

Abstract
Background:
In experimental therapy of cancer, survivin is considered to be one of the well-established targets. Studies have found that it is overexpression in most of the human tumors, but it is rarely found in normal tissues. It is having varied structural and functional role. It controls cell division and cellular stress response and also regulates metastasis and migration of cancerous cells. It has also been recognized as a biomarker which makes it unconventional drug target. In spite of being one of the centrally active components in metastasis and invasion, their clinical use is minimal. To increase the therapeutic efficiency of cancer and its various stages, it is important to survey novel reagents targeting the pathways and mechanism involving survivin.
Objective:
The aim of this study was to identify novel survivin inhibitor candidates using in silico screening.
Materials and Methods:
In this course of work, virtual screening on a dataset of natural compounds retrieved from ZINC and other libraries were performed. Comparative analysis of the protein was done by studying the binding affinity of inhibitors that are already available. The best interacting complex was set for molecular dynamics simulation for 25 ns to validate the stability of system. These molecules were checked for their toxicity and absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties using OSIRIS and pre-ADMET tools.
Results:
We discovered ten such candidates with better binding efficiency with survivin in comparison to marketed chemical against the same. Furthermore, these inhibitor candidates did not induce cell toxicity. Binding affinity of reference molecules was varied from −6.8 to −8.5 kcal/mol while that of top scoring compound ZINC00689728 is −9.3 kcal/mol binding energy. Good placement and strong bond formation of selected molecule was observed during course of work. It is also having permissible ADMET property.
Conclusion:
Considering all the parameters, the screened molecule can be considered as a potential lead compound for designing new drug against survivin. Further investigation and testing will be required to make it to the final stage.
SUMMARY
Survivin is one of the important protein of metastasis. Inhibiting survivin might led to the increased therapeutic efficiency of cancer. In this work we are screening library of natural compounds in view of finding some potent inhibitor against survivin.
Abbreviations used: MD: Molecular dynamics, LogS: Aqueous solubility, Acceptor HB: Hydrogen bond acceptor, Donor HB: Donor hydrogen bond donor, ADMET: Absorption, distribution, metabolism, excretion, and toxicity, RCSB: Research Collaboratory for Structural Bioinformatics, OPLS: Optimized potentials for liquid simulations, RMSD: Root-mean-square deviation.
Keywords: Natural compounds, screening, simulation, survivin, toxicity
INTRODUCTION
Most of the cancer patient's undergone treatment has suffered from relapsing of infection in form of metastatic tumor. Metastatic process comprises cell invasion of tumor from the primary tumor, intravasion, arrest, and extravasation of the circulatory system which is followed by the growth at a distant site and angiogenesis.[1,2] There are various methods of measuring metastatic progression; one involves the measurement of size of large lesions on imaging and by indices of patient survival. Metastasis is a network of various proteins and pathways interweaved in a manner that they affect functioning of each other. There are various proteins that have not been explored as they were expected to be and have potency to be a target for secondary infection that is caused by metastasis. Expression of survivin has reported in almost all the tumors qualifying it as a biomarker of metastatic tumor.[3,4] In malignancies as well as in melanoma, the dual role of survivin has been studied and this includes both promitotic and antiapoptotic one.[5] In various studies conducted on animal model system, downregulation of the survivin or inactivation of its function has shown to inhibit tumor growth. It is evident that protein survivin is an unconventional target and earlier also various approaches had been applied to inhibit the same. These methods include survivin-specific immune response, interference from its expression, interaction, and inhibition with its binding pattern. Direct inhibition of protein through YM155 has also been tested.[6] Considering the importance of survivin in progression of the metastatic movement and considering the fact that there is few computational study on the interactions of natural compounds and survivin, it would be of interest to perform computational studies to screen some potent and effective herbal compounds against survivin. The number of encouraging examples signifies the importance and feasibility of the method. It involves steps that area “nonconventional” in current drug development protocols, but it is clearly worthwhile pursuing.
Apart from providing a beneficiary platform for searching a potent inhibitory molecule, this method also helps in screening of large library of chemicals in a very short span of time, which reduces the cost and energy of finding a new molecule to half of its original cost.
