

Abstract
Background
Loss of paternal methylation (LOM) of the H19/IGF2 intergenic differentially methylated region (H19/IGF2:IG-DMR) causes alteration of H19/IGF2 imprinting and Silver-Russell syndrome (SRS). Recently, internal deletions of the H19/IGF2:IG-DMR have been associated with LOM and SRS when present on the paternal chromosome. In contrast, previously described deletions, most of which cause gain of methylation (GOM) and Beckwith-Wiedemann syndrome (BWS) on maternal transmission, were consistently associated with normal methylation and phenotype if paternally inherited.
Presentation of the hypothesis
The presence of several target sites (ZTSs) and three demonstrated binding regions (BRs) for the imprinting factor ZFP57 in the H19/IGF2:IG-DMR suggest the involvement of this factor in the maintenance of methylation of this locus. By comparing the extension of the H19/IGF2:IG-DMR deletions with the binding profile of ZFP57, we propose that the effect of the deletions on DNA methylation and clinical phenotype is dependent on their interference with ZFP57 binding. Indeed, deletions strongly affecting a ZFP57 BR result in LOM and SRS, while deletions preserving a significant number of ZFPs in each BR do not alter methylation and are associated with normal phenotype.
Testing the hypothesis
The generation of transgenic mouse lines in which the endogenous H19/IGF2:IG-DMR is replaced by the human orthologous locus including the three ZFP57 BRs or their mutant versions will allow to test the role of ZFP57 binding in imprinted methylation and growth phenotype.
Implications of the hypothesis
Similarly to what is proposed for maternally inherited BWS mutations and CTCF and OCT4/SOX2 binding, we suggest that deletions of the H19/IGF2:IG-DMR result in SRS with LOM if ZFP57 binding on the paternal chromosome is affected.
Electronic supplementary material
The online version of this article (10.1186/s13148-018-0454-7) contains supplementary material, which is available to authorized users.
Keywords: Genomic imprinting, Silver-Russell syndrome, DNA methylation, ZFP57, H19/IGF2:IG-DMR deletions, Beckwith-Wiedemann syndrome
Background
The imprinted monoallelic expression of the H19 and IGF2 genes is controlled by differential methylation of the H19/IGF2 intergenic differentially methylated region (H19/IGF2:IG-DMR) on the maternal and paternal chromosome 11p15.5 [1]. DNA methylation abnormalities of the H19/IGF2:IG-DMR cause contrasting growth disorders (i.e., Beckwith-Wiedemann syndrome (BWS, MIM#130650) and Silver-Russell syndrome (SRS, MIM#180860)), if they are present on the maternal or paternal chromosome, respectively [2]. More specifically, 5–10% of BWS cases are caused by gain of methylation (GOM) of the maternal H19/IGF2:IG-DMR resulting in biallelic expression of the growth factor IGF2 and silencing of the growth inhibitor H19, while 40–60% of SRS cases are associated with loss of methylation (LOM) of the paternal H19/IGF2:IG-DMR leading to IGF2 suppression and biallelic H19 expression.
We and others have previously demonstrated that GOM is associated with maternal transmission of internal deletions or single nucleotide variants (SNVs) of the H19/IGF2:IG-DMR in a subgroup of BWS cases [3–13]. These mutations affect target sites for CTCF (CTSs) and/or OCT4/SOX2 (OTSs/STS) that likely result in methylation of the remaining H19/IGF2:IG-DMR sequence on the maternal chromosome and imprinting alteration (Table 1 and Additional file 1: Table S1). These studies also show that paternal transmission of these mutations is consistently associated with normal methylation and clinical phenotype. In apparent contrast with these observations, Abi Habib and co-workers have recently reported new H19/IGF2:IG-DMR deletions that are associated with LOM and SRS, when paternally inherited [14].
Table 1.

