

Abstract
Background
Current technologies in next-generation sequencing are offering high throughput reads at low costs, but still suffer from various sequencing errors. Although pyro- and ion semiconductor sequencing both have the advantage of delivering long and high quality reads, problems might occur when sequencing homopolymer-containing regions, since the repeating identical bases are going to incorporate during the same synthesis cycle, which leads to uncertainty in base calling. The aim of this study was to evaluate the analytical performance of a pyrosequencing-based next-generation sequencing system in detecting homopolymer sequences using homopolymer-preintegrated plasmid constructs and human DNA samples originating from patients with cystic fibrosis.
Results
In the plasmid system average correct genotyping was 95.8% in 4-mers, 87.4% in 5-mers and 72.1% in 6-mers. Despite the experienced low genotyping accuracy in 5- and 6-mers, it was possible to generate amplicons with more than a 90% adequate detection rate in every homopolymer tract. When homopolymers in the CFTR gene were sequenced average accuracy was 89.3%, but varied in a wide range (52.2 – 99.1%). In all but one case, an optimal amplicon-sequencing primer combination could be identified. In that single case (7A tract in exon 14 (c.2046_2052)), none of the tested primer sets produced the required analytical performance.
Conclusions
Our results show that pyrosequencing is the most reliable in case of 4-mers and as homopolymer length gradually increases, accuracy deteriorates. With careful primer selection, the NGS system was able to correctly genotype all but one of the homopolymers in the CFTR gene. In conclusion, we configured a plasmid test system that can be used to assess genotyping accuracy of NGS devices and developed an accurate NGS assay for the molecular diagnosis of CF using self-designed primers for amplification and sequencing.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4544-x) contains supplementary material, which is available to authorized users.
Keywords: Pyrosequencing, Homopolymer detection, Cystic fibrosis
Background
In genomics, a homopolymer (HP) is a sequence of consecutive identical bases. Approximately 1.43 million HPs (also known as mononucleotide microsatellites) exist in the human exome, with the size of 4-mer and up. This also means that an average of eight such HP sequences can be found in every exon. They are believed to play roles in transcriptional regulation and recombination [1, 2], and the vast majority (96.7%) of them are in the range of 4-mer to 6-mer. HP sequences composed of A:T base pairs are over-represented in the human genome compared to G:C HPs [3–5]. Although both pairs show structural stability [6, 7], these loci in the genome are highly mutagenic and have been characterized as hotspots for length change mutations [8–10], which has, presumably, contributed to their reduced occurrence in the exome over time.
Certain sequencing-by-synthesis based next generation sequencing (NGS) techniques have a relatively high error rate in determining homopolymer regions, due to the principles used for detection. In pyrosequencing [11–13] or ion semiconductor sequencing [14], the signal which depends on the emitted light or the concentration of released hydrogen ions, respectively, should be directly proportional to the number of incorporated bases during a single dNTP dispensation. However, since linearity starts to diminish in HP stretches exceeding four bases, erroneous over- and under calls are going to happen [15–17]. Since HPs are more prone to insertion and deletion mutations (indels), problems are going to aggravate, when utilizing pyrosequencers or ion semiconductor chemistry in diagnostic procedures [18].
There are numerous bioinformatic correction tools to separate artifacts from true genetic variations. Some of these algorithms are based on clustering the flowgrams; for example Denoiser, which utilizes rank-abundance distributions, or PyroNoise/AmpliconNoise, which calculates a likelihood using empirically derived error distributions [19]. Acacia’s main focus is on HP sequences and the algorithm uses a dynamically updated cluster consensus when aligning reads [20]. Coral and ECHO are multiple alignment based techniques [21], while HECTOR is a homopolymer spectrum based error corrector, with a multistage correction workflow [22]. Another useful software is FlowClus, which provides feedback on the denoising process, allowing the user to apply more suitable analysis parameters for the particular dataset [23]. The most recent tools, such as NoDe (Noise Detector) and DUDE-Seq are believed to produce even lower error rates and are more time-efficient [24, 25]. Even if sophisticated correction tools [22, 26, 27] are used to overcome the difficulties of detection and significantly improve accuracy, it is still very important to estimate the capability of the corresponding NGS system to correctly determine HPs. To avoid uncertainty in the diagnostic testing of patient samples, it is also recommended that the maximum length of stable HP detection, for reasonable identification of indel mutations in such sequences, be defined before using the NGS instrument in routine clinical practice [28].