MATERIALS AND METHODS
Current work was conducted on a system with 8 GB RAM, Intel(R) Core(Tm), 2GB graphics, and i7-3220 CPU @ 2.30 GHz processor. Maestro suite of Schrödinger was used for screening and docking simulations. Desmond suite was utilized for molecular simulation studies (2015, Schrödinger, LLC, New York, NY, USA), absorption, distribution, metabolism, and excretion (ADME) analysis was performed on an online server pre-absorption, distribution, metabolism, excretion, and toxicity (ADMET) (https://preadmet.bmdrc.kr/), and for toxicity prediction, OSIRIS Property Explorer was used (http://www.organicchemistry.org/prog/peo/).
Selection and preparation of target
In this course of work, crystal structure of human survivin molecule was retrieved from Research Collaboratory for Structural Bioinformatics (RCSB) with PDB ID: 1F3H [Figure 1]. Refinement of protein structure was done through protein preparation panel of Schrödinger. Missing bond orders were added to the crystal structure of molecule. Bond orders were assigned, and constraint of root-mean-square deviation (RMSD) and optimized potentials for liquid simulation force field was used for restrained minimization. GLIDE v6.7 2015 Schrödinger, LLC, New York, NY, USA, was utilized for grid generation.
Figure 1.
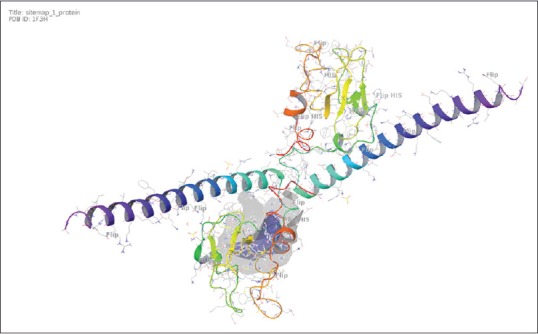
Three-dimensional structure of protein survivin with its active site
Preparation of ligand and virtual screening studies
Screening of libraries is a common trend in computational drug designing. Low-energy binding positions of drug candidates are searched through signifying a binding position. Virtual screening reduces the size of chemical compound repository to manageable size leading to synthesis of only few molecules to the subsequent screening against the chosen targets. Dataset of all the natural products from Zinc library[9] and dataset from Drugbank Drug were retrieved in SDF format and set for virtual screening GLIDE v6.7, 2015, Schrödinger, LLC, New York, NY, USA. Virtual screening by GLIDE is based on three layers of docking, i.e., high-throughput virtual screening (HTVS), standard precision (SP), and extra precision (XP). All compounds of the dataset were subjected for HTVS docking first and around 10% of the datasets move to SP docking and again precised amount of molecule further move to final XP docking. High-ranked top 11 compounds along with their binding affinity and other entities are mentioned in Table 1. Detailed two-dimensional (2D) structures of top 11 compounds are shown in Figure 2.
Table 1.
Top eleven screened compounds with their database ID, Docking score, number of Interacting hydrogen bonds, interacting residues, and other possible interactions
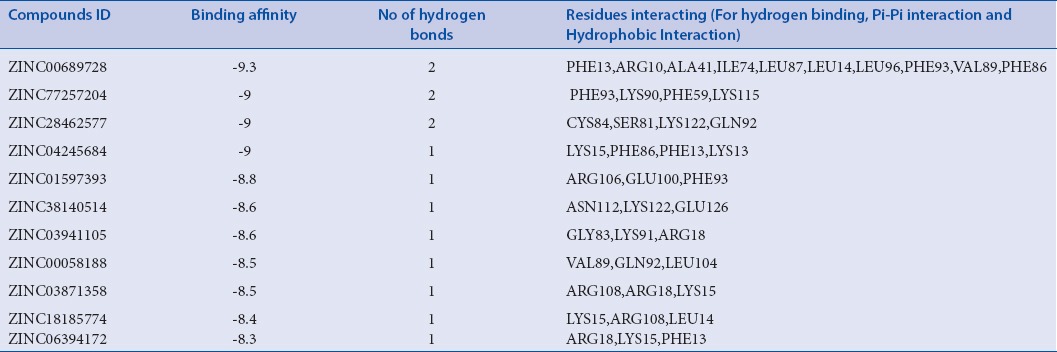
Figure 2.
