

Abstract
Paraphrasing Dobzhansky’s famous dictum, I discuss how interrogating cancer through the lens of evolution has transformed our understanding of its development, causality and treatment resistance. The emerging picture of cancer captures its extensive diversity and therapeutic resilience, highlighting the need for more innovative approaches to control.
Evolutionary biology and medicine
Dobzhansky’s insight applies not just to biology but to much in medicine. For example, our vulnerability to many chronic diseases in modern societies probably owes much to a mismatch between contemporary lifestyles and historical, evolutionary adaptations [1, 2]. Another potent example is with the development of drug resistance in microbes and parasites which is contingent upon clonal, evolutionary selection [3]. Similarly, the emergence of new or more virulent microbial pathogens reflects the outcome of evolutionary arms races between the immune system’s pathogen recognition repertoire and the high mutability of viruses, parasites and bacteria [4]. It’s a travesty that it is still possible to obtain a medical degree whilst in denial, or lacking understanding, of the essential tenets of evolutionary biology [5]. But, it is also likely that some evolutionary biologists are unaware of the medical implications of their field.
Cancer provides a paradigm for the applicability of evolutionary principles to a medical problem [6]. An appreciation that cancer clones develop, or evolve, in the context of a complex tissue ecosystem has transformed our understanding of cancer biology and highlighted the need for more innovative approaches to therapy that can thwart evolutionary resilience [7–9]. An evolutionary logic pervades all major areas of cancer sciences including causation, cancer clone development and resistance to therapies [10] (Fig. 1).
Fig. 1.
Nothing in cancer makes sense except in the light of evolution. Images: Charles Darwin (Cambridge University Library), breast cancer cell (National Cancer Institute [83])
Cancer clone evolution
The natural history of cancer is illustrated in a very simplified fashion in Fig. 2. The evolutionary trajectory of a cancer clone, starting from a single mutant cell and progressing to a malignant and metastatic clone of ~ 1011 cells, can have very variable dynamics, with time frames ranging from a few months (some aggressive paediatric tumours) to one or several decades (many adult epithelial carcinomas). The tempo of cell population dynamics can be steady or proceed in jumps—punctuated equilibrium [11]. The majority of initiated tumour clones never evolve to fully fledged malignant clones [12, 13] but for those that do, the end game is dissemination in the body, or metastasis, a territorial hijack with onboard therapeutic resistance. It’s an evolutionary process, not just in terms of change over time but in the true Darwinian sense of random genetic variation and natural selection of the best-adapted or fittest variants. Cancer clone progression is equivalent to fast track evolution of an asexual species of unicellular organisms. But it’s fuelled by the recombinatorial genetic diversity normally acquired via sexual reproduction.
Fig. 2.
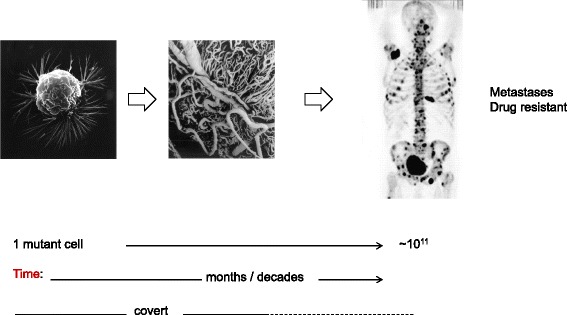
Natural history of cancer. Left: Breast cancer cell (National Cancer Institute [83])
Middle: Stereoscan image showing neovascularisation around an in situ carcinoma (angiogenesis). Photo provided by Professor M A Konerding. Right: PET scan showing cancer disseminated throughout the body (dark patches). Image originally published in JNM [84] and reproduced with permission: Even-Sapir E, Metser U, Mishani E, Gennady Lievshitz G, Lerman H, Leibovitch I. The detection of bone metastases in patients with high-risk prostate cancer: 99mTc-MDP planar bone scintigraphy, single- and multi-field-of-view SPECT, 18F–fluoride PET, and 18F–fluoride PET/CT. J Nucl Med. 2006;47:287–97. © by the Society of Nuclear Medicine and Molecular Imaging, Inc. Most of this evolutionary process is clinically silent or covert
These ideas first emerged in the 1970s [14]. The evidence then was based on observations of serial changes, over time, in gross chromosomal structural alterations in cells. The current perspective is more detailed and contextual [7] with cancer cells subject to whole genome sequencing [15]. Single cell genetic scrutiny [16–20] or analysis of small micro-dissected regions of tumours [21–23] identifies sub-clonal architectures from which phylogenic relationships can be inferred.