The coding region of the cystic fibrosis transmembrane regulator gene (CFTR) contains 24 homopolymer stretches, involving 17 out of 27 exons. In conjunction with the whole genome, T and A homopolymers vastly outnumber G and C homopolymers; 14 thymine, 8 adenine, 2 guanine and no cytosine HPs are present. In exon 14 there is a seven adenine long homopolymer region (c.2046_2052) and genetic alterations affecting this region could create poly-A tracts of different sizes, e.g. the pathogenic mutation c.2051_2052delAAinsG (2183delAAinsG) results in a five, while c.2052delA (2184delA) results in a six adenine long homopolymer segment. In case of the relatively frequent c.2052_2053insA (2184insA) mutation an eight adenine long homopolymer stretch is formed.
To assess the analytical validity of pyrosequencing-based NGS technology and to test the optimization possibilities, two experimental test systems were developed. In the first, homopolymer sessions were introduced into plasmid vectors and the resultant plasmids were used as templates for subsequent sequencing experiments. Next we analyzed the CFTR gene to also assess the analytical performance of a newly developed pyrosequencing-based assay on patient samples. In this second experiment, all homopolymer-containing and non-HP CFTR exons were tested. These samples originated from cystic fibrosis (CF) patients with known CFTR mutation status. Assessment of the Ion Torrent platform was carried out in a similar way and the results were recently published [29].
Another reason for investigating the CFTR gene was that the above mentioned CF producing mutation (2184insA) is very common in certain geographical regions of Europe [30], including Hungary [31, 32]. Despite, current CF mutation detection kits do not cover this particular mutation site.
Methods
The two assay systems, which were used during the experiments are shown on Fig. 1. To test the analytical performance of the Roche pyrosequencing-based benchtop NGS system (Roche 454 Life Sciences, Branford, CT, USA) in assessing homopolymer sequences, a series of plasmid vectors were generated using pcDNA3.1 as a template (Invitrogen, Life Technologies, Carlsbad, CA). Altogether 12 clones were produced (4-mer, 5-mer, and 6-mer homopolymers of all four nucleotides) using site-directed mutagenesis and following the manufacturer’s instructions (Quikchange II, Agilent Technologies, Santa Clara, CA). In order to avoid the possible generation of length-change mutations during the plasmid replication and the amplification of the specific fragments, PicoMaxx enzyme mix that contains Pfu DNA polymerase (Agilent Technologies, Santa Clara, CA) was utilized. As a quality control step, two colonies of the site-directed mutagenesis were analyzed and confirmed by Sanger sequencing in case of all mutations (Additional file 1: Figure S1).
Fig. 1.
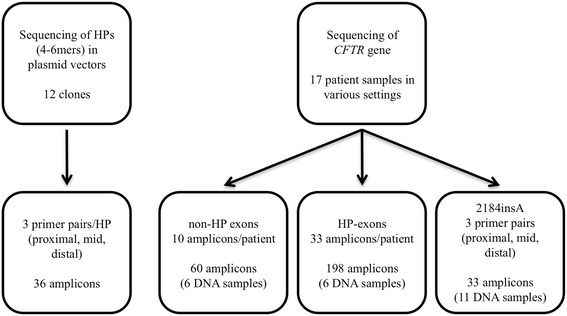
Experimental setup. In the plasmid system 4-to 6-mer homopolymers (HPs) were generated and tested using different primer sets. In the CFTR assay, 17 samples with known genotype were tested. This test system was divided into three parts, exons containing no HPs, HP-containing exons and the 2184 insA mutation
Each homopolymer tract in the plasmid system was investigated using three pairs of primers (Fig. 1) in order to test the hypothesis, that in the beginning of the sequencing reaction a sufficiently high signal-to-noise ratio might enable precise HP length detection. Our primer design was as follows: i) the homopolymer was located in the vicinity of the forward amplification/sequencing primer in 3′ direction, ii) the analyzed homopolymers were located approximately in the middle of the amplicon and iii) the reverse amplification/sequencing primer’s 3′ end was generated to be as close as possible to the HP segment. Primers used for mutagenesis, amplification, and sequencing are listed in Additional file 2: Table S1. The size of the homopolymer clones varied between 366 and 387 bp, depending on the HP length and the primers used.