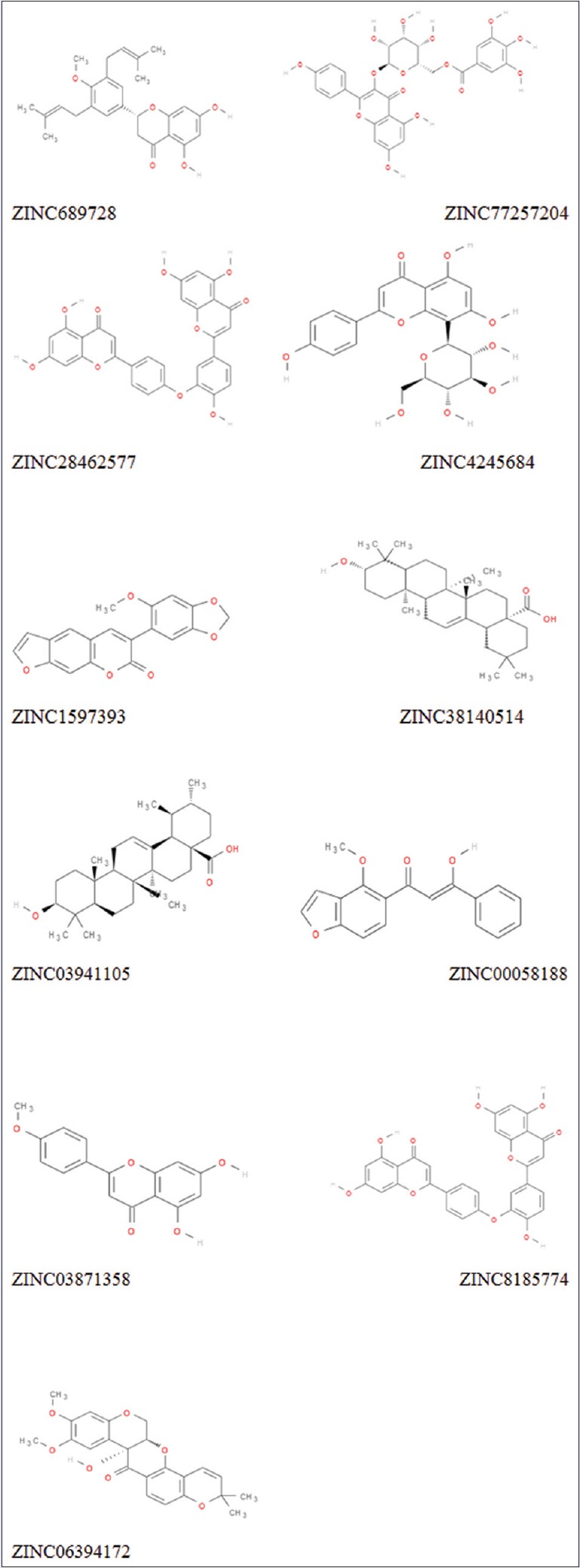
Two-dimensional structure of top 11 screened inhibitors (ZINC689728, ZINC77257204, ZINC28462577, ZINC4245684, ZINC1597393, ZINC38140514, ZINC03941105, ZINC00058188, ZINC03871358, ZINC8185774, and ZINC06394172)
Retrieving reference molecules
Various known inhibitors of survivin were downloaded from repositories, and their docking studies were performed with that of 1F3H on the same grid which was used for virtual screening, by GLIDE module of 2015, Schrödinger, LLC, New York, NY, USA.
Molecular dynamics simulation
Desmond suite of Schrodinger was used for molecular dynamics (MD) simulation, among all the 11 screened ligands, one with maximum binding affinity, maximum hydrogen bonds, and with allowed ADME, and toxicity values were chosen for MD simulations. Further, RMSD and root-mean-square fluctuation (RMSF) plot of protein–ligand complex was analyzed to ensure the stability and all other conformational changes of the setup or system during whole process of 25 ns simulation.
System building step through Schrodinger
Preprocessing of the complex was done through Protein Preparation Wizard of Maestro. It was done by modifying all the side chains of complex and through putting it for minimization processing before MD simulation. There are various missing atoms with complex, and during preparation of complex, these missing atoms were added (2015 Schrödinger, LLC, New York, NY, USA). Specific size of continuous repeating units (10 × 10 × 10) Å was chosen for orthorhombic box-shaped boundary.
Molecular dynamics simulation through Schrodinger
Once the process of system building was done, MD was performed. Bad contacts found in residues were removed through minimizing its energy with hybrid steepest descent method and the Limited-memory Broyden–Fletcher–Goldfarb–Shanno algorithms.