Clonal phylogenies for cancer cells can reveal early or founder genetic lesions (present in all cells) and time-ordered sequences of subsequent mutations. In most cases, sub-clonal architectures are branching rather than linear [15–18, 21–24], reminiscent of Charles Darwin’s iconic ‘I think’ drawing in which he imagined how different species might evolve from a common ancestor (Fig. 3). Side branches of individual cancers often have independent mutations in the same genes, reflecting parallel or convergent evolution and prevailing selective pressures on all sub-clones. This new, evolutionary portrait of cancer cell diversity and its variegated genetics [24] has considerable practical implications for patient prognosis, monitoring and treatment [8–10].
Fig. 3.
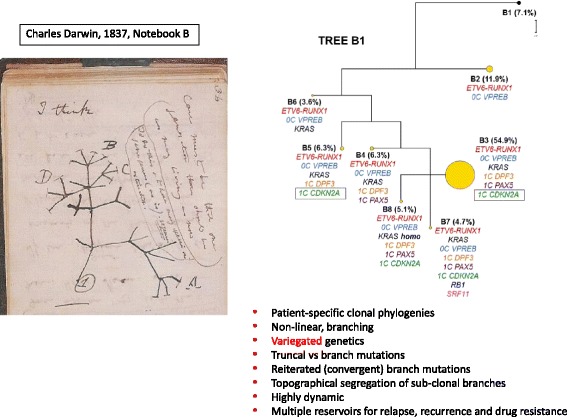
Critical features of cancer clone phylogenetics. Left: Charles Darwin’s iconic ‘I think’ drawing of a phylogenetic tree from his 1837 Notebook (B) [85]. Right: Example of subclonal phylogeny based on single cell genetics (in leukemia), taken from author’s own research in [17]. Seven subclones shown (B2–8), each with mutations listed. B1 (7.1%) are normal cells. CDKN2A in green in box: reiterated mutation of same gene is different branches or subclones. One subclone (B3) is numerically dominant (at 54.9%)
There are caveats to these analyses. Cancer cell phylogenetic constructs are often based on single time point snapshots and miss the dynamic shifts in sub-clonal population structure that occur at early pre-clinical time points, over time with progression of disease and in recurrence or relapse. The depth of genomic sequencing is still limited in most cases and, as a consequence, minor sub-clones are invisible and the extent of diversity under-estimated [25].
Although some of the sub-clonal architecture in cancer derives from neutral evolution or drift [26], particularly in early phases with low cell numbers [27], a prevailing view is that cancer cell populations undergo positive, or, occasionally, negative, selection via tissue ecosystem pressures [7, 28]. In this sense, the highly recurrent genetic changes in gene copy number or single nucleotide variants can be seen as adaptive, being selected, in a Darwinian sense, as a consequence of the fitness benefit they provide [10]. Fitness is expressed via so-called ‘hallmark’ phenotypic features of cancer cells [29], which include enhanced proliferation, resistance to signals for cell death or senescence, metabolic changes and epigenetic shifts favouring self-renewal of stem/progenitor cells at the expense of differentiation. All of which impact, directly or indirectly, on reproductive fitness. Some cancers exhibit massive genomic instability [15] but even this can be considered as an adaptive strategy, gambling on rare ‘winners’, as similarly employed by bacteria under potent metabolic stress [30].
The cancer ecosystem
The cancer tissue ecosystem is itself complex and dynamic and is altered as a consequence of the invasion of cancerous cells. Understanding the network of regulatory interactions within the cancer ecosystem, involving stromal cells, the vasculature, and invasive inflammatory cells, is in its infancy. Nevertheless, there is accumulating evidence that features of the cancer microenvironment, including hypoxia and acidosis, diversity of inflammatory cell infiltrates, activated stromal cells and patterns of vascularisation, are major drivers of cancer clone progression, impacting on clinical outcome [24, 27, 31–35]. Ecosystem variables may also provide novel opportunities for therapy [36].
These considerations suggest that it might be possible to define an evolutionary and ecological index of individual cancers that is predictive of the likelihood of progression, metastasis and drug resistance and which could, in the future, guide critical patient management decisions [37].
Not all cancer cells have equivalent proliferative capacity. Many cancers, if not all, have sub-populations of cells with stem cell-like features or self-renewal capacity, i.e. they reproduce themselves at the expense of differentiation [38]. The frequency or proportion of these cells within a cancer cell population varies greatly, as do their other phenotypic features, which has led to some confusion on their relevance. However, most cancer sub-clones contain cells with extensive propagating or stem cell function [24, 39–41]. It is likely that self-renewing, or stem, cells are the major cellular substrate for the selective processes that underlie clonal architecture, progression of disease, metastasis, recurrence and drug resistance. As such, they provide both the evolutionary units of selection [42–44] in cancer and the ultimate targets for therapeutic control or cure [10, 38, 45, 46].