Only deletions involving ZTSs, with reported phenotype on paternal transmission (in gray shades), were considered for this study
CTSs CTCF target sites, OTSs OCT4 target sites, STS SOX2 target site, ZTSs ZFP57 target sites, GOM gain of methylation, LOM loss of methylation, NM normal methylation, nr not reported, BWS Beckwith-Wiedemann syndrome, SRS Silver-Russell syndrome
Presentation of the hypothesis
The H19/IGF2:IG-DMR acquires DNA methylation in male germ cells and maintains it on the paternal allele of somatic cells throughout development despite the intense epigenetic reprogramming occurring post-fertilization [15, 16]. In mouse embryos and ESCs, maintenance of methylation at the orthologous H19/Igf2:IG-DMR as well as other imprinted DMRs is ensured by binding of the zinc-finger protein ZFP57, which is needed to recruit a number of heterochromatin-associated factors, including the corepressor KAP1, DNA methyltransferases, and histone methyltransferases [17–19]. Differently from CTCF and OCT4/SOX2 that bind the unmethylated maternal allele, ZFP57 recognizes a methylated TGCCGC motif on the paternal allele, present in multiple copies in the H19/Igf2:IG-DMR [20].
The human H19/IGF2:IG-DMR has a repetitive structure and includes two units of tandem repeats each composed of shorter repetitive modules (Fig. 1a). Twelve potential ZFP57 target sites (ZTSs) are present within the DMR, either as single isolated hexameric motifs or as closely spaced doublets of hexamers (Additional file 1: Table S1). In particular, the telomeric unit includes three singlets and the centromeric unit three doublets. Notably, ZTS doublets appear to be particularly important for ZFP57 binding in the mouse [20]. The genomic profile of ZFP57 binding, which has recently been obtained in a human cell line, is consistent with the location of the ZTSs [21]. Indeed, three ZFP57 binding regions (ZFP57 BRs) were demonstrated in the H19/IGF2:IG-DMR, with the largest one corresponding to the three ZTS doublets of the centromeric unit (Fig. 1a). Based on the comparison of the H19/IGF2:IG-DMR deletions (Fig. 1b and Table 1) with the ZFP57 binding profile and target sites, we propose that ZFP57 binding can explain the differential effects of these mutations on paternal transmission and that only the deletions interfering with ZFP57 result in SRS.
Fig. 1.
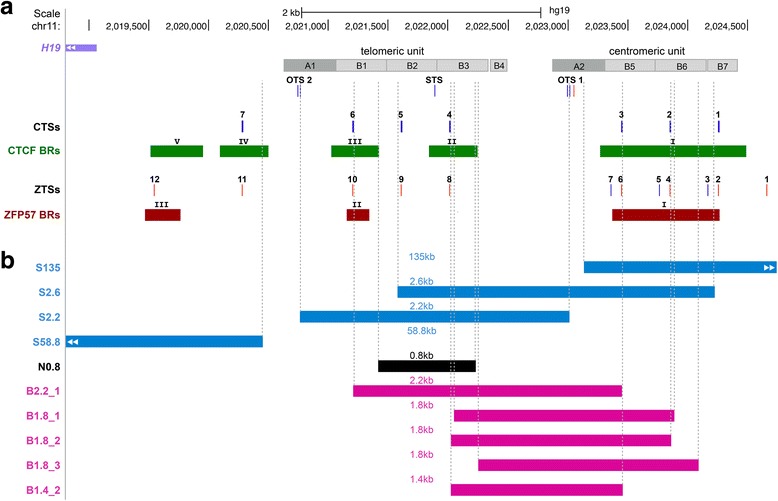
Paternal H19/IGF2:IG-DMR deletions and ZFP57 binding. a Schematic representation of the H19/IGF2:IG-DMR. The genomic region corresponds to GRCh37/hg19 chr11:2,018,812–2,024,740 [28] on the UCSC Genome Browser. The A- and B-type repetitive modules are represented as gray boxes. Target sites for OCT4 (OTSs), SOX2 (STS), CTCF (CTSs), and ZFP57 (ZTSs) are indicated by vertical bars; motif sequences present in the forward strand are in red, those present in the reverse strand are in blue. The CTCF BRs (transcription factor ChIP-seq, ENCODE data) are indicated as green bars. The ZFP57 BRs demonstrated in HEK293T cells (GSM2466451 [21]) are indicated as brown bars. b Paternally inherited H19/IGF2:IG-DMR deletions. Deletions associated with SRS and LOM on paternal transmission [14, 22] are represented in blue; the deletion associated with normal methylation and normal phenotype on paternal transmission [11] is in black; deletions associated with BWS and GOM on maternal transmission and normal phenotype on paternal transmission [3, 4, 6, 7, 11] are in pink. Breakpoints, molecular, and phenotypic effects of all the deletions are reported in Table 1
