

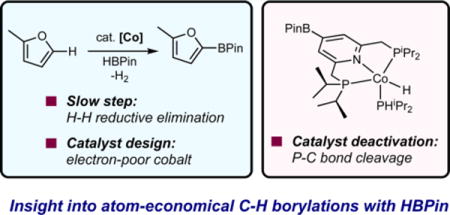
Abstract
Studies into the mechanism of cobalt-catalyzed C(sp2)−H borylation of five-membered heteroarenes with pinacolborane (HBPin) as the boron source established the catalyst resting state as the trans-cobalt(III) dihydride boryl, (iPrPNP)Co(H)2(BPin) (iPrPNP = 2,6-(iPr2PCH2)2(C5H3N)), at both low and high substrate conversions. The overall first-order rate law and observation of a normal deuterium kinetic isotope effect on the borylation of benzofuran versus benzofuran-2-d1 support H2 reductive elimination from the cobalt(III) dihydride boryl as the turnover-limiting step. These findings stand in contrast to that established previously for the borylation of 2,6-lutidine with the same cobalt precatalyst, where borylation of the 4-position of the pincer occurred faster than the substrate turnover and arene C−H activation by a cobalt(I) boryl is turnover-limiting. Evaluation of the catalytic activity of different cobalt precursors in the C−H borylation of benzofuran with HBPin established that the ligand design principles for C− H borylation depend on the identities of both the arene and the boron reagent used: electron-donating groups improve catalytic activity of the borylation of pyridines and arenes with B2Pin2, whereas electron-withdrawing groups improve catalytic activity of the borylation of five-membered heteroarenes with HBPin. Catalyst deactivation by P−C bond cleavage from a cobalt(I) hydride was observed in the C−H borylation of arene substrates with C−H bonds that are less acidic than those of five-membered heteroarenes using HBPin and explains the requirement of B2Pin2 to achieve synthetically useful yields with these arene substrates.
Keywords: borylation, C−H activation, pinacolborane, mechanism, cobalt
Graphical Abstract
INTRODUCTION
Transition-metal-catalyzed arene C(sp2)−H borylation1 has emerged as one of the most widely used C−H functionalization reactions, both in academia2 and industry,3 because of the versatility of the resulting products. Although highly predictable chemo- and regioselectivities are observed in these functionalizations using iridium catalysis, the high cost and low natural abundance of precious-metal catalysts motivate the search for base-metal alternatives that exhibit superior or complementary selectivity in the C(sp2)−H borylation of arenes. In ongoing efforts toward this aim,4 our laboratory5 and others6 have explored earth-abundant first-row transition-metal catalysts for the C(sp2)−H borylation of arenes. Among these, cobalt complexes bearing bis(phosphino)pyridine (iPrPNP) ligands7 are some of the most active and synthetically useful.5a
Our recent mechanistic investigations on the borylation of pyridines and unactivated arenes with (iPrPNP)CoCH2SiMe3 (Scheme 1) as the precatalyst and bis(pinacolato) diboron (B2Pin2) as the boron reagent established a Co(I)−Co(III) redox couple with turnover-limiting C−H activation promoted by a cobalt(I) boryl.8a However, under these conditions, borylation of the 4-position of the pincer ligand of the cobalt catalyst occurred prior to turnover, rendering the metal relatively electron poor and slowing the rate of oxidative addition of the arene C−H bond. These findings inspired the synthesis of second-generation catalysts where methyl and pyrrolidinyl groups were installed in the 4-position of the pincer to block unwanted borylation and increase the electron density of the cobalt center (Scheme 1).8a The increased activity of these catalysts was applied to the borylation of fluoroarenes where unprecedented levels of selectivity for sites ortho to fluorine were observed and used in a streamlined synthesis of the anti-inflammatory drug, Flurbiprofen.8b
Scheme 1.