In the second set of experiments, a CFTR gene mutation detection system was developed and analyzed in detail. 17 clinical samples were used with known CFTR mutation status determined by an in vitro diagnostic assay (Elucigene CF29v2, Elucigene Diagnostics, Manchester, UK) and Sanger sequencing. Representative electropherograms of the clinical samples are shown in Additional file 3 Figure S2 and Additional file 4 Figure S3 (all HP-containing exons of the CFTR gene and a 2184insA mutation in heterozygous form, respectively, as tested by Sanger sequencing). Primer design for CFTR mutation analysis was similar to the plasmid system’s described above, except for the “HP in the middle” type amplicons, which were left out of this experiment (primers listed in Additional file 5: Table S2). Exons e3, e14, e15, and e24 have multiple HP sections, most of which could only be covered within the same amplicons; therefore altogether 33 HP-containing amplicons were tested per patient. When designing the primers, all known single-nucleotide polymorphisms (SNPs) were taken into consideration to maximize annealing efficiency and minimize allele drop-out, which was shown to be an issue in a previous test system [29]. We also designed primers for CFTR exons that do not contain homopolymer stretches (Additional file 6: Table S3) to be able to analyze the complete gene. The most crucial section of the gene (within exon 14) was further tested using 11 human DNA samples; including four wild type and seven 2184insA heterozygotes. As with the plasmid system, we used three additional primer pairs to generate amplicons for NGS sequencing with “proximal,” “mid,” and “distal” HP locations. “Proximal” primers were located at a distance of 5 and 11 base pairs from the poly-A tract. In all samples, PicoMaxx enzyme mix was used for the amplification processes.
All human participants gave informed consent for diagnostic genetic analysis. In this study DNA samples were then applied anonymously and procedures were in accordance with the current revision of the Helsinki Declaration. The laboratory is approved by the National Public Health and Medical Officer Service (approval number: 094025024).
Sequencing data of individual reads were evaluated using Amplicon Variant Analyzer software. We differentiated between “proximal”, “mid” and “distal” type of reads referring to HP to sequencing primer distance. Statistics were done using GraphPad Prism v5.03. Numbering of the CFTR exons is based on current recommendations (Ensembl ENSG00000001626). In mutation nomenclature both Human Genome Variation Society (HGVS) and legacy names are used, as suggested [33]. Genotyping accuracy was defined by the percent of correct reads in samples with known genotypes. Acceptable genotyping accuracy was defined to have at least 75% accurate reads of all reads.
Results
Quality control of the template preparation
The site-directed mutagenesis experiments were controlled using Sanger sequencing that were performed in duplicates in case of all mutations. No discrepancy was found in any of the introduced mutations (Additional file 1: Figure S1).
Sequencing homopolymer containing plasmids
When sequencing homopolymer-containing plasmids, mean coverage was 479 ± 145 and an evident negative correlation was observed between homopolymer length and read accuracy (Fig. 2). The average correct genotyping rate of all four nucleotides combined was 95.8, 87.4 and 72.1% in 4-mers, 5-mers, and 6-mers, respectively (with a 79.6–99.3% range in 4-mers, 36.9–98.4% in 5-mers and 14.5–93.8% in 6-mers). While the pyrosequencing-based NGS system was able to detect poly-A 6-mers reliably (with a mean of 76.4%), detection rates fell under 75% for poly-C, poly-G and poly-T 6-mers (means: 71.5, 68.3 and 63.4%, respectively). In general, longer HPs had lower genotyping accuracy, although, the most accurate reads still reached 98.4% for 5-mers and 93.8% for 6-mers, indicating that careful optimization in a given sequence context might help to skip the poor performing primers and find the most functioning ones for the analysis. After testing, primer localization failed to show any association with genotyping accuracy (Fig. 3).
Fig. 2.
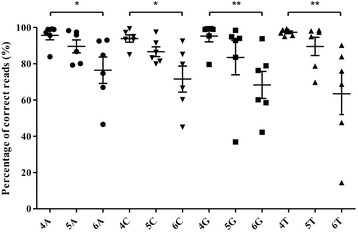
Detection of homopolymers in a plasmid-based system. Read accuracy is expressed as percentage of correct reads. (Kruskal-Wallis test followed by Dunn’s Multiple Comparison Test, * P < 0.05 and ** P < 0.01)
Fig. 3.
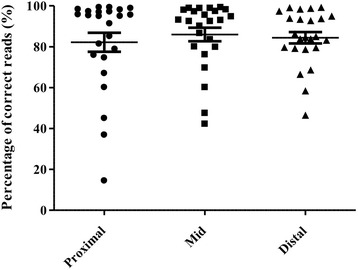
Sequencing primer distance has no effect on homopolymer genotyping. Read accuracy is expressed as average percentage of correct reads. Proximal, equal and distal localization of the homopolymer from the sequencing primer yielded no significant variance in read accuracy. (Kruskal-Wallis test followed by Dunn’s Multiple Comparison Test)
Sequencing human DNA samples
Coding regions and exon-intron boundaries in the CFTR gene from 6 cystic fibrosis patients with a known mutation status were sequenced. Mean base coverage was 263 ± 178 using our in-house assay. Altogether 246 amplicons were included in the data analysis (> 40 reads).