The three deletions reported by Abi Habib et al. (S135, S2.6, and S2.2) affect different portions of the DMR (Fig. 1b). S135 eliminates the ZFP57 BR-I and all the ZTSs (2–7) of the centromeric unit. S2.6 also affects BR-I with ZTSs 3–7, and in addition, removes two ZTSs (8, 9) from the telomeric unit. S2.2 abolishes BR-II with all the ZTSs (8–10) of the telomeric unit. All three deletions are associated with LOM, but a more severe hypomethylation was reported for S135 and S2.6, suggesting that the centromeric unit plays a major role in methylation maintenance [14]. The presence of a large ZFP57 binding region (BR-I) and a higher number of ZTSs within this region (Fig. 1a) is consistent with this observation.
In 2011, Grønskov and co-workers [22] described another SRS case with a paternal deletion (S58.8) associated with LOM of the H19/IGF2:IG-DMR (Fig. 1b). In addition to the H19 promoter, gene, and telomeric enhancers, this mutation removed the ZFP57 BR-III with ZTSs 11 and 12, indicating that also this region is involved in methylation maintenance (Fig. 1a, b).
How to explain the normal phenotype associated with the other deletions? Why they do not cause LOM on paternal transmission? N0.8 does not affect any ZFP57 BR (Fig. 1b). B2.2_1, B1.8_1–3, and B1.4_2 only partially affect either BR-I or BR-II. In particular, differently from the SRS-associated deletions, they never include the ZTS doublet 2–3 of BR-I and ZTS 10 of BR-II, which may be sufficient for ZFP57 binding.
There is a possible limitation in our hypothesis. ZFP57 BRs were demonstrated in a transformed cell system (HEK293T cells) overexpressing the human protein, and they might not faithfully represent all the BRs of the endogenous ZFP57 in its biological relevant tissues/developmental time points. However, if we take into consideration the ZTSs not included in BRs, an important role of ZTSs 8 and 9 is excluded by normal methylation of the N0.8 deletion, while a possible contribution of ZTS 1 and ZTS 11 to ZFP57 binding would not affect our hypothesis (Fig. 1a, b). A further possible criticism in our hypothesis concerns the normal methylation of the H19/IGF2:IG-DMR in transient neonatal diabetes mellitus 1 (TNDM1, OMIM 601410) patients with loss of function mutations of ZFP57 and multi-locus imprinting disturbances (MLID) [23, 24]. However, it is possible that this methylation pattern results from phenotypic selection of affected loci and that this might not necessary represent all the loci regulated by ZFP57 in humans. Moreover, although no ZFP57 mutation has been demonstrated, a defect of this gene in SRS cannot be excluded because only a few cases with MLID have been screened, so far [25, 26].
Testing the hypothesis
We recently described a transgenic mouse line in which the endogenous mouse H19/Igf2 IG-DMR was replaced by the orthologous human sequence (chr11:2,019,934–2,024,611) including ZTSs 2–11 [27]. While its function was conserved upon maternal transmission, the humanized locus was not properly methylated in sperms and methylation not maintained in somatic cells on paternal transmission. We now observe that this transgene was lacking BR-III that could be necessary for methylation maintenance. To test this hypothesis, we propose to generate a new knock-in mouse carrying the complete human H19/IGF2:IG-DMR sequence with all its ZFP57 BRs, which we expect to establish and maintain the imprinted methylation correctly. Similarly, mutants of the most relevant ZFP57 target sites (ZTSs 2–3, ZTS 10, and ZTS 12) can be generated to test their specific role in SRS etiology.
