

Abstract
Naringin is one of the most interesting phytopharmaceuticals that has been widely investigated for various biological actions. Yet, its low water solubility, limited permeability, and suboptimal bioavailability limited its use. Therefore, in this study, polymeric micelles of naringin based on pluronic F68 (PF68) were developed, fully characterized, and optimized. The optimized formula was investigated regarding in vitro release, storage stability, and in vitro cytotoxicity vs different cell lines. Also, cytoprotection against ethanol-induced ulcer in rats and antitumor activity against Ehrlich ascites carcinoma in mice were investigated. Nanoscopic and nearly spherical 1:50 micelles with the mean diameter of 74.80±6.56 nm and narrow size distribution were obtained. These micelles showed the highest entrapment efficiency (EE%; 96.14±2.29). The micelles exhibited prolonged release up to 48 vs 10 h for free naringin. The stability of micelles was confirmed by insignificant changes in drug entrapment, particle size, and retention (%) (91.99±3.24). At lower dose than free naringin, effective cytoprotection of 1:50 micelles against ethanol-induced ulcer in rat model has been indicated by significant reduction in mucosal damage, gastric level of malondialdehyde, gastric expression of tumor necrosis factor-alpha, caspase-3, nuclear factor kappa-light-chain-enhancer of activated B cells, and interleukin-6 with the elevation of gastric reduced glutathione and superoxide dismutase when compared with the positive control group. As well, these micelles provoked pronounced antitumor activity assessed by potentiated in vitro cytotoxicity particularly against colorectal carcinoma cells and tumor growth inhibition when compared with free naringin. In conclusion, 1:50 naringin–PF68 micelles can be represented as a potential stable nanodrug delivery system with prolonged release and enhanced antiulcer as well as antitumor activities.
Keywords: naringin, pluronic F68, polymeric micelles, in vitro cytotoxicity, antiulcer, antitumor activity
Introduction
Naringin (Figure 1A) is present in grape and citrus fruits, cherries, beans, and oregano.1 It is found in grapefruit juice up to concentrations of 800 mg/L.2 Naringin is a promising phytopharmaceutical that exerts versatile pharmacological effects such as antioxidant, antiulcer, antiallergic, anticancer, and blood lipid-lowering activities.3,4 The intake of grapes and citrus fruits by humans for a long time without development of side effects has empirically proven the safety of naringin.5 Naringin has been practically nontoxic for Sprague Dawley rats in oral subchronic toxicity study for 13 consecutive weeks, and no adverse effects were observed in rats up to 1,250 mg/kg/day.6
Figure 1.
Structures of the drug and the copolymer.
Notes: (A) Naringin and (B) PF68.
Abbreviations: EO, ethylene oxide; PF68, pluronic F68; PO, propylene oxide.
Gastric ulcer results when some aggressive factors are not balanced by the defensive action of some endogenous factors.7,8 Endogenous aggressive factors include overproduction of pepsin and hydrochloric acid, refluxed bile, leukotrienes, and reactive and stress oxygen species. The defensive mechanisms to protect the gastric mucosa are the surface mucus, bicarbonate, the regulation of gastric mucosal blood flow, surface active phospholipids, antioxidants, the preservation of epithelial homeostasis, and the acceleration of epithelial regeneration. For decades, gastric ulcer therapy based on the reduction of gastric acid secretion using anticholinergic drugs, antacids, histamine H2 receptor antagonists, and proton pump inhibitors since excessive gastric acid secretion was thought to be the main cause of the gastric ulcer. Nevertheless, most of the treatment regimens currently used showed limited efficacy and several adverse reactions, which may limit their use.9 Hence, there is a great need for safe, economic, and efficient antiulcer agents. Natural products emerged as an interesting source of compounds with potential antiulcer activities. The cytoprotection of naringin against ethanol-induced ulcers in rats has been reported; however, a large dose of 400 mg/kg was required.10,11 A significant reduction in the ulcer index and the protection of gastric mucosa in acetylsalicylic acid-induced ulcer in rats have been documented for naringin (200 mg/kg).12 The cytoprotection by naringin against ulcer has been referred to higher glycoprotein secretion and more viscous gastric mucosa as well as superoxide anion scavenger and antioxidant properties.10,11 The anti-inflammatory activity of naringin has been attributed to the suppression of tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), caspase-3, and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in macrophages.1,13
The nonselectivity of the cytotoxic action and the narrow therapeutic index are the main limitations of the majority of currently available anticancers.14 The use of naturally occurring biocompatible anticancer agents can be represented as a safe promising alternative. Naringin has induced cytotoxicity via apoptosis in mouse leukemia P388 cells.13 A possibility of suppression of colon cancer development by naringin has been documented.15 It has been reported that naringin could inhibit the growth potential of breast cancer cells by modulating β-catenin pathway.16,17 This drug (200 mg/kg) suppressed proliferation and increased apoptosis of colon epithelial cells.15 Naringin (40 mg/kg) exhibited significant protection in N-nitrosodiethylamine-induced liver carcinogenesis in rats.18 Selective inhibition of nodulogenesis by naringin may explain cytotoxic potential against hepatocellular carcinoma.19 Also, free radical scavenging and antioxidant potential of this drug may also explain its efficacy against liver cancer.20