Although individual read accuracy varied in a wide range (52.2 – 99.1%), average accuracy was generally excellent using the assay (89.3%). The assay was able to detect all small-scale genetic alterations (missense, nonsense, splice site mutations, frameshift/in-frame deletions and insertions) previously identified by Sanger sequencing, providing 100% sensitivity and specificity (Table 1). Regarding the 24 HP stretches the self-designed primer set yielded good performance with more than 80% genotyping accuracy in all but one HP (data not shown). The exception was a 7A HP tract (c.2046_2052) with a 52.2% average correctness.
Table 1.
Performance of the assay in detecting small-scale alterations in the CFTR gene
| Location | cDNA position | Legacy name | Mutation class | Sensitivity |
|---|---|---|---|---|
| i6 | c.654-10delAGTT | 786-10delAGTT | Splice site | 1/1 |
| i6 | c.743 + 40A > G | 875 + 40A/G | Splice site | 1/1 |
| i7 | c.869 + 11C > T | 1001 + 11C > T | Splice site | 1/1 |
| e8 | c.926C > G | A309G | Missense | 1/1 |
| e11 | c.1394C > T | T465I | Missense | 1/1 |
| e11 | c.1397C > G | S466X | Nonsense | 1/1 |
| e11 | c.1521_1523delCTT | F508del | In-frame deletion* | 6/6 |
| e12 | c.1624G > T | G542X | Nonsense* | 1/1 |
| e14 | c.2012delT | 2143delT | Frameshift deletion | 2/2 |
| e14 | c.2051_2052delAAinsG | 2183AA > G | Frameshift insdel* | 1/1 |
| e14 | c.2052_2053insA | 2184insA | Frameshift insertion | 6/6 |
| e14 | c.2052delA | 2184delA | Frameshift deletion* | 1/1 |
| e15 | c.2562 T > G | 2694 T/G | Synonymous | 4/4 |
| i16 | c.2657 + 5G > A | 2789 + 5G > A | Splice site* | 1/1 |
| e17 | c.2856G > C | M952I | Missense | 1/1 |
| e21 | c.3454G > C | D1152H | Missense* | 1/1 |
| e23 | c.3846G > A | W1282X | Nonsense* | 1/1 |
| e27 | c.4389G > A | 4521G/A | Synonymous | 1/1 |
Mutations that are included in the Elucigene CF29v2 kit are labelled with an asterisk
Therefore a common mutation in this 7A HP (c.2052_2053insA, a 8-mer) was further analyzed using three primer sets (Fig. 1, primer sequences are shown in Additional file 5: Table S2). Depending on the primer used, correct detection of 7A in four wild type DNA samples reached 81%, the percentage of correct reads is shown in Fig. 4. In patients who were heterozygous for the c.2052_2053insA mutation, we calculated the percentage of detected 7A and 8A signals, which, theoretically, should have been 50% for each. The detected genotype frequencies varied in a wide range (16–49%, on average), but the results greatly depended on the primers used for sequence analysis. Two primer pairs (Fig. 5b and c) proved to be rather poor performers with 45–50% irrelevant nucleotide calls. On the other hand, sequencing with a third set of primers, correct 7A and proximal 8A calls were detected with satisfactory accuracy (Fig. 5a), having 27% misreads on average, but even this set could not identify 8 adenines from an approximate distance of 200 bp.
Fig. 4.
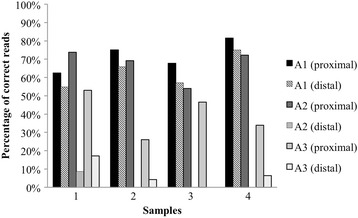
Investigating detection accuracy for 7-mers in exon 14, using three different amplicons for sequencing. The percentage of valid reads in 4 wild type samples is shown from both directions
Fig. 5.