Implications of the hypothesis
Our hypothesis implies that the effect of H19/IGF2:IG-DMR deletions depends on two factors: the parental origin of the mutation and the transcription factors whose binding is affected. So far, maternal deletions, which are all associated with BWS, have been proposed to alter the binding of CTCF and/or OCT4/SOX2 resulting in DMR hypermethylation. Conversely, we propose that paternal deletions are associated with SRS when ZFP57 binding is affected and this happens when one of the three ZFP57 BRs is lost. Since the role of some ZTSs appears critical for methylation maintenance, SNVs affecting these binding sites may be present in deletion-negative SRS cases with H19/IGF2:IG-DMR LOM.
Additional file
Table S1. List of CTCF, OCT4/SOX2 and ZFP57 binding regions and potential target sites in the H19/IGF2:IG-DMR. Genomic positions of the binding regions (if demonstrated) and potential target sites and motifs of all these factors have been listed. (PDF 86 kb)
Acknowledgements
The authors thank Andrea Freschi and all the other members of Andrea Riccio’s lab for the useful discussion on the hypothesis.
Funding
This study was funded by the Telethon-Italia grant no. GGP15131 (AR).
Availability of data and materials
Not applicable.
Abbreviations
- BR
Binding region
- BWS
Beckwith-Wiedemann syndrome
- CTS
CTCF target site
- ESC
Embryonic stem cell
- GOM
Gain of methylation
- H19/IGF2:IG-DMR H19/IGF2
Intergenic differentially methylated region
- LOM
Loss of methylation
- MLID
Multi-locus imprinting disturbances
- NM
Normal methylation
- nr
Not reported
- OTS
OCT4 target site
- SNV
Single nucleotide variation
- SRS
Silver-Russell syndrome
- STS
SOX2 target site
- TNDM1
Transient neonatal diabetes mellitus 1
- ZTS
ZFP57 target site
Authors’ contributions
AS and FC carried out the study. AS and AR contributed to the interpretation of data, hypothesis formulation, manuscript writing, and revision. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13148-018-0454-7) contains supplementary material, which is available to authorized users.
Contributor Information
Angela Sparago, Email: angela.sparago@unicampania.it.
Flavia Cerrato, Email: flavia.cerrato@unicampania.it.
Andrea Riccio, Email: andrea.riccio@unicampania.it.
References
- 1.Kalish JM, Jiang C, Bartolomei MS. Epigenetics and imprinting in human disease. Int J Dev Biol. 2014;58(2–4):291–298. doi: 10.1387/ijdb.140077mb. [DOI] [PubMed] [Google Scholar]
- 2.Soellner L, Begemann M, Mackay DJ, Grønskov K, Tümer Z, Maher ER, et al. Recent advances in imprinting disorders. Clin Genet. 2017;91(1):3–13. doi: 10.1111/cge.12827. [DOI] [PubMed] [Google Scholar]
- 3.Sparago A, Cerrato F, Vernucci M, Ferrero GB, Silengo MC, Riccio A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nat Genet. 2004;36(9):958–960. doi: 10.1038/ng1410. [DOI] [PubMed] [Google Scholar]
- 4.Prawitt D, Enklaar T, Gärtner-Rupprecht B, Spangenberg C, Oswald M, Lausch E, et al. Microdeletion of target sites for insulator protein CTCF in a chromosome 11p15 imprinting center in Beckwith-Wiedemann syndrome and Wilms' tumor. Proc Natl Acad Sci U S A. 2005;102(11):4085–4090. doi: 10.1073/pnas.0500037102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cerrato F, Sparago A, Farina L, Ferrero GB, Cirillo-Silengo M, Riccio A. Reply to “Microdeletion and IGF2 loss of imprinting in a cascade causing Beckwith-Wiedemann syndrome with Wilms’ tumor”. Nat Genet. 2005;37(8):786–787. doi: 10.1038/ng0805-786. [DOI] [PubMed] [Google Scholar]