Prime MM/GBSA calculations
Prime/MM/GBSA module of the Schrodinger was used for calculating relative binding energy of the chosen ligands. XP output file pv.maegz was used for this study. Further, active site of the protein was set for the self-adjustment to itself up to 5 Å for ligand accordingly. The equation for the calculation of Delta G can be sum up as _ Gbind = Ecomplex (minimized) − [Eligand (unbound, minimized) + Ereceptor (unbound, minimized)].
Calculation of absorption, distribution, metabolism, and excretion property
Druglikeness characteristics of the top 11 screened compounds were analyzed by pre-ADMET to predict the pharmacokinetics, druglikeness, and biochemical properties. There is certain range which is acceptable for each of the property, and these features were justified over that range to categorize the leads druggable.
Toxicity prediction
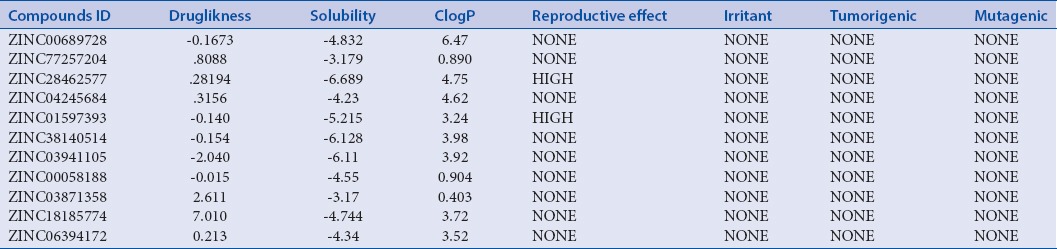
OSIRIS is an online tool for predicting toxicity; it was used to explore the toxicity of the best 11 screened out compounds. Best scored 11 compounds were taken for these investigations. OSIRIS (http://www.organic-chemistry.org) gives information about various biochemical and biophysical properties of lead that helps in categorizing it under drug-like compound. These properties include druglikeness, drug score, mutagenicity, irritancy, reproductive effect, solubility, and ClogP.
RESULTS
Receptor and analysis of its active site
1F3H survivin was taken as a receptor and its 3D structure was retrieved from RCSB. Submitted structure was obtained from X-ray diffraction method with resolution of 2.58 Å. Active site of protein was determined through sitemap module of Schrödinger.[7] It consists of two chains A and B. Important residues consist of PHE13, LEU14, LYS15, ARG18, GLU40, PHE58, PHE59, PHE86, VAL89, LYS90, LYS91, GLN92, PHE93, LEU96, LEU104, THR48, GLU51, MET54, LYS62, GLU63, LEU64, GLU65, LYS115, ASN118, ASN119, LYS122, GLU123, and GLU126.
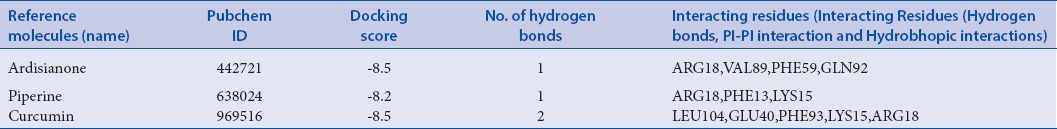
Reference molecules and its analysis
Binding affinity of reference molecules is varied from −6.8 to −8.5 kcal/mol [Tables 2 and 3]. Reference molecule processing has intended to select the most efficient binding site before conducting the virtual screening study.
Table 2.
Known Reference molecules and their PubChem IDs, docking scores, and interacting residues

Table 3.