In spite of this great therapeutic potential, the cleavage of naringin in the lumen or in the cell gut at the harsh pH and enzymatic conditions of the gastrointestinal tract has been reported.21,22 Consequently, the oral bioavailability of naringin is low (~8%) and its half-life has been estimated to be 2.6 h.23 In addition, its poor solubility in water hindered its formulation for parenteral use.24 Thus, alternative naringin formulations such as nanoparticulate systems would be essential to protect the drug against degradation in the gut, improve its bioavailability, and provide a sustained delivery of the drug in the purpose of potentiating its therapeutic efficacy. Nano-sized drug delivery systems were successful to improve the therapeutic effectiveness of some phytopharmaceuticals. For example, nanocapsulated quercetin employing poly(d,l-lactic-co-glycolic acid) showed higher efficacy over free quercetin in the prevention of ethanol-induced gastric ulcer in rats.25 Potentiated anticancer activity of naringenin against human cervical (HeLa) cancer cells upon loading into Eudragit E nanoparticles has been documented in the literature.26
Polymeric micelles have been emerged as a successful approach for the site-specific delivery of various drugs. Upon dilution, polymeric micelles are highly more stable than surfactant micelles mainly due to a relatively low critical micelle concentration (CMC) of the former.27 Therefore, they have been widely employed to deliver chemotherapeutic agents, such as docetaxel alone and in combination with other drugs.28–30 As well, antitumor activity of doxorubicin has been considerably enhanced through different micellar nanoparticles.31–33 Pluronics represent a class of block copolymers formed of hydrophilic blocks of poly(ethylene oxide) (PEO) and hydrophobic poly(propylene oxide) (PPO) in tri-block arrangement of PEO–PPO–PEO (Figure 1B).34 Pluronics form micelles with a hydrophobic PPO core within a hydrophilic PEO shell in aqueous solutions above CMC. Pluronic nanomicelles showed a noticeable improvement in stability, solubility, biodistribution, and pharmacokinetics of encapsulated drugs. It has been reported that nanoscopic particles showed an increased disposition in inflamed tissue as ulcerative colon of rats being 5- to 6.5-fold higher than in the healthy control.35 The author suggested that the selective accumulation in the ulcerative areas and the surrounding tissue may be due to either the increased sticky mucus secretion or the particles uptake into the macrophages highly present in the inflamed tissue. The increased residence time at the inflammation sites would permit higher therapeutic effectiveness as well as subsequent dose reduction and cost-effectiveness particularly on large scale.35
Pluronics can inhibit the drug efflux by the reduction of adenosine triphosphate (ATP) formation in drug-resistant cells following incorporation into cells membrane and then into cells. Consequently, these copolymers have been potentially employed to counteract tumor multidrug resistance (MDR) against numerous anticancer agents.34,36–40 Highly limited drug access into normal tissues and an enhanced passive drug targeting to tumors possibly due to excessive permeability of blood vessels of cancer cells can be obtained on drug encapsulation within micellar formulations.37 This effect is called enhanced permeation and retention (EPR). The reduced extravasation of blood vessels of normal cells and the diminished clearance by kidney can account for the prolonged circulation time of micelles.38
In the light of the abovementioned facts, it was worth to prepare, characterize, and optimize polymeric micellar delivery systems of naringin with the relatively inexpensive and biocompatible pluronic F68 (PF68). The effects of polymeric micellization on cytoprotective activity of naringin against ethanol-induced ulcer in rats were investigated. Additionally, to evaluate the influence of micellization of naringin on its antitumor activity, the in vitro cytotoxicity of the selected naringin–PF68 micelles against three cell lines, such as human hepatocellular liver carcinoma (HepG2), breast cancer (MCF-7), and colorectal carcinoma (Caco-2), was evaluated. Finally, in vivo antitumor activity was assessed in Ehrlich ascites carcinoma (EAC)-bearing mice by recording tumor growth and percentage tumor inhibition.
Materials and methods
Materials
Naringin, PF68, dimethylsulfoxide (DMSO), thiobarbituric acid, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide, pyrogallol, and reduced glutathione (GSH) Ellman’s reagent were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). NF-κB activation and extraction kits were obtained from Abcam (Cambridge, MA, USA). Enzyme-linked immunosorbent assay (ELISA) kit for IL-6 was purchased from R&D Systems, Inc. (Minneapolis, MN, USA). TNF-α antibodies were provided by (dilution 1/100; Boster Biological Technology, Pleasanton, CA, USA), and those of caspase-3 were obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Cisplatin (5 mg/mL vial; Bristol-Myers Squibb, New York, NY, USA) was diluted with isotonic saline. Dulbecco’s Modified Eagle’s Medium (DMEM) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Fetal calf serum (FCS) was purchased from Hyclone (Logan, UT, USA). All other chemicals were of analytical grade.