- 6.Sparago A, Russo S, Cerrato F, Ferraiuolo S, Castorina P, Selicorni A, et al. Mechanisms causing imprinting defects in familial Beckwith-Wiedemann syndrome with Wilms’ tumour. Hum Mol Genet. 2007;16(3):254–264. doi: 10.1093/hmg/ddl448. [DOI] [PubMed] [Google Scholar]
- 7.Demars J, Shmela ME, Rossignol S, Okabe J, Netchine I, Azzi S, et al. Analysis of the IGF2/H19 imprinting control region uncovers new genetic defects, including mutations of OCT-binding sequences, in patients with 11p15 fetal growth disorders. Hum Mol Genet. 2010;19(5):803–814. doi: 10.1093/hmg/ddp549. [DOI] [PubMed] [Google Scholar]
- 8.De Crescenzo A, Coppola F, Falco P, Bernardo I, Ausanio G, Cerrato F, et al. A novel microdeletion in the IGF2/H19 imprinting centre region defines a recurrent mutation mechanism in familial Beckwith-Wiedemann syndrome. Eur J Med Genet. 2011;54(4):e451–e454. doi: 10.1016/j.ejmg.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Demars J, Rossignol S, Netchine I, Lee KS, Shmela M, Faivre L, et al. New insights into the pathogenesis of Beckwith-Wiedemann and Silver-Russell syndromes: contribution of small copy number variations to 11p15 imprinting defects. Hum Mutat. 2011;32(10):1171–1182. doi: 10.1002/humu.21558. [DOI] [PubMed] [Google Scholar]
- 10.Poole RL, Leith DJ, Docherty LE, Shmela ME, Gicquel C, Splitt M, et al. Beckwith-Wiedemann syndrome caused by maternally inherited mutation of an OCT-binding motif in the IGF2/H19-imprinting control region, ICR1. Eur J Hum Genet. 2012;20(2):240–243. doi: 10.1038/ejhg.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beygo J, Citro V, Sparago A, De Crescenzo A, Cerrato F, Heitmann M, et al. The molecular function and clinical phenotype of partial deletions of the IGF2/H19 imprinting control region depends on the spatial arrangement of the remaining CTCF-binding sites. Hum Mol Genet. 2013;22(3):544–557. doi: 10.1093/hmg/dds465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abi Habib W, Azzi S, Brioude F, Steunou V, Thibaud N, Das Neves C, et al. Extensive investigation of the IGF2/H19 imprinting control region reveals novel OCT4/SOX2 binding site defects associated with specific methylation patterns in Beckwith-Wiedemann syndrome. Hum Mol Genet. 2014;23(21):5763–5773. doi: 10.1093/hmg/ddu290. [DOI] [PubMed] [Google Scholar]
- 13.Higashimoto K, Jozaki K, Kosho T, Matsubara K, Fuke T, Yamada D, et al. A novel de novo point mutation of the OCT-binding site in the IGF2/H19-imprinting control region in a Beckwith-Wiedemann syndrome patient. Clin Genet. 2014;86(6):539–544. doi: 10.1111/cge.12318. [DOI] [PubMed] [Google Scholar]
- 14.Abi Habib W, Brioude F, Azzi S, Salem J, Das Neves C, Personnier C, et al. 11p15 ICR1 partial deletions associated with IGF2/H19 DMR hypomethylation and Silver-Russell syndrome. Hum Mutat. 2017;38(1):105–111. doi: 10.1002/humu.23131. [DOI] [PubMed] [Google Scholar]
