can be found in Table S1. All qRT-PCR experiments were performed at least three times.
Patient database bioinformatics
Gene expression data were obtained from different databases of patients with GBM using GlioVis (http://gliovis.bioinfo.cnio.es) (Bowman et al., 2016). Expression levels were categorized into two groups, taking the median as a reference. The survival times and status for patients in both groups were analyzed using the Kaplan-Meier statistical test via log-rank test.
Flow cytometry
Bulk tumor cells dissociated the day before were analyzed for cell surface expression using fluorophore-conjugated TLR4-PE (Santa Cruz, 1:50). Cells were incubated with these antibodies for 30 min at room temperature, washed with PBS, and analyzed using a BD LSRFORTESSA with DAPI (Sigma, 1:10,000) as a control for live cells and an isotype control for signal specificity. To separate TLR4 high and TLR4 low cells for limiting dilution analysis, the 20% of cells with the highest expression and the 20% of cells with the lowest expression, respectively, were sorted. All flow cytometry experiments were performed at least three times.
Lentiviral shRNA and overexpression construct preparation
Lentiviral constructs were prepared according to modified protocols from Tronolab (http://tronolab.epfl.ch). Using calcium phosphate precipitation, 293FT cells were co-transfected with the packaging vectors psPAX2 and pMD2.G (Addgene, Cambridge, MA, USA) and lentiviral vectors directing the expression of i) MISSION shRNA (Sigma) specific to RBBP5: TRCN0000353567 (KD1) and TRCN0000369176 (KD2)) or a non-targeting control (NT) shRNA (SHC002) and ii) overexpression of TLR4: accession number BC117422 ((LV335816); Applied Biological Materials, Richmond, Canada), overexpression of RBBP5: accession number BC053856 (LV283511), or control vector (LV590) to produce virus. Media on the 293FT cell cultures were changed 18 hr after transfection, and viral supernatants were collected 12, 24, and 36 hr later and concentrated using polyethylene glycol precipitation for immediate use or stored at −80°C for future use. CSCs and non-CSCs were infected with viral supernatants, selected with puromycin for 48 hr, and then used for experiments within five passages.
For TLR4 overexpression in vitro, 5×106 CSCs were nucleofected with 10 μg of pcDNA3-TLR4-YFP (Addgene; plasmid #13018) using the Mouse Neural Stem Cell Nucleofector Kit (Lonza Group, Basel, Switzerland). Cells were pelleted, resuspended in nucleofection buffer with supplement, DNA was added, and the mixture was nucleofected using program A-033. Cells were then cultured in supplemented neurobasal medium for 3 days before being assayed.
Luciferase promoter reporters
A total of 500,000 cells per condition were plated in 6-well plates pretreated with Geltrex (Life Technologies), which was used as an adherence substrate. The following day, cells were transfected with each of the promoter reporters (Genecopoeia) using Lipofectamine 2000 (Life Technologies) with 2 μg of DNA per reaction in Opti-Mem (Lerner Research Institute Media Core) medium. After 6 hr of incubation at 37°C, Opti-Mem was replaced with complete Neurobasal. Forty-eight hours later, medium was collected, and secreted Gaussia luciferase was analyzed using Secrete-Pair Gaussia Luciferase (Genecopoeia) per the manufacturer’s instructions. Ten microliters of supernatant was mixed with 100 μl of working reagent solution in 96-well plates in triplicate. Plates were read within 1 min. All luciferase experiments were performed at least three times.
Bioinformatics of transcription factors regulated by TLR4
We analyzed the promoter regions of SOX2, OCT4 and NANOG using the UCSC genome browser and compiled a list of common transcription factors that bind to these genes. To narrow down the list to a few candidates, we obtained processed RNA-seq data described in Suvàet al. (Suvàet al., 2014) (GEO: GSE54791). For each specimen pair, ratios were taken of the expression of the gene in TPCs relative to that in DGCs. Our list of transcription factors common to the SOX2, OCT4 and NANOG promotor regions was cross-referenced to these ratios, and we chose the transcription factors that had the highest relative expression in TPCs for further analysis. These were then analyzed via qRT-PCR in TLR4-overexpressing cells compared to cells expressing a control vector.