Technical advances in genomics have driven much of this paradigm shift in our understanding of cancer biology. On occasion, this has encouraged a rather gene-centric view of cancer, not altogether dissimilar to Richard Dawkins’ selfish gene perspective on evolution itself. Cancer has, for example, been defined as ‘a disease of the genome’ [15]. Mutated genomes lie at the heart of the emergence and malignant progression of cancer but we should not ignore the critical, contextual role of the ecosystem habitats in which this adaptive process occurs.
Mapping cancer clone complexity and evolution over time is demanding with solid tumours, which, unlike the blood borne leukaemias, often have topographically segregated sub-clones [21–23]. This means that biopsy-based samples can be highly biased [8]. A solution to this challenge is, however, provided by serial screening of cancer-derived DNA fragments in plasma. This allows presumed unbiased, sensitive, serial monitoring of cancer clone evolution, virtually in real time. Applications of this technology are already impacting on patient management, for example by detecting the early, pre-clinical re-emergence of disease or drug-resistant sub-clones [47–49].
Evolutionary origins of vulnerability to cancer
There is less appreciation of the relevance of an evolutionary perspective to causation in cancer. Cancer epidemiologists, understandably, focus on proximate mechanisms that hold the prospect of intervention—for example with cigarette smoking, UVB exposure or viruses. It is now well recognised, from genome-wide association studies (GWAS), that a very large number (~ 100 s) of inherited gene variants impact on cancer risk. Individual variants are mostly in non-coding regulatory regions and, individually, contribute to only a very modest increase in risk, conferring odds ratios of ~ 1.01–1.1 [50].
What is missing from this genetic epidemiology description is an appreciation of vulnerability. Why is it that animals in effectively all phyla can, and do, develop cancer [51], including Cnidaria (e.g. Hydra) at the base of the animal phylogenetic tree [52]? Why is it that cancer risk for ageing humans is now off the scale at close to a 50% lifetime risk?
The ubiquity of cancer can plausibly be ascribed to the intrinsic ‘design’ fallibilities of replicating organisms [53], including the essential mutability of DNA. Many cancer relevant genes recognised as tumour suppressors or oncogenes appeared at that time that multi-cellularity emerged around 700 million years ago [54]. The multi-cellular contract requires compliance of cell behaviour and proliferative restraint. But as many cells, and especially stem cells, retain extensive proliferative or regenerative capacity, opportunities exist for mutant cheaters. And, as in other social groupings [55, 56], cheaters will occasionally succeed in taking these opportunities. Stem cells, in particular, are an evolutionary liability with respect to mutability, positive selection and cancer [57]. Moreover, they function in an environment, endogenous or exogenous, replete with genotoxic challenges that can damage DNA as with, for example, UVB, natural plant toxins or oxidative metabolism.
But we still require a plausible explanation of why humans, at least in modern, developed societies, have such a high lifetime risk of a cancer diagnosis. Some ascribe this to a consequence of ageing itself. As common, historical causes of death—famine, predation, infection, cardio-vascular disease—have come under control, cancer could then be the default health outcome in ageing individuals who are post-reproductive and may have reduced capacity for DNA repair and immune surveillance. Could this just be then the legacy of intrinsic cancer risk leaking through unrestrained? If so, how come ageing and large elephants and whales have rather little cancer (Peto’s paradox) [58, 59]? How come the incidence rates of particular cancers vary substantially (2–400×) between different places and over time, and as seen in migrant groups [60]? The time/place patterns of cancer incidence only make sense if lifestyle factors are a significant component of risk.
One explanation for at least some of the very high risk of cancer in humans that this author favours is that it reflects the impact of chronic exposure to an evolutionary mismatch [6, 53]: a mismatch between our rapid social evolution and ‘modern’ lifestyles versus historical, evolutionary adaptations (Fig. 4). An example of this would be with the risk of skin cancers in white, Caucasian individuals, which is orders of magnitude greater than the risk of individuals with black or pigmented skin. Historically, variable levels of skin pigmentation, via melanin content, have been selected by environmental pressures. Depigmented, pale or white skin was an adaptation to cloudy northern climes with low UVB levels in Europe, improving survival prospects and reproductive fitness probably via enhanced vitamin D levels and, possibly, diminution of the impact of frostbite [61]. Modern lifestyles and foreign exile or travel have disrupted this adaptive context. This is rapid social evolution that outpaces any prospect of genetic, evolutionary adaptations. Similar arguments apply to modern reproductive lifestyles (compounded with diet)—early menarche, delayed first pregnancy and diminished breastfeeding that escalate risk of breast cancer via a mismatch with evolutionary adaptations of non-seasonal oestrus and protracted breastfeeding [62, 63]. Reduced microbial exposures in infancy may underlie the increased risk of childhood acute lymphoblastic leukaemia in affluent, developed societies as they deprive the immune system of the ‘educational’ microbial exposure required for its network settings [63].