Known Natural inhibitors of survivin from literature, and their pubChem ID, d ocking score, and their interacting residues
Different molecular library used in screening
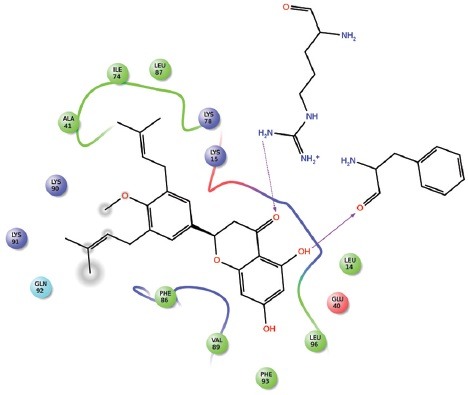
Dataset of all the naturally occurring molecules was taken from ZINC database. It comprises 139079 natural compounds. Apart from ZINC library, 7759 compounds were taken from drug database. In total, 146838 from various datasets were used for performing screening study against protein 1F3H. Cutoff range was fixed for better evaluation of the system. In this case, it was marked to −8.3.00 kcal/mol. Screened compounds were showing better score than either of the synthetic or natural reference compound. Further, top 11 ligands with higher binding affinity and score >−8.3 kcal/mol were chosen for the future analysis. Molecules with their detail interaction including interaction residues, hydrogen bond, and docking score have been mentioned in Table 1. Moreover, the interaction of best compound with that of protein is shown in Figure 3a (2D interaction) and Figure 3b (3D interaction).
Figure 3.

(a) Two-dimensional interaction diagram of ZINC00689728 with protein survivin, 6 hydrogen bonds are forming between chosen ligand and protein. Residues which are involved in bond formation are ARG18, LYS13, PHE93, LYS15, PHE86, and GLU40. (b) Three-dimensional interaction diagram of ligand and surviving
Study of the interaction complex of protein and ligand
Top scoring compound ZINC00689728 is having -9.3kcal/mol binding energy with two hydrogen bonds, where PHE13 and ARG10 of protein is interacting with CH group of benzene ring; there is Pi-Pi stacking observed with ALA41, ILE74, and LEU87. Hydrophobic interaction will include LEU14, LEU96, PHE93, VAL89, and PHE86. Similarly, second-ranked compound in list is ZINC77257204 having binding score of 9 kcal/mol and forming two hydrogen bonds with LYS90 and LYS115 amino acid residues. The third compound in list is ZINC28462577 having binding affinity of −9 kcal/mol with two hydrogen bonds with the LYS122, GLN92 amino acid residues. Score function and all other interactions such as binding affinity and number of hydrogen bond of the all selected 11 compounds such as ZINC00689728, ZINC77257204, ZINC28462577, ZINC04245684, ZINC01597393, ZINC38140514, ZINC03941105, ZINC00058188, ZINC03871358, ZINC18185774, and ZINC06394172 are shown in Table 1.
Molecular dynamics simulation studies
Studying the atomic level perturbation through MD simulation helps in understanding various biological aspects of molecule. These aspects include insights in structural makeup of complex or protein, conformational aspect of protein,[8] and search of unique molecules. A protein–ligand (ZINC00689728 and SURVIVIN) complex was set for MD simulation stability analysis. Once the system reaches its equilibrium stage, the production run was executed. After completion, it generated various interaction diagrams, simulation trajectory, and plots. These plots were put for an analysis for checking the stability of the interaction between ligand and protein. Simulation trajectory was found to behave stably and hence it confirms the appropriate docking of ligand and protein.
Analysis of root-mean-square deviation of protein–ligand complex
Movement of backbone and folding backbone inside the system defines the functional behavior of protein–ligand complex. Now, analysis of RMSD enables to detect the structural changes occurring in backbone of concerned protein and also in other residues of side chain of survivin protein through the complete process of 25 ns. Initially, sharp edges were observed in C-alpha, backbone of the protein converged after around 25 ns [Figure 4]. Later, it is getting stable when moving toward 25 ns and acts continuously till the end of simulation. Initial instabilities are showing that protein is going rigorous conformational changes over the period. Ligands RMSD and RMSF are showing consistent behavior during whole simulation period, which can be infer as stability of the complex.
Figure 4.