Epigenetic analysis
Chromatin immunoprecipitations were performed using 1×107 cells per immunoprecipitation. Cells were fixed with 1% formaldehyde, and chromatin was sheared using a QSonica Q800R1 at 70% amplitude with a setting of 15 s ON and 45 s OFF, for a total of 160 min. A total of 5 μg of each antibody used was bound to Dynabeads (ThermoFisher Scientific, Waltham, MA, USA) in an overnight incubation at 4°C. Dynabeads were then incubated with sheared chromatin overnight at 4°C. Antibody-bound chromatin was washed, de-crosslinked at 65°C for 8 hr, and then eluted from beads. DNA was phenol-chloroform extracted and resuspended in 105 μL of water. A 15 μL qRT-PCR reaction was performed that contained 4 μL of chromatin, 2X FastStart Universal SYBR Green Master-Rox (Roche, Basel, Switzerland), and 2 μM solutions of primer pairs. Antibodies used included anti-H3K4me3, anti-H3K4me2, and anti-H3K4me1 (Abcam, Cambridge, MA, USA). Primer sequences are included in the Key Resources Table.
QUANTIFICATION AND STATISTICAL ANALYSIS
Reported values are mean values ± standard error of the mean from studies performed at least in triplicate. Precise experimental details (number of animals or cells and experimental replication) is provided in the figure legends and in the EXPERIMENTAL MODEL AND SUBJECT DETAILS and METHOD DETAILS sections above. Unless otherwise stated, one-way ANOVA was used to calculate statistical significance, with p values detailed in the text and figure legends. P values < 0.05 were considered significant. Data analysis was done using SigmaStat (Systate Software) or Prism (GraphPad).
Supplementary Material
Highlights.
Glioblastoma cancer stem cells (CSCs) show reduced expression of TLR4
TLR4 overexpression inhibits proliferation and maintenance of CSCs
TLR4 signals via TBK1 to suppress expression of RBBP5
Knockdown of RBBP5 inhibits maintenance of the cancer stem cell state
Acknowledgments
We thank Drs. George Stark, Sarmishtha De, Paul Fox, Noa Noy, J. Mark Brown, and Ofer Reizes (Cleveland Clinic); Monica Venere (Ohio State University); and the members of the Lathia laboratory for insightful discussion and constructive comments on the manuscript. We also thank Dr. Jeremy Rich for many insightful comments and discussions. We thank Amanda Mendelsohn (Cleveland Clinic for Medical Art and Photography) for illustration assistance. We thank Cathy Shemo, Patrick Barrett, Joseph Gerow, and Sage O’Bryant for flow cytometry assistance. We thank Dr. Chris Hubert for assistance with RNA-sequencing data processing. This work used the Leica SP5 confocal/multi-photon microscope that was purchased with partial funding from National Institutes of Health SIG grant 1S10RR026820-01. This work was funded by the Lerner Research Institute (J.D.L.), a Distinguished Scientist Award from the Sontag Foundation (J.D.L.), and immunotherapy grants from Blast GBM (J.D.L. and M.A.V.) and the Cleveland Clinic VeloSano Bike Race (J.D.L. and M.A.V.). The Lathia laboratory also receives funding from National Institutes of Health grants NS089641, NS083629, CA191263, and CA157948; a Research Scholar Award from the American Cancer Society; and the Case Comprehensive Cancer Center.
Footnotes
Supplemental Information includes five figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.stem.2016.12.001.
AUTHOR CONTRIBUTIONS
Conception and Design, A.G.A. and J.D.L.; Financial Support, J.D.L.; Collection and/or Assembly of Data, A.G.A., P.S.T., J.S.H., T.J.A., D.J.S., S.M.T., and M.S.L.; Data Analysis and Interpretation, A.G.A., P.S.T., J.S.H., T.J.A., D.J.S., E.E.M.-H., A.J., O.R., M.S.L., M.A.V., and J.D.L.; Manuscript Writing, A.G.A. and J.D.L.; Final Approval of Manuscript, all authors.