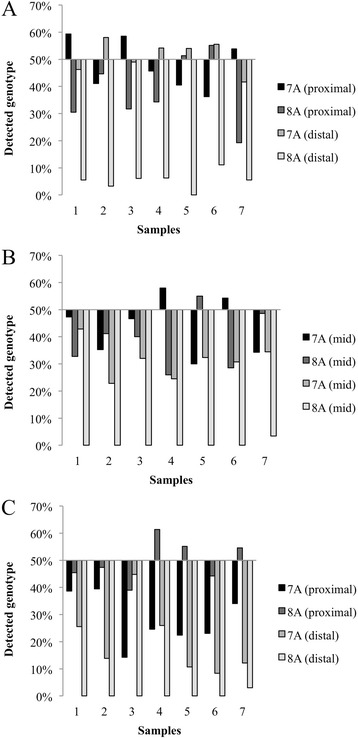
Investigating detection accuracy for 7- and 8-mers in exon 14 using three different amplicons for sequencing. Seven patients heterozygous for 2184insA were analyzed, a, b and c show the degree of deviation of the detected genotypes (7A and 8A) from the theoretical 50%
Discussion
Compared to the Sanger approach, recent next generation sequencing platforms provide deeper coverage and unprecedented throughput. However, inherent drawbacks in terms of read length or false calls could be an issue in some situations [34, 35], which is why careful correction of sequencing errors is crucial [26, 27]. Despite being one of the most common NGS techniques [36], pyrosequencing suffers from over- or undercalls in HP runs, which was described previously [37, 38]. New methods in processing pyrosequencing data might improve the correct analysis of indel mutations even in homopolymer regions [39–41], but it is prudent to be aware of the limitations of medical instrumentation used for investigating patient samples.
In order to assess the accuracy of pyrosequencing in homopolymer regions, first we established a plasmid-based test system. The resulting plasmids contained the most common homopolymers (i.e. 4-mer to 6-mer) as judged by Sanger sequencing. We found that pyrosequencing was more reliable in the case of 4-mers and sequencing of longer HP tracts was increasingly less reliable (see Fig. 2). Reliability unambiguously decreased with the growing number of nucleotides in the homopolymer. Based on our previous experiences, we hypothesized that in the initial phase of the sequencing reaction (immediately following the primers) the signal-to-noise ratio could be acceptably high for the appropriate determination of subtle HP sections. Our results using the plasmid system described was unable to confirm our hypothesis (see Fig. 3). Despite the great variability in sequencing accuracy, we found usable combinations of primers, for all sized homopolymers tested, which highlights the importance of careful primer selection and testing before being used for routine genetic diagnostics.
In the second part we focused on a molecular genetic diagnostic assay development and optimization, where pyrosequencing of the entire coding region of the CFTR was carried out. Here we used clinical samples that had been tested using molecular diagnostic-level assays (a CE-IVD kit and Sanger sequencing). The assay showed excellent sensitivity and specificity, because all benign and pathogenic variants (missense, nonsense, splice site alterations, out-of-frame or in-frame deletions and insertions previously identified by Sanger sequencing) were confirmed by pyrosequencing using the in-house designed primer set both in HP and non-HP containing regions. Except for a unique 7A tract, sequencing of 24 HP stretches yielded good performance with genotyping accuracy greater than 80%, including the detection of another 7-mer (7 T) in exon 1 with 83% accuracy. Therefore, it is likely, that homopolymer detection depends not only on HP length or primer distance, but also on several other factors, such as the nucleotide microenvironment in the DNA sequence or the spatial location of beads on PicoTiter plates [42].
It is extremely important to identify even small scale insertions or deletions in homopolymer sections with high reliability. 2184insA, a frequent CF-causing mutation in Hungary and Western Ukraine [30–32] is an eight-adenine long HP region was further analyzed by using 11 DNA samples from CF patients. Other mutations are also known (2184delA and 2183delAAinsG) to affect the same poly-A tract, creating five or six adenine long HPs. We found that mutations that lead to the formation of 5-mers and 6-mers, can be detected with high specificity. While genotyping of the 8-mer c.2052_2053insA is reproducible, the detection rate using pyrosequencing chemistry fails to reach 75% most of the time, therefore this region of the gene still needs to be Sanger sequenced when testing patient samples in routine diagnostic procedures.
Conclusion
Reliable mutation detection plays a key role when using new NGS techniques for routine clinical diagnostics. In order to reach the required analytical sensitivity and specificity, some NGS methods might need more complex workflow (i.e. the addition of fragment analysis or supplementary Sanger sequencing of certain nucleotide regions). We have developed a plasmid test system that can be used to assess genotyping accuracy of next generation sequencing systems, which could be very useful during the validation of those methods relative to correct homopolymer detection. Through careful planning of PCR primers, we developed an amplicon-based NGS assay that can be used for detection of small-scale CFTR mutations and showed, that its analytical validity in the clinical setting is acceptable for 4- to 6-mer HPs, but not for 7-mers and beyond. At the same time, investigation of the 2184insA mutation clearly shows that Sanger sequencing is still required in specific situations and thus, cannot yet be eliminated from the molecular diagnostic workflow.