- 15.Kelsey G, Feil R. New insights into establishment and maintenance of DNA methylation imprints in mammals. Philos Trans R Soc Lond Ser B Biol Sci. 2013;368(1609):20110336. doi: 10.1098/rstb.2011.0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monk D. Germline-derived DNA methylation and early embryo epigenetic reprogramming: the selected survival of imprints. Int J Biochem Cell Biol. 2015;67:128–138. doi: 10.1016/j.biocel.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, et al. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell. 2008;15(4):547–557. doi: 10.1016/j.devcel.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell. 2011;44(3):361–372. doi: 10.1016/j.molcel.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riso V, Cammisa M, Kukreja H, Anvar Z, Verde G, Sparago A, et al. ZFP57 maintains the parent-of-origin-specific expression of the imprinted genes and differentially affects non-imprinted targets in mouse embryonic stem cells. Nucleic Acids Res. 2016;44(17):8165–8178. doi: 10.1093/nar/gkw505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anvar Z, Cammisa M, Riso V, Baglivo I, Kukreja H, Sparago A, et al. ZFP57 recognizes multiple and closely spaced sequence motif variants to maintain repressive epigenetic marks in mouse embryonic stem cells. Nucleic Acids Res. 2016;44(3):1118–1132. doi: 10.1093/nar/gkv1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imbeault M, Helleboid PY, Trono D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature. 2017;543(7646):550–554. doi: 10.1038/nature21683. [DOI] [PubMed] [Google Scholar]
- 22.Grønskov K, Poole RL, Hahnemann JM, Thomson J, Tümer Z, Brøndum-Nielsen K, et al. Deletions and rearrangements of the H19/IGF2 enhancer region in patients with Silver-Russell syndrome and growth retardation. J Med Genet. 2011;48(5):308–311. doi: 10.1136/jmg.2010.086504. [DOI] [PubMed] [Google Scholar]
- 23.Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40(8):949–951. doi: 10.1038/ng.187. [DOI] [PubMed] [Google Scholar]
- 24.Boonen SE, Mackay DJ, Hahnemann JM, Docherty L, Grønskov K, Lehmann A, et al. Transient neonatal diabetes, ZFP57, and hypomethylation of multiple imprinted loci: a detailed follow-up. Diabetes Care. 2013;36(3):505–512. doi: 10.2337/dc12-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spengler S, Gogiel M, Schönherr N, Binder G, Eggermann T. Screening for genomic variants in ZFP57 in Silver-Russell syndrome patients with 11p15 epimutations. Eur J Med Genet. 2009;52(6):415–416. doi: 10.1016/j.ejmg.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 26.Docherty LE, Rezwan FI, Poole RL, Turner CL, Kivuva E, Maher ER, et al. Mutations in NLRP5 are associated with reproductive wastage and multilocus imprinting disorders in humans. Nat Commun. 2015;6:8086. doi: 10.1038/ncomms9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hur SK, Freschi A, Ideraabdullah F, Thorvaldsen JL, Luense LJ, Weller AH, et al. Humanized H19/Igf2 locus reveals diverged imprinting mechanism between mouse and human and reflects Silver-Russell syndrome phenotypes. Proc Natl Acad Sci U S A. 2016;113(39):10938–10943. doi: 10.1073/pnas.1603066113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monk D, Morales J, den Dunnen JT, Russo S, Court F, Prawitt D, et al. Recommendations for a nomenclature system for reporting methylation aberrations in imprinted domains. Epigenetics. 2016; Dec 2:1–5. [DOI] [PMC free article] [PubMed]
- 29.Scott RH, Douglas J, Baskcomb L, Huxter N, Barker K, Hanks S, et al. Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat Genet. 2008;40(11):1329–1334. doi: 10.1038/ng.243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of CTCF, OCT4/SOX2 and ZFP57 binding regions and potential target sites in the H19/IGF2:IG-DMR. Genomic positions of the binding regions (if demonstrated) and potential target sites and motifs of all these factors have been listed. (PDF 86 kb)
Data Availability Statement
Not applicable.