References
- Anido J, Sáez-Borderías A, Gonzàlez-Juncà A, Rodón L, Folch G, Carmona MA, Prieto-Sánchez RM, Barba I, Martínez-Sáez E, Prudkin L, et al. TGF-β receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell. 2010;18:655–668. doi: 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, Holland EC. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4:226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman RL, Wang Q, Carro A, Verhaak RGW, Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-Oncol. 2016 doi: 10.1093/neuonc/now247. Published online November 9, 2016. http://dx.doi.org/10.1093/neuonc/now247. [DOI] [PMC free article] [PubMed]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah MT, Chen JY, Sahoo D, Contreras-Trujillo H, Volkmer AK, Scheeren FA, Volkmer JP, Weissman IL. CD14-expressing cancer cells establish the inflammatory and proliferative tumor microenvironment in bladder cancer. Proc Natl Acad Sci U S A. 2015;112:4725–4730. doi: 10.1073/pnas.1424795112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Tsukamoto H, Liu JC, Kashiwabara C, Feldman D, Sher L, Dooley S, French SW, Mishra L, Petrovic L, et al. Reciprocal regulation by TLR4 and TGF-β in tumor-initiating stem-like cells. J Clin Invest. 2013;123:2832–2849. doi: 10.1172/JCI65859. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Chicoine MR, Won EK, Zahner MC. Intratumoral injection of lipopolysaccharide causes regression of subcutaneously implanted mouse glioblastoma multiforme. Neurosurgery. 2001;48:607–614. doi: 10.1097/00006123-200103000-00032. [DOI] [PubMed] [Google Scholar]
- Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, Edwards MR, Michelsen KS, Kroeger KM, Liu C, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6:e10. doi: 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, Caviglia JM, Khiabanian H, Adeyemi A, Bataller R, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21:504–516. doi: 10.1016/j.ccr.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Zhou H, DeSantis D, Croniger CM, Li X, Stark GR. Erlotinib protects against LPS-induced endotoxicity because TLR4 needs EGFR to signal. Proc Natl Acad Sci U S A. 2015;112:9680–9685. doi: 10.1073/pnas.1511794112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, Zhu S, Qiao Y, Liu YJ, Chen W, Zhao G, Chen J. Recent advances in the role of toll-like receptors and TLR agonists in immunotherapy for human glioma. Protein Cell. 2014;5:899–911. doi: 10.1007/s13238-014-0112-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13:759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- Ernst P, Vakoc CR. WRAD: enabler of the SET1-family of H3K4 methyltransferases. Brief Funct Genomics. 2012;11:217–226. doi: 10.1093/bfgp/els017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyler CE, Foo WC, LaFiura KM, McLendon RE, Hjelmeland AB, Rich JN. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells. 2008;26:3027–3036. doi: 10.1634/stemcells.2007-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo M, Ho J, Coutinho FJ, Vanner R, Lee L, Head R, Ling EK, Clarke ID, Dirks PB. A tumorigenic MLL-homeobox network in human glioblastoma stem cells. Cancer Res. 2013;73:417–427. doi: 10.1158/0008-5472.CAN-12-1881. [DOI] [PubMed] [Google Scholar]
- Grimm M, Kim M, Rosenwald A, Heemann U, Germer CT, Waaga-Gasser AM, Gasser M. Toll-like receptor (TLR) 7 and TLR8 expression on CD133+ cells in colorectal cancer points to a specific role for inflammation-induced TLRs in tumourigenesis and tumour progression. Eur J Cancer. 2010;46:2849–2857. doi: 10.1016/j.ejca.2010.07.017. [DOI] [PubMed] [Google Scholar]
- Heddleston JM, Wu Q, Rivera M, Minhas S, Lathia JD, Sloan AE, Iliopoulos O, Hjelmeland AB, Rich JN. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ. 2012;19:428–439. doi: 10.1038/cdd.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann A, Cherryholmes G, Schroeder A, Phallen J, Alizadeh D, Xin H, Wang T, Lee H, Lahtz C, Swiderski P, et al. TLR9 is critical for glioma stem cell maintenance and targeting. Cancer Res. 2014;74:5218–5228. doi: 10.1158/0008-5472.CAN-14-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004;117:5965–5973. doi: 10.1242/jcs.01589. [DOI] [PubMed] [Google Scholar]
- Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, MacSwords J, Lathia JD, McLendon R, Lindner D, Sloan A, Rich JN. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011;18:829–840. doi: 10.1038/cdd.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Hu F, a Dzaye OD, Hahn A, Yu Y, Scavetta RJ, Dittmar G, Kaczmarek AK, Dunning KR, Ricciardelli C, Rinnenthal JL, et al. Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro-Oncol. 2015;17:200–210. doi: 10.1093/neuonc/nou324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- Kitange GJ, Mladek AC, Schroeder MA, Pokorny JC, Carlson BL, Zhang Y, Nair AA, Lee JH, Yan H, Decker PA, et al. Retinoblastoma binding protein 4 modulates temozolomide sensitivity in glioblastoma by regulating DNA repair proteins. Cell Rep. 2016;14:2587–2598. doi: 10.1016/j.celrep.2016.