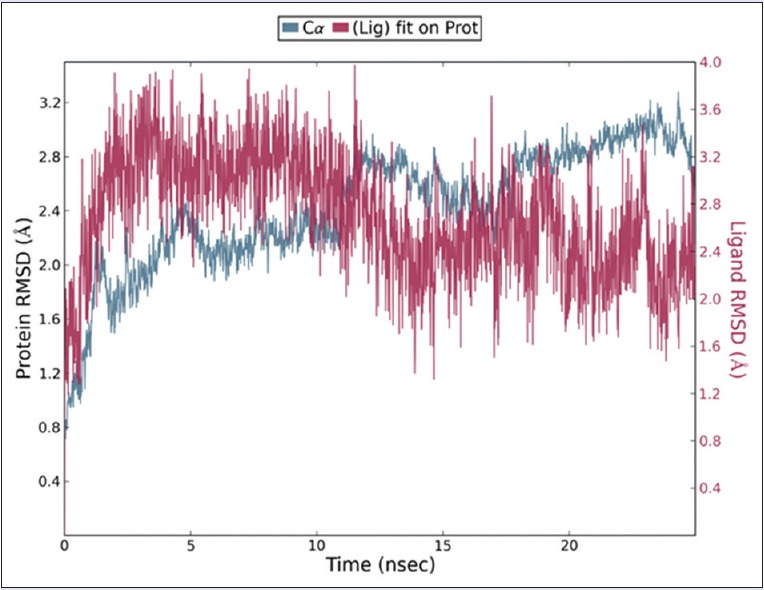
Root-mean-square deviation plot of C-alpha of protein and ligand at 25 ns. It is depicting the quality of the pose in respect to the time. Stability is being confirmed at the end of 25 ns simulation
Analysis of root-mean-square fluctuations of the protein
Deviation of various residues of protein in comparison to the chosen reference molecule is mainly analyzed by RMSF as shown in Figure 5.
Figure 5.
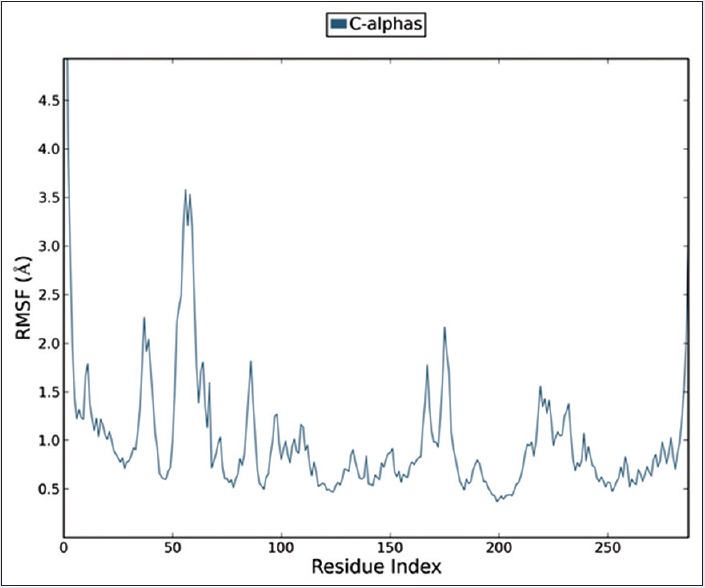
Root-mean-square fluctuation plot of residue number and C-alpha backbone of protein survivin at 25 ns simulation. It predicts the fluctuation of the backbone atoms; herein, the diagram protein is getting less fluctuating thus restoring the stability of backbone
Analysis of complex after simulation
Figure 3 is showing simulation analysis of protein survivin and ligand ZINC00689728 interaction. RMSD analysis of the complex confirms that the complex is structurally stable throughout the 25 ns simulation.
Result of MM/GBSA calculations
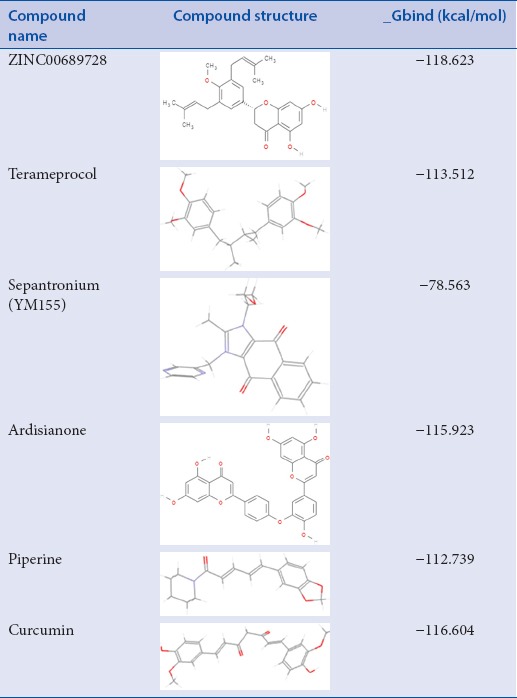
Estimation of relative binding energy was done through implying MM/GBSA study. Prime module (Jacobson et al. 2004) of Schrodinger was used for the study. Molecules with best ranking score in XP mode were subjected for the calculation. These studies were subjected to access the relative binding affinity of ligand toward receptor. Results are reported in Table 4.