Preparation of naringin–PF68 micelles
Thin-film hydration method has been used to prepare pluronic micelles.41,42 Ten milliliters methanol were used to dissolve mixtures of naringin (10 mg) and PF68 at the three respective weight ratios of 1:30, 1:40, and 1:50. Methanol was evaporated using rotary evaporator (Rotavapor®; Buchi, Flawil, Swizerland) at 50°C for 1 h to form a thin film of naringin–PF68 matrix. The formed film was hydrated with 10 mL deionized water by stirring for 30 min at 60°C. The resultant solution was filtered through 0.2-µm filter to get rid of the unincorporated drug and finally lyophilized (SIM, FD8-8T, Newark, NJ, USA). The same procedure was employed to prepare the plain micelles except that the drug was not included.
Characterization of naringin–PF68 micelles
Determination of loading efficiency (LE%) and entrapment efficiency (EE%)
Methanol was used to disrupt the micellar structure to be then analyzed spectrophotometrically (ultraviolet/visible [UV/VIS] spectrophotometer; JASCO, Tokyo, Japan) for drug content at 281 nm vs plain micelles treated the same. The equations used for the estimation of LE% and EE% of naringin in PF68 micelles were as follows:43
Further characterization of polymeric micelles with PF68 showing the highest EE% would be carried out.
Particle size measurements
Dynamic light scattering (DLS) (Malvern Instruments, Malvern, UK) was used to record the size distribution of the studied micelles. The lyophilized micelles were reconstituted in double deionized water, properly diluted, and sonicated to obtain uniformly distributed micelles.
Morphological analysis
Examination of the selected pluronic micelles morphology was accomplished using transmission electron microscope (TEM-2100; JEOL, Tokyo, Japan) operated at an accelerating voltage of 160 kV. A drop of the aqueous solution obtained by reconstitution of lyophilized naringin–PF68 micelles was placed on a carbon grid to dry slowly in air before examination under TEM.
Fourier transform-infrared (FT-IR) spectroscopy
FT-IR spectra of naringin, PF68, plain micelles, and naringin–PF68 micelles were analyzed using an FT-IR spectrophotometer (Thermo Fisher Scientific). Disk of each sample with potassium bromide was individually scanned over a wavenumber range of 500–4,000/cm.
Differential scanning colorimetry (DSC)
DSC (model DSC-4, PerkinElmer Inc., Waltham, MA, USA) was utilized to study the thermal characteristics of naringin, PF68, plain micelles, and optimized drug–PF68 micelles. Heating of the examined samples (4 mg) was carried under nitrogen gas flow over a temperature range of 50°C–350°C at a rate of 10°C/min. Indium with the purity of 99.99% and the melting point of 156.6°C was used to calibrate the temperature.
X-ray diffractometry (XRD)
XRD analysis (X-ray diffractometer; Diano, Woburn, MA, USA) of naringin, PF68, plain micelles, and naringin–PF68 micelles was carried out at 45 kV, 9 mA, and an angle of 2θ.
In vitro drug release study
Dialysis method was utilized to study in vitro release of naringin from optimized micelles with PF68 compared to drug solution in water:methanol (9:1) mixture (1 mg/mL) as a control. Four milliliters of naringin micellar aqueous solution or naringin control solution were placed in a dialysis membrane bags (Spectra/Por®, 12–14 kDa M Wt cutoff, Canada). The dialysis bag was immersed into 50 mL release medium and incubated at 37°C±0.5°C under gentle agitation (75 rpm) (GFL, Gesellschaft fur Labortechnik GmbH, Burgwedel, Germany). It has been documented that the average residence time of a formulation in stomach is 2 h.44 Therefore, in the present study, the release medium of simulated gastric fluid (SGF), pH 1.2, for 2 h and then simulated intestinal fluid (SIF), pH 6.8, was used to predict the drug release profile following oral administration.45 Phosphate-buffered saline (PBS), pH 7.4, was also utilized to examine the drug release pattern possibly obtained after parenteral administration. Sodium lauryl sulfate (0.25%, w/v) was added to each dissolution medium to obtain the sink condition. At certain time intervals, aliquots of the release medium (1 mL) were withdrawn. An equal volume of the fresh medium was added to replace the withdrawn sample. Standard calibration curves of naringin at different release media have been constructed following its quantification spectrophotometrically at 281 nm (UV/VIS spectrophotometer). Plots of mean cumulative percent release of naringin vs time were constructed.