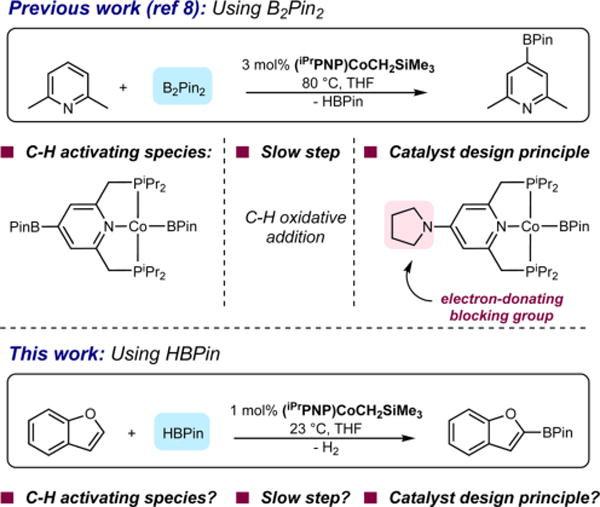
Influence of Arene and Boron Source on the Mechanism of Cobalt-Catalyzed C(sp2)−H Borylation
Catalytic arene C−H borylation methods that can employ HBPin as the stoichiometric boron source are particularly attractive because of their improved atom economy over variants that require B2Pin2.9 However, despite the mature understanding of the mechanisms of iridium-10–16 and cobalt-catalyzed C(sp2)−H borylation using B2Pin2 as the boron reagent,8a open questions remain about the operative pathways in cobalt-catalyzed reactions when HBPin is used as the stoichiometric boron source. With (iPrPNP) cobalt precatalysts, the success of the catalytic C−H borylation depends on the identity of the boron reagent. While HBPin is an effective reagent for the borylation of five-membered heteroarenes, it is far less effective with pyridines and unactivated arenes. Obtaining insights into the turnover-limiting step and nature of catalyst deactivation pathways in this class of C−H functionalization reaction is crucial for the rational design of next generation catalysts. Information on the speciation of iridium,17,18 ruthenium,19 iron,6a,d heterobimetallic copper,6c,20 and frustrated Lewis pair (FLP)21 catalysts in the presence of HBPin have been reported; however, the turnover-limiting step and possible catalyst deactivation pathways have not been identified, and principles for future catalyst development are not evident.
Here we describe a detailed study of the mechanism of cobalt-catalyzed C−H borylation of five-membered heteroarenes using HBPin as the borylating reagent. The catalyst resting state, identity of the C−H activating species, and the turnover-limiting step have been identified, and these findings demonstrate that the catalyst design principles used for optimal performance differ depending on the identity of the arene and boron reagent used. Catalyst deactivation pathways for the C−H borylation of pyridines and unactivated arenes with HBPin were also identified and provide insight why HBPin is ineffective for these substrates and why B2Pin2 is necessary for successful C−H borylation.
RESULTS AND DISCUSSION
Determination of the Catalyst Resting State
In our initial communication, trans-(iPrPNP)Co(H)2BPin was observed as the catalyst resting state during the C(sp2)−H borylation of 2-methylfuran with HBPin at 23 °C. The data were collected at 1 h of reaction time, corresponding to approximately 12% conversion.5a Monitoring the progress of the catalytic reaction over the time scale required for complete conversion has since been carried out using (iPrPNP)-CoCH2SiMe3 as the precatalyst. 2-Methylfuran was selected as the substrate because Co-catalyzed borylation of this arene with HBPin occurred at a rate that was conveniently monitored by 31P NMR spectroscopy. As shown in Figure 1, only trans-(iPrPNP)Co(H)2BPin5a,8 was detected over the course of the reaction, and neither 4-BPin-(iPrPNP)Co(N2)BPin8a nor 4-BPin-(iPrPNP)Co(H)2BPin8a was observed, demonstrating that the 4-position of the pincer does not undergo competitive borylation with the substrate.
Figure 1.
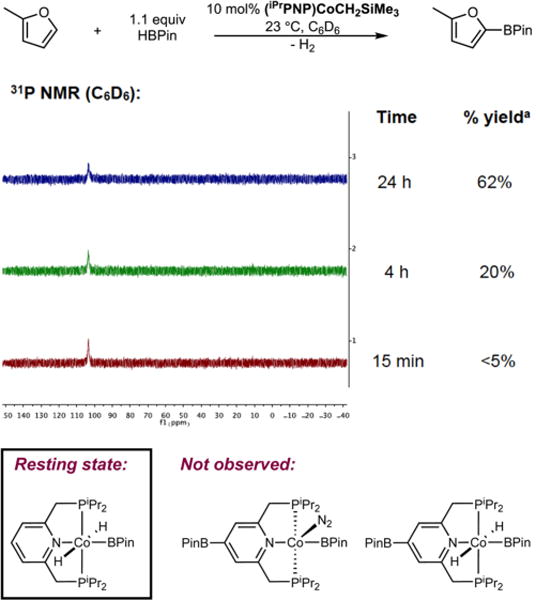
31P NMR spectrum of the reaction mixture of 2-methylfuran and HBPin in the presence of 10 mol % of (iPrPNP)CoCH2SiMe3 in benzene-d6 at 23 °C. The peak at 103.2 ppm in the 31P NMR spectrum corresponds to trans-(iPrPNP)Co(H)2BPin. aNMR yield of borylated product.