Fig. 4.

Evolutionary origins of vulnerability to cancer. See [63] for detailed discussion of this argument
Ways of escape
When cancer is lethal, this is usually because of two reasons. Firstly, the cancer clones disseminate or metastasize to ecotopic tissues, compromising essential functions. And, secondly, at this advanced stage cancer cells are almost invariably robust and resistant to therapy. Thwarting the evolutionary resilience of cancer can then be seen as the major therapeutic challenge [10, 64–66].
The emergence of drug resistance makes sense, and only makes sense, in an evolutionary context. Several distinct routes to therapeutic escape are employed and each has an evolutionary rationale (Table 1). The ‘classic’ route to drug (or immunotherapy) resistance in cancer is, essentially, via the same mechanisms as seen with drug resistance in bacteria, malaria or HIV [3, 67, 68]; namely the positive selection (by therapy) of pre-existing variants that can evade the drug, or immune predation, via the serendipitous possession of mutations in the target pathway. We don’t yet have a Luria-Delbrück fluctuation test for this. Nevertheless, with highly targeted therapy, escapee cells spawning recurrence of disease have mutations in drug binding sites and can be backtracked, in some instances, to tissue prior to drug exposure [69–71]. Given enough cells and a reasonable mutation rate, this route to escape is inevitable.
Table 1.
Routes to escape from targeted therapy in cancer
| • Genetic variation | i. ‘Target’ segregated in sub-clones, not truncal |
| ii. ‘Target’ mutated and impervious to drugs | |
| • Epigenetic plasticity | i. Inhibited target bypassed by signal redundancy in network |
| ii. Quiescent/dormant stem cells intrinsically resistant |
If the therapeutic target, e.g. a mutant kinase, is in a side branch of the cancer clone’s evolutionary tree rather than truncal, then targeted therapy with a tyrosine kinase inhibitor (TKI) cannot be curative [72]. It’s then equivalent to pollarding trees or pruning plants—the plant, or cancer, shrinks in size and then rebounds with a flourish. Therapies targeted at critical signalling molecules such as kinases are also readily bypassed by signal redundancy [73, 74], a longstanding, networked feature of eukaryotic cells [75].
As might have been anticipated from an evolutionary perspective, cancer stem cells can evade therapeutic predation via other routes. Normal stem cells spend most of their time reversibly quiescent and out of proliferative cell cycle, which renders them less liable to mutation and damage [76]. They are also well equipped with efflux pumps for noxious, drug-like natural chemicals. Stem cells are vital to life—and limited in number. It is unsurprising that they will have acquired multiple protective mechanisms; mechanisms that can be readily co-opted by cancer stem cells under therapeutic assault [77]. It is now clear that quiescent or dormant cancer stem cells are intrinsically resistant to drugs and radiotherapy [38]. They can ‘hunker’ down, using a ploy long adopted by bacteria and other, eukaryotic, unicellular species [78]. Dormant cancer stem cells can re-enter a proliferative cycle and regenerate a malignant clone after two decades or more of ‘sleep’ [79].
Collectively, these escape routes equip cancer cells, especially in advanced disease with high turnover burdens and high mutation rates (or genetic instability), with great resilience (Table 1). Like a tardigrade [80], they can survive almost any insult.
The one real success in targeted therapy, small molecule tyrosine kinase inhibitors (TKIs) in chronic myeloid leukaemia (CML) [81], is contingent upon the target kinase (ABL1) being the founder lesion in every cell and the clones having minimal genetic diversity. Resistant mutants with altered ABL1 kinase do occur (prior to treatment) but many patients achieve sustained remissions or re-enter remission following a switch to an alternative TKI [71]. And when patients are effectively ‘cured’, the mutant leukaemia stem cells may persist for a protracted period of time, but in a dormant state. CML stem cells that are quiescent, or dormant, appear to have no dependence on the mutated ABL1 kinase. The CML clone isn’t then eradicated but kept under control, its capacity for evolutionary progression blocked.