Additional files
Figure S1. Representative Sanger electropherograms of the generated homopolymers. 4-mers, 5-mers, and 6-mers are shown in the left, middle, and right columns, respectively. (PDF 176 kb)
Table S1. Mutagenesis, amplification and sequencing primers in the plasmid system. Amplification/sequencing primers contain a starting “Tag sequence,” which was separated by a space within the primer sequence. (DOCX 35 kb)
Figure S2. Representative Sanger electropherograms of all the HP-containing exons of the CFTR gene (clinical sample number 50615). The electropherograms show the exonic sequences including two nucleotides of the introns. (PDF 1945 kb)
Figure S3. Sanger electropherogram of a sample of a patient with a CFTR 2184insA mutation in heterozygous form. (PDF 45 kb)
Table S2. Self-designed primers used for assessing HP regions in the CFTR gene. Flopping bases on known SNPs are in brackets. “Tag sequences” also included in the beginning of the primers, separated by a space. (DOCX 58 kb)
Table S3. Self-designed primers used for assessing non- HP regions in the CFTR gene. “Tag sequences” also included in the beginning of the primers, separated by a space. (DOCX 31 kb)
Acknowledgements
Not applicable.
Funding
This study was supported by the Hungarian Research Fund (K109076) and Ministry of National Economy, Hungary (GINOP-2.3.2–15–2016-00039) to IB, 00064203/6003, CZ.2.16/3.1.00/24022OPPK from the Czech Ministry of Health and CZ.02.1.01/0.0/0.0/16_013/0001634 and LM2015091 from the Czech Ministry of Youth, Education and Sports to MM, and the New National Excellence Program of the Ministry of Human Capacities (UNKP-16-3 IV/4) to IG. Research funding played no role in the study design; in the data collection and analysis and interpretation.
Availability of data and materials
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Abbreviations
- CF
Cystic fibrosis
- CFTR
Cystic fibrosis transmembrane regulator gene
- HGVS
Human genome variation society
- HP
Homopolymer
- NGS
Next generation sequencing
- SNP
Single-nucleotide polymorphism
Authors’ contributions
LM, ED and AH: analysis and acquisition of data, KK, JK, VK, and MM Jr.: design of the study, interpretation of data and revising the manuscript. IB: design of the study, interpretation of data and writing the manuscript. GI: design of the study, analysis, acquisition and interpretation of data, writing the manuscript. All authors have read and approved the manuscript.
Ethics approval and consent to participate
All human participants gave informed consent for diagnostic genetic analysis. In this study DNA samples were then applied anonymously and procedures were in accordance with the current revision of the Helsinki Declaration. The laboratory is approved by the National Public Health and Medical Officer Service (approval number: 094025024).
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4544-x) contains supplementary material, which is available to authorized users.
Contributor Information
Gergely Ivády, Email: ivady@med.unideb.hu.
László Madar, Email: mlaszlo0911@gmail.com.
Erika Dzsudzsák, Email: dzserka@gmail.com.
Katalin Koczok, Email: koczok@med.unideb.hu.
János Kappelmayer, Email: kappelmayer@med.unideb.hu.
Veronika Krulisova, Email: veronika.krulisova@fmotol.cz.
Milan Macek, Jr, Email: milan.macek.jr@fmotol.cuni.cz.
Attila Horváth, Email: horvath.attila4444@gmail.com.
István Balogh, Phone: +36 52 340006, Email: balogh@med.unideb.hu.
References
- 1.Templeton AR, Clark AG, Weiss KM, Nickerson DA, Boerwinkle E, Sing CF. Recombinational and mutational hotspots within the human lipoprotein lipase gene. Am J Hum Genet. 2000;66:69–83. doi: 10.1086/302699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brahmachari SK, Sarkar PS, Raghavan S, Narayan M, Maiti AK. Polypurine/polypyrimidine sequences as cis-acting transcriptional regulators. Gene. 1997;190:17–26. doi: 10.1016/S0378-1119(97)00034-6. [DOI] [PubMed] [Google Scholar]
- 3.Dechering KJ, Cuelenaere K, Konings RN, Leunissen JA. Distinct frequency-distributions of homopolymeric DNA tracts in different genomes. Nucleic Acids Res. 1998;26:4056–4062. doi: 10.1093/nar/26.17.4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toth G, Gaspari Z, Jurka J. Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res. 2000;10:967–981. doi: 10.1101/gr.10.7.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Denver DR, Morris K, Kewalramani A, Harris KE, Chow A, Estes S, et al. Abundance, distribution, and mutation rates of homopolymeric nucleotide runs in the genome of Caenorhabditis Elegans. J Mol Evol. 2004;58:584–595. doi: 10.1007/s00239-004-2580-4. [DOI] [PubMed] [Google Scholar]