Having identified the catalyst resting state, attention was devoted to elucidating the identity of the cobalt compound responsible for activation of the C(sp2)−H bond. The catalyst resting state, trans-(iPrPNP)Co(H)2BPin, is coordinatively saturated (an 18-electron Co(III) compound) and likely requires either the reductive elimination of two of its anionic ligands (−H or -BPin) or the dissociation of one of its neutral ligands (pyridine or phosphine) to open a coordination site for C−H bond activation. Pathways involving reductive elimination of either H2 or HBPin seem the most plausible as Co(I)−Co(III) redox cycles are amply precedented in [(PNP)Co] chemistry.8a,22,23 As presented in Scheme 2, the “hydride pathway” generates (iPrPNP)CoH following reductive elimination of HBPin, whereas the alternative “boryl pathway” involves H2 loss to access (iPrPNP)CoBPin, analogous to the intermediate responsible for C−H activation during the borylation of 2,6-lutidine with B2Pin2.8a Because of the trans disposition of the hydride ligands in trans-(iPrPNP)Co-(H)2BPin, isomerization to the cis isomer either by phosphine dissociation24 or HBPin reductive coupling is required to achieve an appropriate geometry for H2 loss. Mechanistic experiments were conducted to distinguish the hydride and the boryl pathways (vide infra).
Scheme 2.
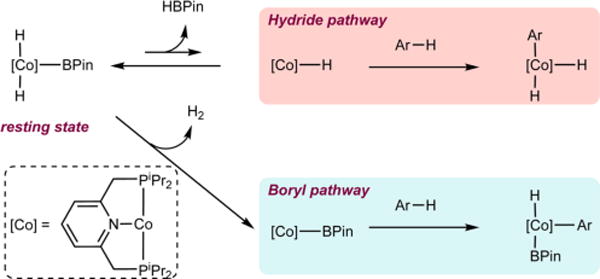
Possible Scenarios for the Generation of the C−H Activating Species from trans-(iPrPNP)Co(H)2BPin
Kinetic Isotope Effect (KIE) and Rate Law Measurements
To determine whether C−H bond cleavage is involved in the turnover-limiting step of the catalytic cycle, the kinetic isotope effect was measured by comparison of the initial rates25 (up to 10% conversion) for the C(sp2)−H borylation of benzofuran and benzofuran-2-d1 measured in two separate vessels. Benzofuran was chosen for the KIE and rate law measurements because of its low volatility compared to 2-methylfuran in order to avoid loss via evaporation during the reaction. With 1 mol % of (iPrPNP)CoCH2SiMe3 as the precatalyst with HBPin at 23 °C, comparison of the relative initial rates of the two reactions yielded a normal, primary deuterium kinetic isotope effect of 1.9(1) (Scheme 3). This value is lower than those measured for the cobalt-catalyzed borylation of 2,6-lutidine (2.9(1) at 80 °C)8a and iridium-catalyzed borylation of 1,2-dichlorobenzene and 1,2-dichlorobenzene-d4 with B2Pin2 (3.3(6) at 25 °C).10 In both reported cases, KIEs of this magnitude support oxidative addition of the C−H bond as the turnover-limiting step during catalysis. The reduced magnitude of the KIE could arise either from turnover-limiting C−H bond activation with an earlier or later transition state than the previously reported examples or a from a change in turnover-limiting step.
Scheme 3.
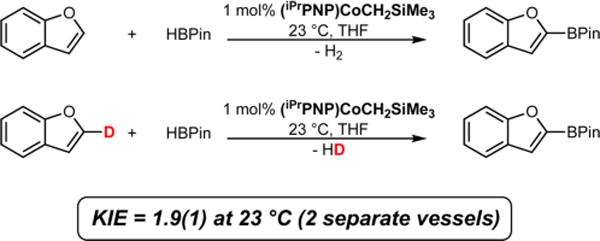
Determination of the Deuterium Kinetic Isotope Effect for the Borylation of Benzofuran and Benzofuran-2-d1 with HBPin as the Boron Source and (iPrPNP)CoCH2SiMe3 as the Pre-Catalyst
The experimental rate law for the C(sp2)−H borylation of benzofuran with HBPin using (iPrPNP)CoCH2SiMe3 was determined using the method of initial rates25 (Table 1) with the goal of distinguishing the pathways in Scheme 2 and providing insight into the turnover-limiting step of the catalytic reaction.
Table 1.
Determination of the Rate Law of the Borylation of Benzofuran with HBPin
|
| |||
|---|---|---|---|
| [Benzofuran] | [HBPin] | [(iPrPNP)CoCH2SiMe3] | Initial rate (M/s) |
| 0.14 M | 0.14 M | 7.00 × 10−4 M | 1.2 ± 0.1 × 10−5 |
| 0.14 M | 0.14 M | 1.40 × 10−3 M | 2.3 ± 0.2 × 10−5 |
| 0.14 M | 0.28 M | 1.40 × 10−3 M | 2.3 ± 0.1 × 10−5 |
| 0.28 M | 0.14 M | 1.40 × 10−3 M | 2.4 ± 0.1 × 10−5 |
| 0.14 M | 0.14 M | 3.50 × 10−4 M | 5.2 ± 0.7 × 10−6 |
| 0.14 M | 0.14 M | 1.05 × 10−3 M | 1.9 ± 0.1 × 10−5 |
|
| |||
The observed rate law was first-order with respect to cobalt precatalyst (Figure 2) and zero-order with respect to both the heteroarene substrate and HBPin. The zero-order behavior with respect to benzofuran concentration eliminates C−H oxidative addition as the turnover-limiting step in the catalytic cycle. In addition, the experimentally determined rate law also excludes the hydride pathway as a mechanistic possibility. If (iPrPNP)CoH were responsible for C−H activation and C−B bond formation via reductive elimination to release the product is turnover-limiting, the reaction would be expected to exhibit a first-order dependence on heteroarene substrate (see Figure 3, Hydride pathway 1). Similarly, a hydride pathway where H−H reductive elimination is the slow step is also unlikely as this regime would result in a first-order dependence on the heteroarene and an inverse first-order dependence on HBPin (see Figure 3, Hydride pathway 2).