Release kinetics
First-order, zero-order, and diffusion-controlled release models were used to analyze in vitro release data.46 To verify the release mechanism, Korsmayer–Peppas kinetic model (mt/m∞=ktn) was also applied as a logarithmic relation of the fraction of drug released (mt/m∞) against the release time (t), where k is the kinetic constant and n is the slope of log mt/m∞ vs log t representing the diffusional exponent for drug release.47 The model showing the greatest correlation coefficient (r2) was suggested to explain naringin release mechanism from PF68 micelles.
Stability study
The storage stability of the lyophilized selected micelles exhibiting the highest drug encapsulation was evaluated over 3 months at room temperature (25°C±1°C). The lyophilized micelles were reconstituted in double deionized water to examine EE% and the size distribution at the beginning (0 months) and the end of the 3 months as previously described. The mean drug retention (%) of the stored micelles was also calculated at the end of the study.
Animals
All animal procedures and handling were accomplished according to the US National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH publication no 85-23, revised 1996). The protocol was approved by the Ethical Committee of Faculty of Pharmacy, Mansoura University, Egypt. Animals were kept under regular 12 h light/12 h dark cycles at a temperature of 24°C±1°C and a relative humidity of 55%±5%. The animals had free access to standard laboratory food and water.
Evaluation of cytoprotective activity against ethanol-induced ulcer in rats
Induction of ulcer
Male Sprague Dawley rats of a weight range of 180–220 g were used to investigate the cytoprotective activity of naringin–pluronic micelles compared with the free drug. Five groups, each consisted of six rats, were employed. Animals of groups I (normal control) and II (positive control) did not receive any treatment for 5 days. The three remaining groups orally pretreated for successive 5 days with either free naringin at two doses of 100 mg/kg (group III) and 200 mg/kg (group IV) or naringin–PF68 1:50 micelles (equivalent to 100 mg/kg of naringin, group V). On the fifth day, all animals had free access to water but were deprived of food for 24 h. On the sixth day, ulcer was induced in rats of groups II (positive control), III, IV, and V by a single intragastric instillation of 70% ethanol (10 mL/kg).48,49
Tissue collection and preparation
Two hours after ethanol instillation, euthanizing animals under deep ether anesthesia was followed by immediate laparotomy to separate stomachs to be opened along the greater curvature and then washed with normal saline to get rid of blood clots and gastric content. Stomachs were macroscopically examined for ulceration and erosion and photographed. Then, each stomach was divided into two portions. The first portion was kept in 10% (v/v) buffered formalin solution for histopathological and immunohistochemical evaluations of TNF-α and caspase-3. The second portion was divided into two parts that were stored at −80°C. One part was homogenized in 10 volumes of lysis buffer (200 mM NaCl, 5 mM ethylenediaminetetraacetic acid, 10 mM Tris, 10% glycerine, 1 mM PMSF, 1 mg/mL leupeptin, and 28 mg/mL aprotinin, pH 7.4) for the determination of cytokine levels including NF-κB and IL-6. The second part was weighed and homogenized in 10 volumes of ice-cold phosphate buffer (100 mM, pH 7.4) for the assessment of oxidative stress markers including malondialdehyde (MDA), superoxide dismutase (SOD), and GSH.49,50
Macroscopic evaluation of gastric ulceration
The gastric tissues were examined by an observer that did not recognize the identity of samples for gastric mucosal lesions. Hemorrhage, linear streaks (erosions), and damage to the mucosal surface were recognized as gross mucosal lesions. Paul’s index as an integral indicator of the number of lesions induced per treatment group and antiulcer activity (AA) were estimated to evaluate the cytoprotective activity of naringin against ulcer.51–53 Paul’s index was calculated by multiplying the mean number of ulcers and the percentage of rats with ulcers to be then divided by 100. As well, AA was calculated by dividing Paul’s index of the positive control group (II) by that of each of the treatment groups. The AA was recognized if AA was at least of two units.
Histopathological examination
Gastric tissues fixed in 10% (v/v) buffered formalin solution were washed. This was followed by dehydration by alcohol, clearing in xylene, and embedding in paraffin in hot air oven (56°C) for 24 h. The obtained paraffin blocks were then cut into 5 µm sections to be deparaffinized and stained with hematoxylin and eosin to be examined using the light microscope (Leica Microsystems, Wetzlar, Germany).54,55 The histopathological examination was performed by an observer unaware of the specimens’ identity to avoid any bias.
Immunohistochemical localization of TNF-α and caspase-3
Paraffin blocks from rats’ gastric tissues were investigated for the immunohistochemical evaluation of TNF-α. Tissue sections were cut and fixed on Superfrost™ Plus Microscope Slides (Thermo Fisher Scientific). Detection was done using the Histostain Bulk kit Invitrogen LAB-SA system. Manual immunohistochemistry stainer was utilized to prepare the slides following previously reported procedure.56 PBS was used to deparaffinize the sections. Antigen retrieval was done by CCl standard (citrate buffer pH 6.0). Blocking the 1,4-dideoxy-1,4-imino-d-arabinitol (DAB) inhibitor (3% H2O2 endogenous peroxidase) was done for 5 min at room temperature. Tested sections were incubated with the employed antibodies of TNF-α or caspase-3 at 25°C±1°C for a period of 40 min and then incubated with the secondary antibody of universal horseradish peroxidase multimer at 37°C for 8 min. Treatment of slides with DAB + H2O2 substrate for 8 min was followed by hematoxylin and the bluing reagent counterstain at 37°C. Washing solution was reaction buffer (PBS). Controls were prepared by staining without primary antibody. Staining degree of positively stained cells was evaluated using a digital camera (Olympus Corporation, Tokyo, Japan) placed on a microscope (Leica Microsystems). Specimen examination was done by a senior pathologist. Immunoreactive cells for TNF-α were counted using the ImageJ analysis.