- 6.Nelson HC, Finch JT, Luisi BF, Klug A. The structure of an oligo(dA).Oligo(dT) tract and its biological implications. Nature. 1987;330:221–226. doi: 10.1038/330221a0. [DOI] [PubMed] [Google Scholar]
- 7.Sen D, Gilbert W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature. 1988;334:364–366. doi: 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- 8.Denver DR, Morris K, Lynch M, Vassilieva LL, Thomas WK. High direct estimate of the mutation rate in the mitochondrial genome of Caenorhabditis Elegans. Science. 2000;289:2342–2344. doi: 10.1126/science.289.5488.2342. [DOI] [PubMed] [Google Scholar]
- 9.Kunkel TA. The mutational specificity of DNA polymerases-alpha and -gamma during in vitro DNA synthesis. J Biol Chem. 1985;260:12866–12874. [PubMed] [Google Scholar]
- 10.Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol. 1997;17:2859–2865. doi: 10.1128/MCB.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hyman ED. A new method of sequencing DNA. Anal Biochem. 1988;174:423–436. doi: 10.1016/0003-2697(88)90041-3. [DOI] [PubMed] [Google Scholar]
- 12.Ronaghi M, Uhlen M, Nyren P. A sequencing method based on real-time pyrophosphate. Science. 1998;281:363–365. doi: 10.1126/science.281.5375.363. [DOI] [PubMed] [Google Scholar]
- 13.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 14.Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475:348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 15.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 16.Balzer S, Malde K, Jonassen I. Systematic exploration of error sources in pyrosequencing flowgram data. Bioinformatics. 2011;27:304–309. doi: 10.1093/bioinformatics/btr251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8:143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seneca S, Vancampenhout K, Van Coster R, Smet J, Lissens W, Vanlander A, et al. Analysis of the whole mitochondrial genome: translation of the ion torrent personal genome machine system to the diagnostic bench? Eur J Hum Genet. 2015;23:41–48. doi: 10.1038/ejhg.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ. Removing Noise From Pyrosequenced Amplicons. BMC Bioinformatics. 2011;12(1):38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bragg L, Stone G, Imelfort M, Hugenholtz P, Tyson GW. Fast, accurate errorcorrection of amplicon pyrosequences using Acacia. Nat Methods. 2012;9(5):425–426. doi: 10.1038/nmeth.1990. [DOI] [PubMed] [Google Scholar]
- 21.Saha S, Rajasekaran S. EC: an efficient error correction algorithm for short reads. BMC Bioinformatics. 2015;16(Suppl 17):S2. doi: 10.1186/1471-2105-16-S17-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wirawan A, Harris RS, Liu Y, Schmidt B, Schroder J. HECTOR: a parallel multistage homopolymer spectrum based error corrector for 454 sequencing data. BMC Bioinformatics. 2014;15:131. doi: 10.1186/1471-2105-15-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaspar JM, Thomas WK. FlowClus: efficiently filtering and denoising pyrosequenced amplicons. BMC Bioinformatics. 2015;16(1):105. doi: 10.1186/s12859-015-0532-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mysara M, Leys N, Raes J, Monsieurs P. NoDe: a fast error-correction algorithm for pyrosequencing amplicon reads. BMC Bioinformatics. 2015;16(1):88. doi: 10.1186/s12859-015-0520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee B, Moon T, Yoon S, Weissman T, Wang J. DUDE-Seq: Fast, flexible, and robust denoising for targeted amplicon sequencing. PLoS One. 2017;12(7):e0181463. doi: 10.1371/journal.pone.0181463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeng F, Jiang R, Chen T. PyroHMMsnp: an SNP caller for ion torrent and 454 sequencing data. Nucleic Acids Res. 2013;41:e136. doi: 10.1093/nar/gkt372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang X, Chockalingam SP, Aluru S. A survey of error-correction methods for next-generation sequencing. Brief Bioinform. 2013;14:56–66. doi: 10.1093/bib/bbs015. [DOI] [PubMed] [Google Scholar]