Outlook: strategies for control
The extent of cellular and genetic diversity in cancer, both between and within individual patients, is daunting but has a logic in terms of normal tissue developmental biology and evolutionary processes of drift and selection [10, 28]. We now see that the major challenge in cancer control is how to thwart the evolutionary resilience of the disease, especially when it is detected relatively late in its trajectory, as with pancreatic, CNS, lung and ovarian cancers. In principle, several strategies are available to us, all endorsed by the National Cancer Institute in the USA and Cancer Research UK. First, despite the contentious argument that many cancers arise via spontaneous mutation [82], the majority are potentially preventable. For example by prudent avoidance (e.g. cigarettes, solar UVB), modified behaviour (e.g. diet/exercise balance) or prophylaxis (e.g. virally induced cancers such as HPV/cervical cancer). Secondly most cancers are curable by surgery or radiotherapy if detected early when localised.
The main challenge of contemporary cancer therapeutics is with advanced or metastatic disease. The third essential strategy is to design drug combinations, including immunotherapy, and schedules that can thwart the emergence of resistance in established disease [64] by either eliminating all cancer stem cells, steering them into more benign fitness peaks or applying an evolutionary break [65]. ‘Taming’ may be a more achievable objective than elimination for metastatic disease [64, 65]. Having a better understanding of the cancer ecosystem and its selective pressures might facilitate novel approaches to control that do not solicit emergence of resistant sub-clones.
There is not, and will not be, a magic bullet or penicillin equivalent for cancer. We need to intervene at all three time points in cancer’s evolutionary trajectory (Fig. 5) if we are to erode further its impact on society.
Fig. 5.
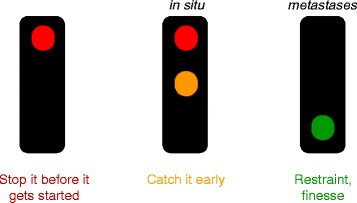
Traffic control of cancer cell evolution. Red: Stop it before it gets started: prophylactic intervention (e.g. HPV vaccines) or prudent avoidance (e.g. cigarette tar, UBV). Red + amber: Catch it early: by surveillance of populations or at risk individuals and early intervention by surgery or radiotherapy. Green: The cancer has already escaped. Employ therapeutic strategies to finesse or restrain continued growth and drug resistance. In situ refers to tumours confined to primary tissue site. Mets metastatic lesions – cancers disseminated to ectopic tissue sites, e.g. breast to brain, prostate to bone. Adapted from author’s reference [10]
Acknowledgements
The author is supported by the Wellcome Trust (105104/Z/14/Z).
Authors’ contributions
MG read and approved the final manuscript.
Competing interests
The author declares that he/she has no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Nesse RM, Stearns SC. The great opportunity: evolutionary applications to medicine and public health. Evol Applications. 2008;1:28–48. doi: 10.1111/j.1752-4571.2007.00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nesse RM, Williams G. Evolution and healing. The new science of Darwinian medicine. London: Weidenfeld & Nicolson; 1995.
- 3.Hughes D, Andersson DI. Evolutionary consequences of drug resistance: shared principles across diverse targets and organisms. Nat Rev Genet. 2015;16(8):459–471. doi: 10.1038/nrg3922. [DOI] [PubMed] [Google Scholar]
- 4.Ewald PW. Evolution of infectious disease. Oxford: Oxford University Press; 1994. [Google Scholar]
- 5.Nesse RM, Schiffman JD. Evolutionary biology in the medical school curriculum. Bioscience. 2003;53(6):585–587. doi: 10.1641/0006-3568(2003)053[0585:EBITMS]2.0.CO;2. [DOI] [Google Scholar]
- 6.Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer. 2006;6:193–203. doi: 10.1038/nrc1816. [DOI] [PubMed] [Google Scholar]
- 7.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27(1):15–26. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Aparicio S, Caldas C. The implications of clonal genome evolution for cancer medicine. N Engl J Med. 2013;368:842–851. doi: 10.1056/NEJMra1204892. [DOI] [PubMed] [Google Scholar]
- 10.Greaves M. Evolutionary determinants of cancer. Cancer Discov. 2015;5(8):806–820. doi: 10.1158/2159-8290.CD-15-0439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–677. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greaves M. Does everyone develop covert cancer? Nat Rev Cancer. 2014;14:209–210. doi: 10.1038/nrc3703. [DOI] [PubMed] [Google Scholar]