Table 4.
Two-dimensional structure of compounds along with their MM/GBSA score
Analysis of absorption, distribution, metabolism, excretion, and toxicity property
Top 11 molecules were put for pharmacokinetic descriptor calculation to evaluate the screened compounds on the parameter of druglikeness. A total of 10 descriptors were taken in account; ADME properties and other parameters for each inhibitor are given in Table 4. From analysis, it can be depicted that all the top 11 screened molecules are falling in adequate range of these ADMET descriptors.
Prediction of toxicity
No toxicity has been noticed for the selected compounds. Detail calculation of various parameters has been listed in Table 5 and 6.
Table 5.
Principal descriptors, their acceptable ranges, and values of the selected inhibitory molecules: Eleven top molecule
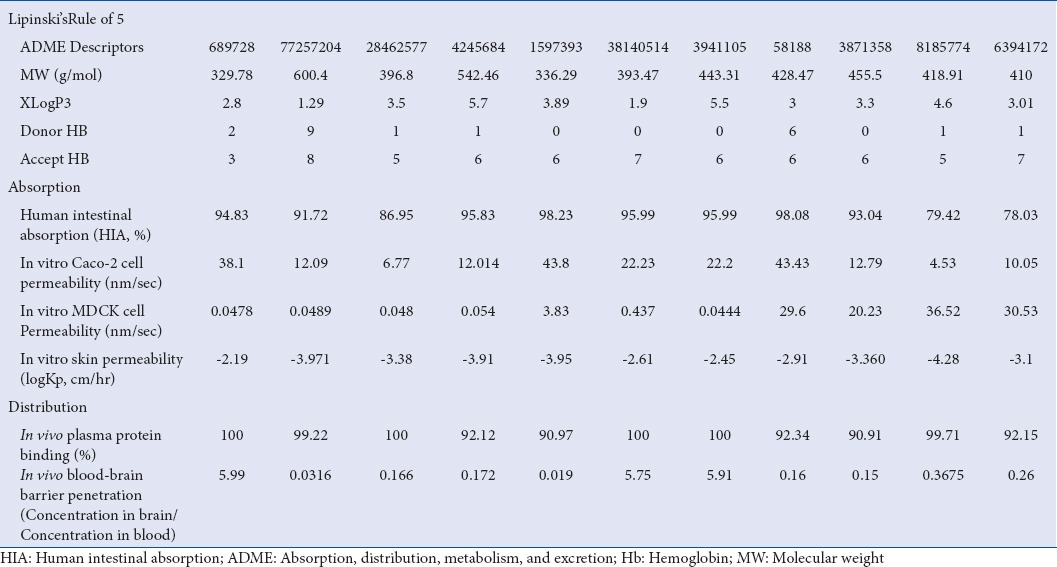
Table 6.
Toxicity parameters of the selected compounds
DISCUSSION
At present, the standard treatment of cancer involves chemotherapy, radiation, and precise medicines. Although these treatments have been shown to be effective to a certain extent, it causes several side effects. Available drug targets specific protein which is beneficial to control the cancer but fails to restrict its movement to other parts. Treatment with surgery also poses problems of relapsing of the disease and formation of new lesions. Treatment with available herbal molecules has its own limitations in terms of efficiency and effectiveness. Taking these current issues into account, our primary goal was to identify a novel marker which expresses itself in diseased condition and can be targeted to localize the movement of cancerous cells. The second motive with which this work was proceeded was to identify novel chemical reagents which can restrict the target with better efficiency. Prior studies have identified one compound that targets the survivin, YM155. It is in its second phase of testing; YM155 is a first-in-class survivin inhibitor currently in phase I/II clinical trials as monotherapy or combination therapy for a variety of human malignancies.[9,10,11,12,13,14] Focusing on same our work has proposed more compounds against survivin with better efficiency than YM155.
Further, we simulated the docking process for survivin and best zinc compound obtained through screening and also calculated MM/GBSA energy. MM/GBSA energy is attractive approaches owing to their modular nature and that they do not require calculations on a training set. MM/GBSA energy and docking simulation showed favorable docking stability.
CONCLUSION
The main purpose of this paper was to screen natural libraries against protein survivin. After implying all the tools, we propose that natural compounds are better in terms of binding affinity and compatible on parameter of ADMET and toxicity studies. Further studies of proposed inhibitors need to be carried out.