Figure 2.
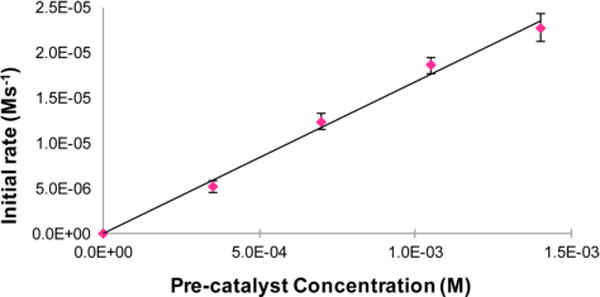
First-order dependence of the initial rate for the formation of 2-BPin-benzofuran on the concentration of the cobalt precatalyst, (iPrPNP)CoCH2SiMe3.
Figure 3.
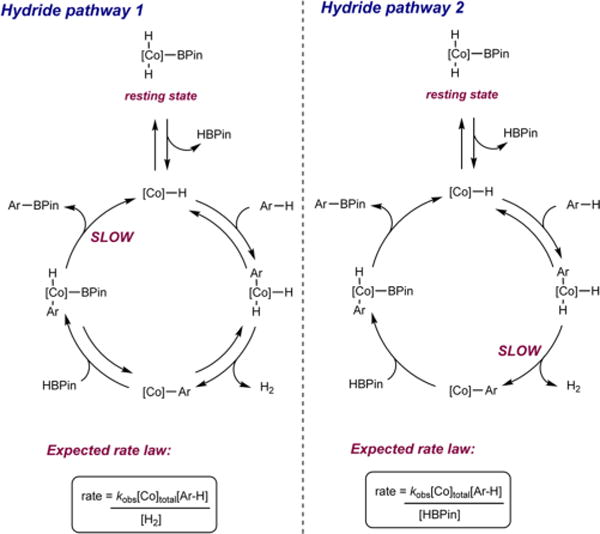
Hydride pathway 1 with turnover-limiting C−B reductive elimination (left) and Hydride pathway 2 with turnover-limiting H−H reductive elimination (right). See Figure S4 and Figure S5 for the derivation of the rate law.
Proposed Mechanism
A mechanism that is consistent with the experimentally measured data is presented in Scheme 4 and is reminiscent of the boryl pathway presented in Scheme 2. The cobalt precatalyst enters the catalytic cycle by reaction with HBPin to form an equivalent of PinBCH2SiMe3 and (iPrPNP)CoH (step 1).5a Oxidative addition of a second equivalent of HBPin to (iPrPNP)CoH generates the catalyst resting state, trans-(iPrPNP)Co(H)2BPin (step 2).5a,26 Turnover-limiting H2 reductive elimination (step 3), either through dissociation of a phosphine24 or reductive coupling of HBPin, generates the C−H activating species, (iPrPNP)CoBPin. Heteroarene C−H oxidative addition (step 4), followed by C−B reductive elimination to release the heteroarylboronate ester product and regenerate (iPrPNP)CoH (step 5) completes the catalytic cycle. This mechanistic scenario is different from that of the borylation of pyridines and arenes with B2Pin2, where catalyst self-borylation is competitive with arene borylation and arene C−H activation by a cobalt(I) boryl is turnover-limiting.
Scheme 4.