Evaluation of gastric MDA, GSH, and SOD
MDA is a thiobarbituric acid reactive substance (TBARS), so its levels in gastric homogenates were determined according to the reported TBARS test.57
Colorimetric assessment of GSH concentrations (nanomoles per milligram protein) in the gastric tissue homogenate was carried out at 412 nm following GSH reaction with Ellman’s reagent (5,5′-dithio-bis[2-nitrobenzoicacid]) after protein precipitation with trichloroacetic acid.58
The enzyme amount that produced 50% inhibition of pyrogallol auto-oxidation was recognized as one unit of SOD activity.59 The activity of SOD was monitored at 420 nm and expressed as unit per milligram of protein.
ELISA of NF-κB and IL-6 in gastric tissue homogenate
ELISA technique was used to evaluate the gastric homogenate levels of NF-κB and IL-6 according to the manufacturer’s directions of the employed kits. Optical density was normalized as nanogram (NF-κB) or picogram (IL-6) per gram of nuclear extract tissue protein.
Evaluation of naringin antitumor activity
Cell cultures
Cell lines of HepG2, Caco-2, and MCF-7 were provided by American Type Culture Collection through VACSERA (Cairo, Egypt). The cells’ growth was accomplished in DMEM containing NaHCO3 (3.7 g/L), d-glucose (4.5 g/L), streptomycin (100 µg/mL), and penicillin (100 U/mL) provided with 10% FCS at 37°C in a humidified 5% CO2 and 95% air atmosphere.
In vitro cytotoxicity study
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) colorimetric assay was used to investigate in vitro cytotoxicity of free naringin, naringin–PF68 micelles, a negative control of plain micelles, and a positive control of cisplatin against HepG2, Caco-2, and MCF-7 cell lines. Seeding of cultured cells maintained in DMEM was done in 96-well culture plates at 1×105/well followed by incubation for 24 h. This was followed by treatment with increasing concentrations of the examined samples and incubation for another 72 h at 37°C in 5% CO2 atmosphere. DMSO was used to dissolve each of free naringin and cisplatin to be diluted with the experiments’ media. Lyophilized plain and medicated micelles were reconstituted in a culture medium and then filtered through a 0.2-µm membrane filter as a mean of sterilization. Wells were rinsed with PBS after the incubation period. Afterward, MTT solution (0.5 mg/mL) was added to each well and incubation was lasted for 4 h at 37°C. Formazan crystals formed due to MTT reduction by mitochondria of viable cells were dissolved by the addition of DMSO to each well and shaking for 15 min. Spectrophotometric analysis at 570 nm using microplate reader (Dynatech, Melville, NY, USA) was applied to estimate the optical absorbance of each well. Influences of tested samples on cells proliferation were evaluated by the calculation of cell viability (%) using the following equation:
In vitro cytotoxic activity was evaluated by the calculation of the mean values of the concentration of the drug required to kill 50% of cells relative to the untreated cultures known as the half maximal inhibitory concentration (IC50).
In vivo antitumor activity
Female Swiss albino mice weighing 20–30 g were used to investigate the antitumor activity of naringin–pluronic micelles in comparison to the free drug. Mice were obtained from Urology and Nephrology Center of Mansoura University, Mansoura, Egypt. Netherlands Cancer Institute established EAC cells. Serial intraperitoneal (ip) injections at 7 days intervals were employed to maintain Ehrlich tumor line at the laboratory of Faculty of Pharmacy, Mansoura University.