- 13.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348(6237):880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 15.Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet. 2012;13:795–806. doi: 10.1038/nrg3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Potter NE, Ermini L, Papaemmanuil E, Cazzaniga G, Vijayaraghavan G, Titley I, Ford A, Campbell P, Kearney L, Greaves M. Single-cell mutational profiling and clonal phylogeny in cancer. Genome Res. 2013;23(12):2115–2125. doi: 10.1101/gr.159913.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, Chen K, Scheet P, Vattathil S, Liang H, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512(7513):155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baslan T, Hicks J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat Rev Cancer. 2017;17(9):557–569. doi: 10.1038/nrc.2017.58. [DOI] [PubMed] [Google Scholar]
- 20.Zahn H, Steif A, Laks E, Eirew P, VanInsberghe M, Shah SP, Aparicio S, Hansen CL. Scalable whole-genome single-cell library preparation without preamplification. Nat Methods. 2017;14(2):167–173. doi: 10.1038/nmeth.4140. [DOI] [PubMed] [Google Scholar]
- 21.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225–233. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, Marjoram P, Siegmund K, Press MF, Shibata D, et al. A Big Bang model of human colorectal tumor growth. Nat Genet. 2015;47(3):209–216. doi: 10.1038/ng.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, Jamal-Hanjani M, Shafi S, Murugaesu N, Rowan AJ, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346(6206):251–256. doi: 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, Kempski H, Moorman AV, Titley I, Swansbury J, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–361. doi: 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- 25.Griffith M, Miller CA, Griffith OL, Krysiak K, Skidmore ZL, Ramu A, Walker JR, Dang HX, Trani L, Larson DE, et al. Optimizing cancer genome sequencing and analysis. Cell Syst. 2015;1(3):210–223. doi: 10.1016/j.cels.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams MJ, Werner B, Barnes CP, Graham TA, Sottoriva A. Identification of neutral tumor evolution across cancer types. Nat Genet. 2016;48(3):238–244. doi: 10.1038/ng.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vermeulen L, Morrissey E, van der Heijden M, Nicholson AM, Sottoriva A, Buczacki S, Kemp R, Tavare S, Winton DJ. Defining stem cell dynamics in models of intestinal tumor initiation. Science. 2013;342(6161):995–998. doi: 10.1126/science.1243148. [DOI] [PubMed] [Google Scholar]
- 28.Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, Davies H, Stratton MR, Campbell PJ. Universal patterns of selection in cancer and somatic tissues. Cell. 2017;171(5):1029–1041. doi: 10.1016/j.cell.2017.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 30.Bjedov I, Tenaillon O, Gerard B, Souza V, Denamur E, Radman M, Taddei F, Matic I. Stress-induced mutagenesis in bacteria. Science. 2003;300(5624):1404–1409. doi: 10.1126/science.1082240. [DOI] [PubMed] [Google Scholar]
- 31.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nature Rev Cancer. 2008;8:56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 32.Anderson ARA, Weaver AM, Cummings PT, Quaranta V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell. 2006;127:905–915. doi: 10.1016/j.cell.2006.09.042. [DOI] [PubMed] [Google Scholar]
- 33.Yuan Y, Failmezger H, Rueda OM, Ali HR, Graf S, Chin SF, Schwarz RF, Curtis C, Dunning MJ, Bardwell H, et al. Quantitative image analysis of cellular heterogeneity in breast tumors complements genomic profiling. Sci Transl Med. 2012;4(157):157ra143. doi: 10.1126/scitranslmed.3004330. [DOI] [PubMed] [Google Scholar]
- 34.Herrera M, Islam AB, Herrera A, Martin P, Garcia V, Silva J, Garcia JM, Salas C, Casal I, de Herreros AG, et al. Functional heterogeneity of cancer-associated fibroblasts from human colon tumors shows specific prognostic gene expression signature. Clin Cancer Res. 2013;19(21):5914–5926. doi: 10.1158/1078-0432.CCR-13-0694. [DOI] [PubMed] [Google Scholar]
- 35.Win T, Miles KA, Janes SM, Ganeshan B, Shastry M, Endozo R, Meagher M, Shortman RI, Wan S, Kayani I, et al. Tumor heterogeneity and permeability as measured on the CT component of PET/CT predict survival in patients with non-small cell lung cancer. Clin Cancer Res. 2013;19(13):3591–3599. doi: 10.1158/1078-0432.CCR-12-1307. [DOI] [PubMed] [Google Scholar]