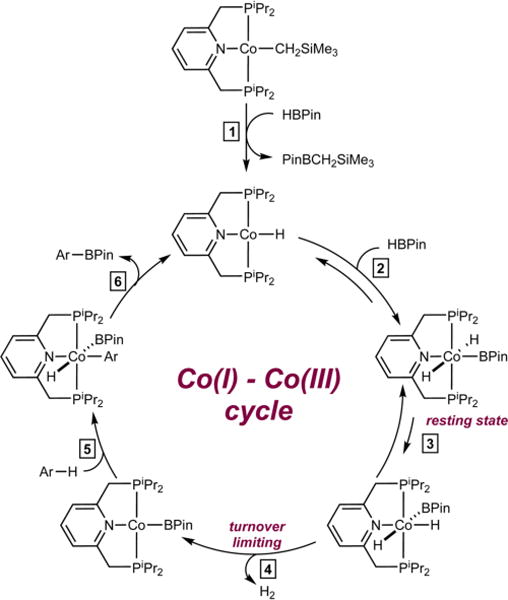
Proposed Mechanism for the Cobalt-Catalyzed C−H Borylation of Five-Membered Heteroarenes with HBPin
Effect of the 4-Substituent on Catalytic Activity
To evaluate the effect of the electron-donating ability of the pincer ligand on the C−H borylation of heteroarenes with HBPin, the catalytic C−H borylation of benzofuran was monitored using different cobalt precatalysts (Figure 4). The reaction profiles in Figure 4 show that the rate of benzofuran borylation with HBPin increases with electron withdrawing groups in the 4-position of the pincer. The origin of this behavior is likely a result of a decreased barrier for turnover-limiting H−H reductive elimination with more electron-poor catalysts. This is opposite of the trend observed for the C(sp2)−H borylation of 2,6-lutidine with B2Pin2 where electron-donating blocking pyrrolidinyl and methyl groups enhanced the rate of C−H borylation by decreasing the barrier for turnover-limiting C−H activation of the arene substrate.8a These findings demonstrate that the turnover-limiting step of the C−H borylation catalytic cycle and hence the catalyst design principles depend on the identity of the substrates.
Figure 4.
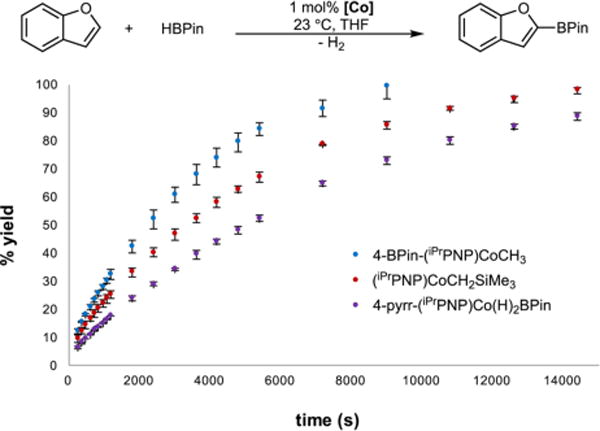
Reaction profiles for the C−H borylation of benzofuran with HBPin using (iPrPNP)CoCH2SiMe35a (red), 4-BPin-( iPrPNP)-CoCH38a (blue), and 4-pyrr-(iPrPNP)Co(H)2BPin8a (purple) as precatalysts.
Determination of Catalyst Deactivation Pathway(s)
Although an effective reagent for the borylation of 5-membered heteroarenes, HBPin proved less effective with pyridines and unactivated arenes and led to overall lower product yields. To probe the origin of this difference, the borylation of 2,6-lutidine was studied with HBPin in the presence of 10 mol % of (iPrPNP)CoCH2SiMe3. Monitoring the catalytic reaction by 31P NMR spectroscopy (Figure 5) revealed significant borylation of the 4-position of the pincer ligand. After 1 h of reaction time, corresponding to <5% conversion, a 2:1 ratio of trans-(iPrPNP)Co(H)2BPin and trans-4-BPin-(iPrPNP)Co-(H)2BPin was observed (red spectrum in Figure 5). Heating the THF-d8 solution of the reaction mixture to 80 °C for 32 h produced 19% yield of the borylated product and a new cobalt compound identified as 4-BPin-(iPrPNP)Co(H)(PHiPr2). This product, arising from P−C bond cleavage of the pincer,22 was independently synthesized from addition of HPiPr2 to 4-BPin-(iPrPNP)CoCH38a under an atmosphere of hydrogen gas (see complete experimental details on page S9) and crystallographically characterized (Figure 6). Performing the C−H borylation of 2,6-lutidine with HBPin in 0.55 M THF solution using 10 mol % of 4-BPin-(iPrPNP)Co(H)(PHiPr2) as the precatalyst resulted in no conversion to the borylated product, establishing that the formation of 4-BPin-(iPrPNP)Co(H)-(PHiPr2) by P−C bond cleavage is a catalyst deactivation pathway (see S10).
Figure 5.
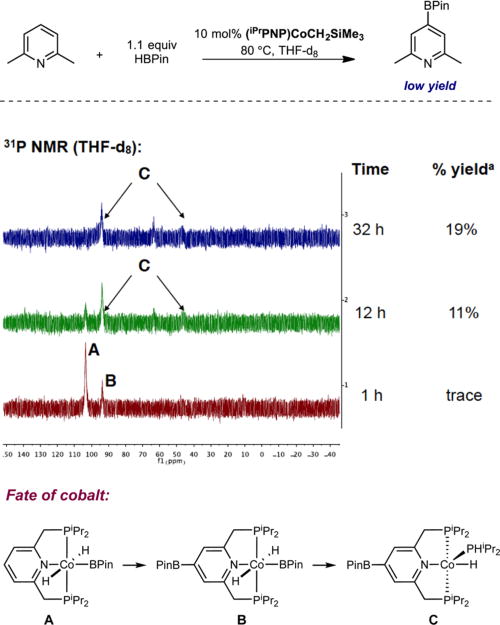
31P NMR spectrum of the reaction mixture of 2,6-lutidine and HBPin in the presence of 10 mol % of (iPrPNP)CoCH2SiMe3 in THF-d8 at 80 °C. aGC yield of borylated product.