Seven days after EAC cells inoculation, needle aspiration from the peritoneal cavity of tumor-bearing mice was done under aseptic conditions to withdraw ascitic fluid to be washed three times using normal saline by centrifugation at 1,000 rpm. Trypan blue was used to test the viability of EAC cells obtained after washing. The cells suspended in normal saline were counted under microscope so that each 0.1 mL contained 5×105 viable EAC cells. Subcutaneous inoculation of 0.1 mL containing 5×105 viable tumor cells into the right thigh of mice lower limb was done for the induction of solid tumors.60 A digital caliper was used to measure the largest diameter of the tumor a and its perpendicular b to evaluate the tumor growth via the equation: tumor size (mm3)=0.5×a×b2.61
When the size of the primary tumor reached 50–100 mm3, 24 mice were divided into four groups of six mice each. Normal saline (5 mL/kg) was given to the first group (EAC-bearing mice). The second group administered cisplatin at a dose of 1 mg/kg ip. The third group received naringin (100 mg/kg ip). The fourth group administered PF68 micelles of naringin at an equivalent amount of 100 mg/kg ip. A negative control group of six mice was also studied. Tumor size was measured 5 days after tumor inoculation and before treatment (day 0) and after 14 days. Changes of tumor size in the treatment group (ΔT) and in the EAC-bearing mice (ΔC) were recorded. The tumor growth was calculated according to the equation ΔT/ΔC×100. The tumor growth inhibition (%) was estimated by subtracting the degree of tumor growth from 100%.61 Survival (%) of each group was calculated at the end of the study (day 14) as the number of mice surviving divided by the number of animals living at the start.62
Statistical analysis
One-way analysis of variance followed by Tukey–Kramer multiple comparisons test was applied to perform statistical analysis of the results at the significance levels of P<0.001, <0.01, and <0.05. Statistical analysis was carried out using the GraphPad Instat 2.04 statistical package (GraphPad Software, Inc., La Jolla, CA, USA).
Results and discussion
Micelle characterization
Drug LE% and EE%
The increase in the polymer content improved EE% of naringin in its micelles possibly due to the higher number of the core-forming blocks allowing encapsulation of more drug (Table 1). It has been reported that several factors govern the LE% and EE% of micelles among them the total polymer content.63 Accordingly, naringin–PF68 1:50 micelles showed the highest EE% (96.14±2.29) among other ratios, hence selected to be characterized and evaluated. These micelles exhibited the LE% of 1.89±0.05.
Table 1.
Drug entrapment and loading efficiencies at different drug-to-polymer ratios
| Preparation | Drug-to-polymer ratio (w/w) | EE% | LE% |
|---|---|---|---|
| Naringin–PF68 micelles | 1:30 | 69.99±2.81 | 2.26±0.09 |
| 1:40 | 83.53±1.74 | 2.04±0.04 | |
| 1:50 | 96.14±2.29 | 1.89±0.05 |
Note: Data are expressed as mean ± SD (n=3).
Abbreviations: EE%, entrapment efficiency; LE%, loading efficiency; PF68, pluronic F68.
Micelles size
The mean diameter as determined by DLS was 74.80±6.56 nm for 1:50 micelles with PF68 with mean value of polydispersity index of 0.30±0.11 indicating a narrow size distribution (Figure 2). It has been reported that both circulation time and biodistribution of the drug are directly affected by the particle size.64 Prolonged circulation times and facilitated access of cells and tissues are expected in case of micelles with diameter <100 nm.63 Unpredictable change in drug’s pharmacokinetics and organ biodistribution may occur due to a broad micelle size distribution. An increased residence time and selective accumulation in inflamed ulcerative tissues can be expected with these nanoscopic particles.35 This would allow dose reduction of naringin and subsequent cost-effectiveness particularly on large-scale production.35 Moreover, these polymeric nanomicelles may selectively accumulate in solid tumors through EPR effect allowing passive targeting of solid tumor and a predictable biodistribution.37,65,66
Figure 2.
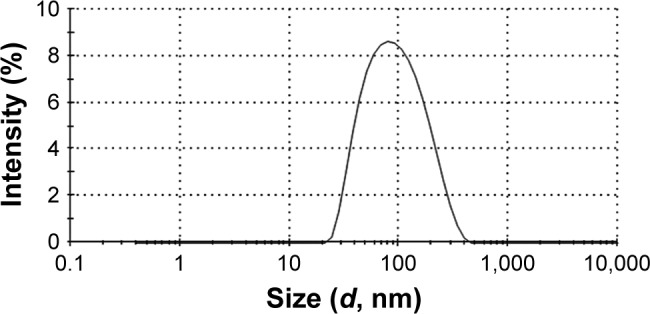
Size distribution curve of naringin–PF68 micelles.
Abbreviation: PF68, pluronic F68.
Morphological analysis
Figure 3 represents TEM image of naringin–PF68 1:50 micelles. In agreement with particle size analysis via DLS, this figure illustrates micelles as well-dispersed and individual spherical particles with diameter <100 nm. However, TEM image shows particles with diameter smaller than that estimated by DLS. This can be explained on the basis that DLS measures the particles hydrodynamic diameter, which includes polymer shells and hydration layer leading to a larger particle size.67,68 In addition, DLS is extremely sensitive to the dispersion/aggregation behavior of the particles in solution.68 Selective accumulation and targeting of naringin in inflamed tissues and tumor cells can still be proposed. Consequently, an enhancement of antiulcer and antitumor activities of naringin can be expected upon micellization with PF68.
Figure 3.

TEM image of naringin–PF68 micelles.
Abbreviations: PF68, pluronic F68; TEM, transmission electron microscope.