- 36.Pienta KJ, McGregor N, Axelrod R, Axelrod DE. Ecological therapy for cancer: defining tumors using an ecosystem paradigm suggests new opportunities for novel cancer treatments. Transl Oncol. 2008;1:158–164. doi: 10.1593/tlo.08178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maley CC, Aktipis A, Graham TA, Sottoriva A, Boddy AM, Janiszewska M, Silva AS, Gerlinger M, Yuan Y, Pienta KJ, et al. Classifying the evolutionary and ecological features of neoplasms. Nat Rev Cancer. 2017;17:605–619. doi: 10.1038/nrc.2017.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 39.Meyer M, Reimand J, Lan X, Head R, Zhu X, Kushida M, Bayani J, Pressey JC, Lionel AC, Clarke ID, et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci U S A. 2015;112(3):851–856. doi: 10.1073/pnas.1320611111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eirew P, Steif A, Khattra J, Ha G, Yap D, Farahani H, Gelmon K, Chia S, Mar C, Wan A, et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature. 2015;518(7539):422–426. doi: 10.1038/nature13952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piccirillo SGM, Colman S, Potter NE, van Delft FW, Lillis S, Carnicer M-J, Kearney L, Watts C, Greaves M. Genetic and functional diversity of propagating cells in glioblastoma. Stem Cell Rep. 2015;4:7–15. doi: 10.1016/j.stemcr.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greaves M. Cancer stem cells as 'units of selection'. Evol Appl. 2013;6(1):102–108. doi: 10.1111/eva.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michod RE. Darwinian dynamics: evolutionary transitions in fitness and individuality. Princeton: Princeton University Press; 1999. [Google Scholar]
- 44.Weissman IL. Stem cells: units of development, units of regeneration, and units in evolution. Cell. 2000;100:157–168. doi: 10.1016/S0092-8674(00)81692-X. [DOI] [PubMed] [Google Scholar]
- 45.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 46.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murtaza M, Dawson S-J, Tsui DWY, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin S-F, Kinsbury Z, Wong ASC, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108–112. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- 48.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra224. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carreira S, Romanel A, Goodall J, Grist E, Ferraldeschi R, Miranda S, Prandi D, Lorente D, Frenel JS, Pezaro C, et al. Tumor clone dynamics in lethal prostate cancer. Sci Transl Med. 2014;6(254):254ra125. doi: 10.1126/scitranslmed.3009448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fletcher O, Houlston RS. Architecture of inherited susceptibility to common cancer. Nat Rev Cancer. 2010;10:353–361. doi: 10.1038/nrc2840. [DOI] [PubMed] [Google Scholar]
- 51.Aktipis CA, Boddy AM, Jansen G, Hibner U, Hochberg ME, Maley CC, Wilkinson GS. Cancer across the tree of life: cooperation and cheating in multicellularity. Philos Trans R Soc Lond Ser B Biol Sci. 2015;370(1673):20140219. doi: 10.1098/rstb.2014.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Domazet-Loso T, Klimovich A, Anokhin B, Anton-Erxleben F, Hamm MJ, Lange C, Bosch TC. Naturally occurring tumours in the basal metazoan Hydra. Nat Commun. 2014;5:4222. doi: 10.1038/ncomms5222. [DOI] [PubMed] [Google Scholar]
- 53.Cancer GM. The evolutionary legacy. Oxford: Oxford University Press; 2000. [Google Scholar]
- 54.Domazet-Loso T, Tautz D. Phylostratigraphic tracking of cancer genes suggests a link to the emergence of multicellularity in metazoa. BMC Biol. 2010;8:66. doi: 10.1186/1741-7007-8-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hughes WO, Boomsma JJ. Genetic royal cheats in leaf-cutting ant societies. Proc Natl Acad Sci U S A. 2008;105(13):5150–5153. doi: 10.1073/pnas.0710262105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gilbert OM, Foster KR, Mehdiabadi NJ, Strassmann JE, Queller DC. High relatedness maintains multicellular cooperation in a social amoeba by controlling cheater mutants. Proc Natl Acad Sci U S A. 2007;104(21):8913–8917. doi: 10.1073/pnas.0702723104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255:197–200. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- 58.Caulin AF, Maley CC. Peto's Paradox: evolution's prescription for cancer prevention. Trends Ecol Evol. 2011;26(4):175–182. doi: 10.1016/j.tree.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abegglen LM, Caulin AF, Chan A, Lee K, Robinson R, Campbell MS, Kiso WK, Schmitt DL, Waddell PJ, Bhaskara S, et al. Potential mechanisms for cancer resistance in elephants and comparative cellular response to DNA damage in humans. JAMA. 2015;314(17):1850–1860. doi: 10.1001/jama.2015.13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stewart BW, Wild CP. World Cancer Report 2014. International Agency for Research on Cancer: Lyon; 2014. [PubMed] [Google Scholar]