Figure 6.
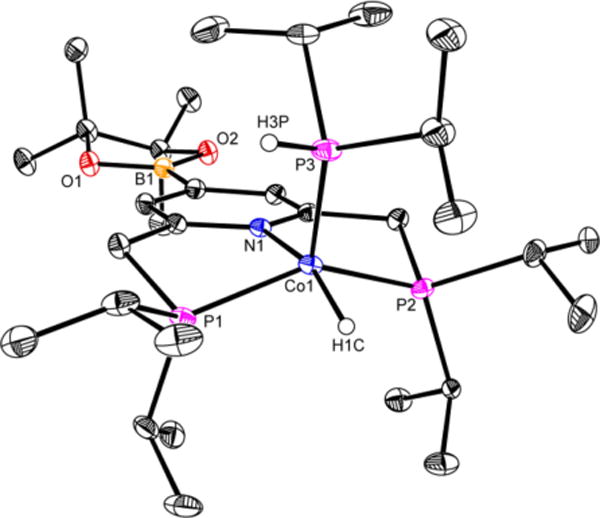
Solid-state structure of 4-BPin-(iPrPNP)CoH(PHiPr2) at 30% probability ellipsoids. Hydrogen atoms, except on the cobalt-hydride and on the secondary phosphine, were omitted for clarity.
Based on these observations, a pathway accounting for the origin of the poor catalytic activity with pyridines and unactivated arenes when HBPin is used as the boron reagent is presented in Scheme 5. Isomerization of trans-4-BPin-(iPrPNP)Co(H)2BPin to the cis isomer (step 1) followed by H2 reductive elimination (step 2) generates the C−H activating intermediate, 4-BPin-(iPrPNP)CoBPin, which undergoes oxidative addition of the arene C(sp2)−H bond (step 3). For pyridines and unactivated arenes where the C−H bond is less acidic than that of five-membered heteroarenes, reductive elimination of HBPin from trans-4-BPin-(iPrPNP)Co- (H)2BPin to generate 4-BPin-(iPrPNP)CoH (step 4) becomes competitive with arene C−H activation (step 3).27 As shown previously with the unmodified version of the ligand,22 the Co(I) hydride undergoes catalyst deactivation by P−C bond cleavage22 to generate 4-BPin-(iPrPNP)Co(H)(PHiPr2) (step 5) and unidentified products. When B2Pin2 is used as the boron reagent, regeneration of the cobalt boryl occurs via the reaction of the cobalt hydride with B2Pin2 (step 6), which decreases the concentration of the cobalt(I) hydride species, or preventing its formation altogether, and thus preventing catalyst deactivation. This explains why the use of B2Pin2 is necessary to achieve synthetically useful yields in the C−H borylation of pyridines and unactivated arenes.
Scheme 5.
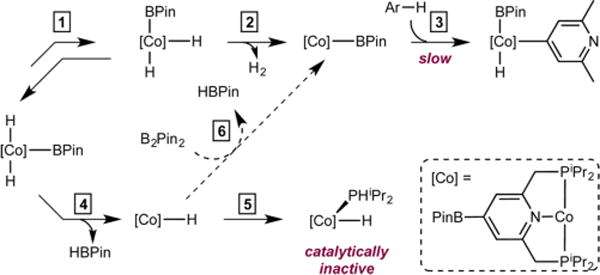
Hypothesis for Why HBPin Is an Ineffective Borylating Reagent for Pyridines and Unactivated Arenes
CONCLUSIONS
Investigations into the mechanism of C−H borylation of five-membered heteroarenes with HBPin using the cobalt precatalyst, (iPrPNP)CoCH2SiMe3, are consistent with reductive elimination of H2 from the cobalt(III) resting state as the turnover-limiting step. This kinetic regime is distinct from the one operative for the borylation of pyridines and arenes with cobalt and iridium catalysts with B2Pin2, where C−H activation is turnover limiting. Comparison of the catalytic activities of different cobalt precatalysts established that the ligand design principles for C−H borylation are dictated by the identity of the arene substrate as well as the boron reagent used as electron-donating groups improve borylation of pyridines and arenes with B2Pin2, whereas electron-withdrawing groups improve borylation of five-membered heteroarenes with HBPin. Monitoring the fate of the cobalt catalyst in the borylation of 2,6-lutidine with HBPin revealed that for arene substrates containing relatively less acidic C−H bonds, catalyst deactivation by P−C bond cleavage becomes competitive with productive catalysis, thus resulting in poor yields. The design of cobalt catalysts that are less susceptible to deactivation by P−C bond cleavage is envisioned to enable more atom-economical C−H borylations of these arene substrates with HBPin.