FT-IR spectroscopy
FT-IR spectra of free naringin, PF68, nonmedicated micelles, and naringin–PF68 micelles are illustrated in Figure 4A. Naringin has polyhydroxy groups, hence O–H bond stretched on exposure to IR and detected as strong signal peak between 3,000 and 3,500/cm.69 Accordingly, FT-IR spectrum of naringin exhibited characteristic bands at 3,444, 3,420, and 3,384 corresponding to more than one O–H aromatic stretching. The bands indicating C=C, C=O, and C–H aliphatic stretching bands appeared at 1,580, 1,683, and 2,931/cm, respectively. The three main peaks of PF68 appeared at 1,111/cm (R–C–O symmetric), 948/cm (R–C–O– asymmetric), and 1,282/cm (–CH2–O–R). The disappearance of drug bands in the spectra of the medicated micelles with PF68 may indicate the drug encapsulation.40
Figure 4.

Solid characterization.
Notes: (A) FT-IR spectra, (B) DSC curves, and (C) XRD patterns of (I) naringin, (II) PF68, (III) plain micelles, and (IV) naringin–PF68 micelles.
Abbreviations: PF68, pluronic F68; FT-IR, Fourier transform infrared spectroscopy; DSC, differential scanning colorimetry; XRD, X-ray diffractometry.
DSC
Figure 4B illustrates DSC thermograms of free naringin, PF68, plain micelles, and naringin–PF68 micelles. The thermogram of naringin showed the phase transition peak at 164.98°C and an endothermic melting peak at 245.8°C with peaks occurring >250°C attributed to naringin decomposition.24,70 The well-recognized endothermic peak of PF68 at 54.68°C has appeared in its DSC thermogram. DSC curves of micelles either plain or medicated showed a characteristic peak of PF68 (55.52°C and 54.72°C, respectively). Interestingly, DSC thermogram of the drug–PF68 micelles did not show the drug endothermic peak suggesting naringin encapsulation in the micelles.
XRD
XRD curves of free naringin exhibited different diffraction peaks reflecting the crystalline nature of the drug (Figure 4C). Two predominant diffraction peaks at 2θ, 19.38 and 23.52, have been identified in PF68 XRD pattern. These peaks were present in the patterns of both lyophilized nonmedicated and naringin–PF68 micellar formulations. The complete absence of the drug diffraction peaks in the XRD pattern describing medicated micelles could indicate the drug encapsulation.
In vitro release study
In vitro release profiles of naringin from its micelles with PF68 at SGF (pH 1.2) for 2 h followed by SIF and that at PBS (pH 7.4) compared with free naringin solution as a control are illustrated in Figure 5A and B, respectively. Cumulative naringin released (%) at different release media were estimated employing the equations describing standard calibration curves at SGF, SIF, and PBS (pH 7.4), which were y=0.0255x+0.0187 (r2=0.9990), y=0.0247x+0.0213 (r2=0.9999), and y=0.0243x+0.0256 (r2=0.9968), respectively.
Figure 5.

In vitro release of naringin from its micelles with PF68 compared with free drug in different dissolution media at 37°C±0.5°C.
Notes: (A) SGF (pH 1.2) (0–2 h) followed by SIF (pH 6.8) and (B) PBS (pH 7.4). Each point represents the mean ± SD (n=3).
Abbreviations: PF68, pluronic F68; SGF, simulated gastric fluid; SIF, simulated intestinal fluid; PBS, phosphate buffered saline.
Naringin release from its control solution at SIF was complete (99.96%±0.62%) within 10 h (Figure 5A). Similarly, the drug was completely released (99.38%±0.71%) after 10 h at PBS (pH 7.4) (Figure 5B), while the gradual release of this drug from the prepared micelles up to 48 h was preceded by a burst release at both media. The burst release was observed in the first 10 h with cumulative drug release of 81.78%±2.58% when SGF was used for 2 h followed by SIF. Similar release pattern was observed at PBS (pH 7.4) as clarified by the cumulative drug release of 83.28%±0.54%. Generally, naringin release continued gradually up to 48 h permitting extended release. Similar biphasic release has been documented for pluronic micelles of chemotherapeutic agents such as camptothecin,71 paclitaxel,42,72 and vorinostat.73 In these studies, the investigated micelles exhibited ≈70%–80% drug release during the burst release phase. The biphasic drug release from the micelles could be explained through its location within the micelles. The initial burst release can be attributed to the drug on the interface of the hydrophilic corona and the micelle hydrophobic core or inside the mobile micelle corona that could be released via hydration of the interfacial drug molecules followed by passive diffusion.68,74 The drug incorporated into the inner core compartment stayed firmly inside the micelles, and hence, it was released slowly with 20%–30% of the initially incorporated drug. Therapeutic concentration may be provided by such initial burst drug release, and the subsequent slow release could keep the drug concentration at the therapeutic levels.75
Release kinetics
Table 2 depicts the results of kinetic analysis of naringin release data from the studied pluronic micelles. The results revealed that in vitro release of naringin from these micelles was best explained by Higuchi model that suggested release controlled by diffusion at SGF/SIF and PBS. However, r2 values of the first and Highuchi models describing drug release during the burst release phase were very close possibly suggesting that the drug release was due to both drug diffusion through the micelles and micelles erosion. In accordance, non-Fickian mechanism was found to best explain the drug release during the burst release phase since n values were 0.702 and 0.647 at SGF/SIF and PBS, respectively. This may confirm the combination of erosion and diffusion during this phase of drug release. Meanwhile, Fickian mechanism representing diffusion-controlled release during the phase of gradual release could be confirmed by the n values of 0.132 and 0.137 at SGF/SIF and PBS, respectively. Fickian and non-Fickian release mechanisms have described curcumin release from pluronic micelles.75
Table 2.