- 28.Feliubadalo L, Lopez-Doriga A, Castellsague E, del Valle J, Menendez M, Tornero E, et al. Next-generation sequencing meets genetic diagnostics: development of a comprehensive workflow for the analysis of BRCA1 and BRCA2 genes. Eur J Hum Genet. 2013;21:864–870. doi: 10.1038/ejhg.2012.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abou Tayoun AN, Tunkey CD, Pugh TJ, Ross T, Shah M, Lee CC, et al. A comprehensive assay for CFTR mutational analysis using next-generation sequencing. Clin Chem. 2013;59:1481–1488. doi: 10.1373/clinchem.2013.206466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Makukh H, Krenkova P, Tyrkus M, Bober L, Hancarova M, Hnateyko O, et al. A high frequency of the cystic fibrosis 2184insA mutation in western Ukraine: genotype-phenotype correlations, relevance for newborn screening and genetic testing. J Cyst Fibros. 2010;9:371–375. doi: 10.1016/j.jcf.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 31.Ivady G, Madar L, Nagy B, Gonczi F, Ajzner E, Dzsudzsak E, et al. Distribution of CFTR mutations in eastern Hungarians: relevance to genetic testing and to the introduction of newborn screening for cystic fibrosis. J Cyst Fibros. 2011;10:217–220. doi: 10.1016/j.jcf.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 32.Ivády G, Koczok K, Madar L, Gombos E, Toth I, Gyori K, et al. Molecular analysis of cystic fibrosis patients in Hungary – an update to the mutational spectrum. J Med Biochem. 2015;34:46–51. doi: 10.2478/jomb-2014-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dequeker E, Stuhrmann M, Morris MA, Casals T, Castellani C, Claustres M, et al. Best practice guidelines for molecular genetic diagnosis of cystic fibrosis and CFTR-related disorders--updated European recommendations. Eur J Hum Genet. 2009;17:51–65. doi: 10.1038/ejhg.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pickrell WO, Rees MI, Chung SK. Next generation sequencing methodologies--an overview. Adv Protein Chem Struct Biol. 2012;89:1–26. doi: 10.1016/B978-0-12-394287-6.00001-X. [DOI] [PubMed] [Google Scholar]
- 35.Rizzo JM, Buck MJ. Key principles and clinical applications of “next-generation” DNA sequencing. Cancer Prev Res (Phila) 2012;5:887–900. doi: 10.1158/1940-6207.CAPR-11-0432. [DOI] [PubMed] [Google Scholar]
- 36.Harrington CT, Lin EI, Olson MT, Eshleman JR. Fundamentals of pyrosequencing. Arch Pathol Lab Med. 2013;137:1296–1303. doi: 10.5858/arpa.2012-0463-RA. [DOI] [PubMed] [Google Scholar]
- 37.Ronaghi M. Improved performance of pyrosequencing using single-stranded DNA-binding protein. Anal Biochem. 2000;286:282–288. doi: 10.1006/abio.2000.4808. [DOI] [PubMed] [Google Scholar]
- 38.Ahmadian A, Ehn M, Hober S. Pyrosequencing: history, biochemistry and future. Clin Chim Acta. 2006;363:83–94. doi: 10.1016/j.cccn.2005.04.038. [DOI] [PubMed] [Google Scholar]
- 39.Deng W, Maust BS, Westfall DH, Chen L, Zhao H, Larsen BB, et al. Indel and carryforward correction (ICC): a new analysis approach for processing 454 pyrosequencing data. Bioinformatics. 2013;29:2402–2409. doi: 10.1093/bioinformatics/btt434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iyer S, Bouzek H, Deng W, Larsen B, Casey E, Mullins JI. Quality score based identification and correction of pyrosequencing errors. PLoS One. 2013;8:e73015. doi: 10.1371/journal.pone.0073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beuf KD, Schrijver JD, Thas O, Criekinge WV, Irizarry RA, Clement L. Improved base-calling and quality scores for 454 sequencing based on a hurdle Poisson model. BMC Bioinformatics. 2012;13:303. doi: 10.1186/1471-2105-13-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gilles A, Meglecz E, Pech N, Ferreira S, Malausa T, Martin JF. Accuracy and quality assessment of 454 GS-FLX titanium pyrosequencing. BMC Genomics. 2011;12:245. doi: 10.1186/1471-2164-12-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative Sanger electropherograms of the generated homopolymers. 4-mers, 5-mers, and 6-mers are shown in the left, middle, and right columns, respectively. (PDF 176 kb)
Table S1. Mutagenesis, amplification and sequencing primers in the plasmid system. Amplification/sequencing primers contain a starting “Tag sequence,” which was separated by a space within the primer sequence. (DOCX 35 kb)
Figure S2. Representative Sanger electropherograms of all the HP-containing exons of the CFTR gene (clinical sample number 50615). The electropherograms show the exonic sequences including two nucleotides of the introns. (PDF 1945 kb)
Figure S3. Sanger electropherogram of a sample of a patient with a CFTR 2184insA mutation in heterozygous form. (PDF 45 kb)
Table S2. Self-designed primers used for assessing HP regions in the CFTR gene. Flopping bases on known SNPs are in brackets. “Tag sequences” also included in the beginning of the primers, separated by a space. (DOCX 58 kb)
Table S3. Self-designed primers used for assessing non- HP regions in the CFTR gene. “Tag sequences” also included in the beginning of the primers, separated by a space. (DOCX 31 kb)
Data Availability Statement
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.