Supplementary Material
Acknowledgments
Financial supported was provided by the National Institutes of Health (1R01GM121441-01). J.V.O. acknowledges the 2015 Howard Hughes Medical Institute International Student Research Fellowship and the 2016 Harold W. Dodds Honorific Fellowship (awarded by the Graduate School at Princeton University). We also thank Máté Bezdek for solving the X-ray structure of 4-BPin-(iPrPNP)Co(H)(PHiPr2), Dr. Scott P. Semproni for helpful discussions and Dr. C. Rose Kennedy for insightful comments and assistance in editing the manuscript.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.7b01151.
Complete experimental details, characterization data, NMR spectroscopic data (PDF)
Crystallographic data (CIF)
Notes
The authors declare no competing financial interest.
References
- 1.Seminal examples:; (a) Iverson CN, Smith MR. J Am Chem Soc. 1999;121:7696–7697. [Google Scholar]; (b) Cho J-Y, Tse MK, Holmes D, Maleczka RE, Jr, Smith MR., III Science. 2002;295:305–308. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]; (c) Ishiyama T, Takagi J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF. J Am Chem Soc. 2002;124:390–391. doi: 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]; (d) Ishiyama T, Takagi J, Hartwig JF, Miyaura N. Angew Chem, Int Ed. 2002;41:3056–3058. doi: 10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 2.(a) Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]; (b) Hartwig JF. Chem Soc Rev. 2011;40:1992–2002. doi: 10.1039/c0cs00156b. [DOI] [PubMed] [Google Scholar]; (c) Hartwig JF. Acc Chem Res. 2012;45:864–873. doi: 10.1021/ar200206a. [DOI] [PubMed] [Google Scholar]
- 3.Campeau L-C, Chen Q, Gauvreau D, Girardin M, Belyk K, Maligres P, Zhou G, Gu C, Zhang W, Tan L, O’Shea PD. Org Process Res Dev. 2016;20:1476–1481. [Google Scholar]
- 4.(a) Chirik PJ, Wieghardt K. Science. 2010;327:794–795. doi: 10.1126/science.1183281. [DOI] [PubMed] [Google Scholar]; (b) Chirik PJ. Acc Chem Res. 2015;48:1687–1695. doi: 10.1021/acs.accounts.5b00134. [DOI] [PubMed] [Google Scholar]; (c) Chirik PJ. Angew Chem, Int Ed. 2017;56:5170–5181. doi: 10.1002/anie.201611959. [DOI] [PubMed] [Google Scholar]
- 5.(a) Obligacion JV, Semproni SP, Chirik PJ. J Am Chem Soc. 2014;136:4133–4136. doi: 10.1021/ja500712z. [DOI] [PubMed] [Google Scholar]; (b) Schaefer BA, Margulieux GW, Small BL, Chirik PJ. Organometallics. 2015;34:1307–1320. [Google Scholar]; (c) Léonard NG, Bezdek MJ, Chirik PJ. Organometallics. 2017;36:142–150. [Google Scholar]
- 6.(a) Hatanaka T, Ohki Y, Tatsumi K. Chem - Asian J. 2010;5:1657–1666. doi: 10.1002/asia.201000140. [DOI] [PubMed] [Google Scholar]; (b) Yan G, Jiang Y, Kuang C, Wang S, Liu H, Zhang Y, Wang J. Chem Commun. 2010;46:3170–3172. doi: 10.1039/b926945b. [DOI] [PubMed] [Google Scholar]; (c) Mazzacano TJ, Mankad NP. J Am Chem Soc. 2013;135:17258–17261. doi: 10.1021/ja408861p. [DOI] [PubMed] [Google Scholar]; (d) Dombray T, Werncke CG, Jiang S, Grellier M, Vendier L, Bontemps S, Sortais JB, Sabo-Etienne S, Darcel C. J Am Chem Soc. 2015;137:4062–4065. doi: 10.1021/jacs.5b00895. [DOI] [PubMed] [Google Scholar]; (e) Furukawa T, Tobisu M, Chatani N. Chem Commun. 2015;51:6508–6511. doi: 10.1039/c5cc01378j. [DOI] [PubMed] [Google Scholar]; (f) Zhang H, Hagihara S, Itami K. Chem Lett. 2015;44:779–781. [Google Scholar]
- 7.Khaskin E, Diskin-Posner Y, Weiner L, Leitus G, Milstein D. Chem Commun. 2013;49:2771–2773. doi: 10.1039/c3cc39049g. [DOI] [PubMed] [Google Scholar]
- 8.(a) Obligacion JV, Semproni SP, Pappas I, Chirik PJ. J Am Chem Soc. 2016;138:10645–10653. doi: 10.1021/jacs.6b06144. [DOI] [PubMed] [Google Scholar]; (b) Obligacion JV, Bezdek MJ, Chirik PJ. J Am Chem Soc. 2017;139:2825–2832. doi: 10.1021/jacs.6b13346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.There are iridium-catalyzed variants of this reaction where only 1/2 equiv of B2Pin2 is used relative to the arene. In this case, the use of B2Pin2 as the boron reagent is more atom-economical because it only generates 1/2 equiv of H2 (instead of 1 equiv. of H2 generated from 1 equiv of HBPin).