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Okun E, Tang SC, Griffioen K, Cheng A, Mughal MR, Laryea G, Selvaraj PK, ffrench-Constant C, Magnus T, et al. Toll-like receptor 3 is a negative regulator of embryonic neural progenitor cell proliferation. J Neurosci. 2008;28:13978–13984. doi: 10.1523/JNEUROSCI.2140-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Hitomi M, Gallagher J, Gadani SP, Adkins J, Vasanji A, Liu L, Eyler CE, Heddleston JM, Wu Q, et al. Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death Dis. 2011;2:e200. doi: 10.1038/cddis.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–1217. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Hong B, Sharabi A, Huang XF, Chen SY. Embryonic stem cells and mammary luminal progenitors directly sense and respond to microbial products. Stem Cells. 2009;27:1604–1615. doi: 10.1002/stem.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoleni S, Politi LS, Pala M, Cominelli M, Franzin A, Sergi Sergi L, Falini A, De Palma M, Bulfone A, Poliani PL, Galli R. Epidermal growth factor receptor expression identifies functionally and molecularly distinct tumor-initiating cells in human glioblastoma multiforme and is required for gliomagenesis. Cancer Res. 2010;70:7500–7513. doi: 10.1158/0008-5472.CAN-10-2353. [DOI] [PubMed] [Google Scholar]
- O’Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors—redefining innate immunity. Nat Rev Immunol. 2013;13:453–460. doi: 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]
- Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain Res Brain Res Rev. 2009;59:278–292. doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun E, Griffioen KJ, Son TG, Lee JH, Roberts NJ, Mughal MR, Hutchison E, Cheng A, Arumugam TV, Lathia JD, et al. TLR2 activation inhibits embryonic neural progenitor cell proliferation. J Neurochem. 2010;114:462–474. doi: 10.1111/j.1471-4159.2010.06778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro-Oncol. 2015;17(Suppl 4):iv1–iv62. doi: 10.1093/neuonc/nov189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradere JP, Dapito DH, Schwabe RF. The yin and yang of Toll-like receptors in cancer. Oncogene. 2014;33:3485–3495. doi: 10.1038/onc.2013.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, Schwartz M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9:1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- Sarrazy V, Vedrenne N, Billet F, Bordeau N, Lepreux S, Vital A, Jauberteau MO, Desmoulière A. TLR4 signal transduction pathways neutralize the effect of Fas signals on glioblastoma cell proliferation and migration. Cancer Lett. 2011;311:195–202. doi: 10.1016/j.canlet.2011.07.018. [DOI] [PubMed] [Google Scholar]
- Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, Hubert CG, Mack SC, Jarrar AM, Karl RT, et al. Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell. 2015;28:441–455. doi: 10.1016/j.ccell.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, Ronen A, Rolls A, London A, Bakalash S, Young MJ, Schwartz M. Toll-like receptor 4 restricts retinal progenitor cell proliferation. J Cell Biol. 2008;183:393–400. doi: 10.1083/jcb.200804010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Suvà ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157:580–594. doi: 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari R, Choudhury SR, Ghosh S, Mehta VS, Sen E. Involvement of TNFα-induced TLR4-NF-κB and TLR4-HIF-1α feed-forward loops in the regulation of inflammatory responses in glioma. J Mol Med (Berl) 2012;90:67–80. doi: 10.1007/s00109-011-0807-6. [DOI] [PubMed] [Google Scholar]
- Uthaya Kumar DB, Chen CL, Liu JC, Feldman DE, Sher LS, French S, DiNorcia J, French SW, Naini BV, Junrungsee S, et al. TLR4 signaling via NANOG cooperates with STAT3 to activate Twist1 and promote formation of tumor-initiating stem-like cells in livers of mice. Gastroenterology. 2016;150:707–719. doi: 10.1053/j.gastro.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Noort JM, Bsibsi M. Toll-like receptors in the CNS: implications for neurodegeneration and repair. Prog Brain Res. 2009;175:139–148. doi: 10.1016/S0079-6123(09)17509-X. [DOI] [PubMed] [Google Scholar]
- Vinnakota K, Hu F, Ku MC, Georgieva PB, Szulzewsky F, Pohlmann A, Waiczies S, Waiczies H, Niendorf T, Lehnardt S, et al. Toll-like receptor 2 mediates microglia/brain macrophage MT1-MMP expression and glioma expansion. Neuro-Oncol. 2013;15:1457–1468. doi: 10.1093/neuonc/not115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717–728. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- Zhao J, Bulek K, Gulen MF, Zepp JA, Karagkounis G, Martin BN, Zhou H, Yu M, Liu X, Huang E, et al. Human colon tumors express a dominant-negative form of SIGIRR that promotes inflammation and colitis-associated colon cancer in mice. Gastroenterology. 2015;149:1860–1871. doi: 10.1053/j.gastro.2015.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.