- 61.Greaves M. Was skin cancer a selective force for black pigmentation in early hominin evolution? Proc Biol Sci B. 2014;281(1781):20132955. doi: 10.1098/rspb.2013.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eaton SB, Pike MC, Short RV, Lee NC, Trussell J, Hatcher RA, Wood JW, Worthman CM, Blurton Jones NG, Konner MJ, et al. Women's reproductive cancers in evolutionary context. Q Rev Biol. 1994;69(3):353–367. doi: 10.1086/418650. [DOI] [PubMed] [Google Scholar]
- 63.Greaves M. Darwinian medicine: a case for cancer. Nat Rev Cancer. 2007;7:213–221. doi: 10.1038/nrc2071. [DOI] [PubMed] [Google Scholar]
- 64.Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30(7):679–692. doi: 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- 65.Gatenby RA, Silva AS, Gillies RJ, Frieden BR. Adaptive therapy. Cancer Res. 2009;69(11):4894–4903. doi: 10.1158/0008-5472.CAN-08-3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target. Cancer Discov. 2017;7(8):805–817. doi: 10.1158/2159-8290.CD-17-0343. [DOI] [PubMed] [Google Scholar]
- 67.Goldberg DE, Siliciano RF, Jacobs WR., Jr Outwitting evolution: fighting drug-resistant TB, malaria, and HIV. Cell. 2012;148:1271–1283. doi: 10.1016/j.cell.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lambert G, Estevez-Salmeron L, Oh S, Liao D, Emerson BM, Tlsty TD, Austin RH. An analogy between the evolution of drug resistance in bacterial communities and malignant tissues. Nat Rev Cancer. 2011;11(5):375–382. doi: 10.1038/nrc3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Diaz LA, Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pfeifer H, Wassmann B, Pavlova A, Wunderle L, Oldenburg J, Binckebanck A, Lange T, Hochhaus A, Wystub S, Brück P, et al. Kinase domain mutations of BCR-ABL frequently precede imatinib-based therapy and give rise to relapse in patients with de novo Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL) Blood. 2007;110(2):727–734. doi: 10.1182/blood-2006-11-052373. [DOI] [PubMed] [Google Scholar]
- 71.Schmitt MW, Loeb LA, Salk JJ. The influence of subclonal resistance mutations on targeted cancer therapy. Nat Rev Clin Oncol. 2016;13(6):335–347. doi: 10.1038/nrclinonc.2015.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yap TA, Gerlinger M, Futreal PA, Pusztai L, Swanton C. Intratumor heterogeneity: seeing the wood for the trees. Sci Transl Med. 2012;4(127):127ps110. doi: 10.1126/scitranslmed.3003854. [DOI] [PubMed] [Google Scholar]
- 73.Turke AB, Zejnullahu K, Wu Y-L, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327(5964):425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant C, Eshkind L, Bockamp E, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135(6):1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 77.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lewis K. Persister cells, dormancy and infectious disease. Nat Rev Microbiol. 2007;5(1):48–56. doi: 10.1038/nrmicro1557. [DOI] [PubMed] [Google Scholar]
- 79.Ford AM, Mansur MB, Furness CL, van Delft FW, Okamura J, Suzuki T, Kobayashi H, Kaneko Y, Greaves M. Protracted dormancy of pre-leukemic stem cells. Leukemia. 2015;29(11):2202–2207. doi: 10.1038/leu.2015.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hashimoto T, Horikawa DD, Saito Y, Kuwahara H, Kozuka-Hata H, Shin IT, Minakuchi Y, Ohishi K, Motoyama A, Aizu T, et al. Extremotolerant tardigrade genome and improved radiotolerance of human cultured cells by tardigrade-unique protein. Nat Commun. 2016;7:12808. doi: 10.1038/ncomms12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112(13):4808–4817. doi: 10.1182/blood-2008-07-077958. [DOI] [PubMed] [Google Scholar]
- 82.Tomasetti C, Vogelstein B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347(6217):78–81. doi: 10.1126/science.1260825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Breast cancer cell. National Cancer Institute, taken by Bruce Wetzel and Harry Schaefer. https://visualsonline.cancer.gov/details.cfm?imageid=1989. Accessed 05 Oct 2017.
- 84.Even-Sapir E, Metser U, Mishani E, Gennady Lievshitz G, Lerman H, Leibovitch I. The detection of bone metastases in patients with high-risk prostate cancer: 99mTc-MDP planar bone scintigraphy, single- and multi-field-of-view SPECT, 18F-fluoride PET, and 18F-fluoride PET/CT. J Nucl Med. 2006;47:287–297. [PubMed] [Google Scholar]
- 85.Barrett PH, Gautrey PJ, Herbet S, Kohn D, Smith S. Charles Darwin's notebooks, 1836–1844: geology, transmutation of species, metaphysical enquiries. 1. Cambridge: Cambridge University Press; 2009. [Google Scholar]