Financial support and sponsorship
It is an original work performed and supported by Facilities of Indian Institute of Information Technology, Allahabad (Department of Applied Sciences).
Conflicts of interest
There are no conflicts of interest.
Acknowledgement
The authors are thankful to Indian Institute of Information Technology, Allahabad, for providing computational facilities to complete this work and to Rashmi Tripathi for their valuable time and advice for completing this paper.
REFERENCES
- 1.Steeg PS. Tumor metastasis: Mechanistic insights and clinical challenges. Nat Med. 2006;12:895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 2.Aplin AE, Kaplan FM, Shao Y. Mechanisms of resistance to RAF inhibitors in melanoma. J Invest Dermatol. 2011;131:1817–20. doi: 10.1038/jid.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeuchi H, Morton DL, Elashoff D, Hoon DS. Survivin expression by metastatic melanoma predicts poor disease outcome in patients receiving adjuvant polyvalent vaccine. Int J Cancer. 2005;117:1032–8. doi: 10.1002/ijc.21267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carrasco RA, Stamm NB, Marcusson E, Sandusky G, Iversen P, Patel BK, et al. Antisense inhibition of survivin expression as a cancer therapeutic. Mol Cancer Ther. 2011;10:221–32. doi: 10.1158/1535-7163.MCT-10-0756. [DOI] [PubMed] [Google Scholar]
- 5.Mita AC, Mita MM, Nawrocki ST, Giles FJ. Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res. 2008;14:5000–5. doi: 10.1158/1078-0432.CCR-08-0746. [DOI] [PubMed] [Google Scholar]
- 6.Minoda M, Kawamoto T, Ueha T, Kamata E, Morishita M, Harada R, et al. Antitumor effect of YM155, a novel small-molecule survivin suppressant, via mitochondrial apoptosis in human MFH/UPS. Int J Oncol. 2015;47:891–9. doi: 10.3892/ijo.2015.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halgren TA. Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model. 2009;49:377–89. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- 8.Balasco N, Barone D, Vitagliano L. Structural conversion of the transformer protein rfaH: New insights derived from protein structure prediction and molecular dynamics simulations. J Biomol Struct Dyn. 2015;33:2173–9. doi: 10.1080/07391102.2014.994188. [DOI] [PubMed] [Google Scholar]
- 9.Tolcher AW, Mita A, Lewis LD, Garrett CR, Till E, Daud AI, et al. Phase I and pharmacokinetic study of YM155, a small-molecule inhibitor of survivin. J Clin Oncol. 2008;26:5198–203. doi: 10.1200/JCO.2008.17.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheson BD, Bartlett NL, Vose JM, Lopez-Hernandez A, Seiz AL, Keating AT, et al. Aphase II study of the survivin suppressant YM155 in patients with refractory diffuse large B-cell lymphoma. Cancer. 2012;118:3128–34. doi: 10.1002/cncr.26510. [DOI] [PubMed] [Google Scholar]
- 11.Giaccone G, Zatloukal P, Roubec J, Floor K, Musil J, Kuta M, et al. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J Clin Oncol. 2009;27:4481–6. doi: 10.1200/JCO.2008.21.1862. [DOI] [PubMed] [Google Scholar]
- 12.Kelly RJ, Thomas A, Rajan A, Chun G, Lopez-Chavez A, Szabo E, et al. A phase I/II study of sepantronium bromide (YM155, survivin suppressor) with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Ann Oncol. 2013;24:2601–6. doi: 10.1093/annonc/mdt249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Satoh T, Okamoto I, Miyazaki M, Morinaga R, Tsuya A, Hasegawa Y, et al. Phase I study of YM155, a novel survivin suppressant, in patients with advanced solid tumors. Clin Cancer Res. 2009;15:3872–80. doi: 10.1158/1078-0432.CCR-08-1946. [DOI] [PubMed] [Google Scholar]
- 14.Tolcher AW, Quinn DI, Ferrari A, Ahmann F, Giaccone G, Drake T, et al. A phase II study of YM155, a novel small-molecule suppressor of survivin, in castration-resistant taxane-pretreated prostate cancer. Ann Oncol. 2012;23:968–73. doi: 10.1093/annonc/mdr353. [DOI] [PubMed] [Google Scholar]