- 10.Boller TM, Murphy JM, Hapke M, Ishiyama T, Miyaura N, Hartwig JF. J Am Chem Soc. 2005;127:14263–14278. doi: 10.1021/ja053433g. [DOI] [PubMed] [Google Scholar]
- 11.Tamura H, Yamazaki H, Sato H, Sakaki S. J Am Chem Soc. 2003;125:16114–16126. doi: 10.1021/ja0302937. [DOI] [PubMed] [Google Scholar]
- 12.Vanchura BA, II, Preshlock SM, Roosen PC, Kallepalli VA, Staples RJ, Maleczka RE, Jr, Singleton DA, Smith MR., III Chem Commun. 2010;46:7724–7726. doi: 10.1039/c0cc02041a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Q, Liskey CW, Hartwig JF. J Am Chem Soc. 2014;136:8755–8765. doi: 10.1021/ja503676d. [DOI] [PubMed] [Google Scholar]
- 14.Larsen MA, Hartwig JF. J Am Chem Soc. 2014;136:4287– 4299. doi: 10.1021/ja412563e. [DOI] [PubMed] [Google Scholar]
- 15.Green AG, Liu P, Merlic CA, Houk KN. J Am Chem Soc. 2014;136:4575–4583. doi: 10.1021/ja411699u. [DOI] [PubMed] [Google Scholar]
- 16.Haines BE, Saito Y, Segawa Y, Itami K, Musaev DG. ACS Catal. 2016;6:7536–7546. [Google Scholar]
- 17.Ghaffari B, Vanchura BA, Chotana GA, Staples RJ, Holmes D, Maleczka RE, Smith MR. Organometallics. 2015;34:4732–4740. doi: 10.1021/acs.organomet.5b00525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Press LP, Kosanovich AJ, McCulloch BJ, Ozerov OV. J Am Chem Soc. 2016;138:9487–9497. doi: 10.1021/jacs.6b03656. [DOI] [PubMed] [Google Scholar]
- 19.Stahl T, Müther K, Ohki Y, Tatsumi K, Oestreich M. J Am Chem Soc. 2013;135:10978–10981. doi: 10.1021/ja405925w. [DOI] [PubMed] [Google Scholar]
- 20.Parmelee SR, Mazzacano TJ, Zhu Y, Mankad NP, Keith JA. ACS Catal. 2015;5:3689–3699. [Google Scholar]
- 21.Légaré MA, Courtemanche MA, Rochette E, Fontaine FG. Science. 2015;349:513–516. doi: 10.1126/science.aab3591. [DOI] [PubMed] [Google Scholar]
- 22.Semproni SP, Hojilla Atienza CCH, Chirik PJ. Chem Sci. 2014;5:1956–1960. [Google Scholar]
- 23.Scheuermann ML, Semproni SP, Pappas I, Chirik PJ. Inorg Chem. 2014;53:9463–9465. doi: 10.1021/ic501901n. [DOI] [PubMed] [Google Scholar]
- 24.Xu H, Bernskoetter W. J Am Chem Soc. 2011;133:14956– 14959. doi: 10.1021/ja2072548. [DOI] [PubMed] [Google Scholar]
- 25.The reaction of (iPrPNP)CoCH2SiMe3 with HBPin happens instantaneously at 23 °C, and thus, precatalyst activation is significantly faster than the time it takes to reach 10% conversion.
- 26.As shown in reference 5a, the C−H borylation of 2-methyl furan with DBPin in the presence of 1 mol% of (iPrPNP)CoCH2SiMe3 resulted in no deuterium incorporation in the unreacted arene, but resulted in the formation of HBPin, consistent with reversible oxidative addition and reductive elimination of B−H(D) bonds.
- 27.Performing the reaction depicted in Figure 5 in the absence of 2,6-lutidine (i.e., addition of 11 equiv of HBPin to a solution of (iPrPNP)CoCH2SiMe3 in THF-d8) instantaneously forms trans- (iPrPNP)Co(H)2BPin (see footnote 25) at 23 °C. Heating this mixture to 80 °C for 32 h resulted in the complete consumption of trans- (iPrPNP)Co(H)2BPin and the formation of trans-4-BPin-(iPrPNP)Co(H)(PHiPr2), thus supporting the hypothesis that P-C bond cleavage is competitive with arene C−H activation when the C− H bond is less acidic than that of five-membered heteroarenes
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.