Kinetic modeling of drug release data
| Release medium | Correlation coefficient (r2)
|
Release order | Korsmeyer–Peppas
|
Main transport mechanism | |||
|---|---|---|---|---|---|---|---|
| Zero order | First order | Higuchi model | r2 | Diffusional exponent (n) | |||
| SGF/SIF Burst release phase |
0.990 | 0.990 | 0.992 | Higuchi | 0.998 | 0.702 | Non-Fickian |
| SGF/SIF Gradual release phase |
0.994 | 0.970 | 0.999 | Higuchi | 0.988 | 0.132 | Fickian |
| PBS (pH 7.4) Burst release phase |
0.977 | 0.994 | 0.997 | Higuchi | 0.998 | 0.647 | Non-Fickian |
| PBS (pH 7.4) Gradual release phase |
0.975 | 0.992 | 0.999 | Higuchi | 0.999 | 0.137 | Fickian |
Abbreviations: SGF, simulated gastric fluid; SIF, simulated intestinal fluid; PBS, phosphate-buffered saline.
Stability study
Large-scale production of pharmaceutical products should be preceded by the assessment of storage stability. Freeze drying is a reliable technique to ensure a long-term stability of these products. Stability of lyophilized naringin–PF68 micelles respective weight ratio 1:50 was studied. The micelles were stable on storage at room temperature for 3 months as reflected by the slight lowering in EE% and the small increase in the average micelles diameter that remained <100 nm (Table 3). Additionally, naringin retention (%) in PF68 micelles was 91.99±3.24 at the end of the 3 months. It has been reported that pluronic micelles were stable after lyophilization without need of cryoprotectant possibly due to the cryoprotectant long PEO chains in pluronic.73,75,76
Table 3.
Storage stability of lyophilized naringin micelles at room temperature 25°C±1°C
| Parameter | 0 month | 3 months |
|---|---|---|
| LE% | 2.26±0.09 | 1.73±0.05 |
| EE% | 96.14±2.29 | 88.41±2.66 |
| Mean particle size, nm | 74.80±6.56 | 80.50±0.70 |
| PDI | 0.30±0.11 | 0.23±0.02 |
| Drug retention (%) | – | 91.99±3.24 |
Note: Data are expressed as mean ± SD (n=3).
Abbreviations: EE%, entrapment efficiency; LE%, loading efficiency; PDI, polydispersity index.
Evaluation of cytoprotective activity against ulcer
Macroscopic evaluation of gastric ulceration
Figure 6 represents gross appearance of freshly excised stomach tissue of different rat groups. Parameters of macroscopic evaluation of ulceration are illustrated in Table 4. Stomach of group I (normal control) showed normal gastric mucosa (neither inflammation nor hemorrhage) (Figure 6A). However, the mucosa of stomach in group II (positive control), which received ethanol (70%, 10 mL/kg) orally, showed multiple gastric ulcers and intensely hemorrhagic mucosa (Figure 6B). Intragastric ethanol administration in rat models resulted in noticeable alteration in membrane damage, cellular levels, exfoliation, and epithelial erosion, as well as cell death.52 Accordingly, rats of this group experienced a high incidence of ulcer (100%), hence the highest Paul’s index of 26.5 (Table 4). When free naringin (100 mg/kg) was given for 5 successive days before ethanol administration (group III), reduced inflammation and hemorrhage were observed (Figure 6C). Accordingly, this group showed significantly (P<0.05) lower mean ulcers number as well as smaller Paul’s index and percentage incidence when compared with positive control (II). Significantly diminished mean ulcers number has been resulted on pretreatment with 200 mg/kg free naringin (IV) relative to positive control group (II, P<0.001) and that received 100 mg/kg free naringin (III, P<0.01). This finding was manifested by lower percentage incidence of rats with ulcers (66.67%) and Paul’s index (5.00) in case of group IV. Oral pretreatment with naringin micelle with PF68 (V) at an amount equivalent to 100 mg/kg of the drug has resulted in a significant decrease in mean ulcers number in comparison with positive control group (II, P<0.001) and that pretreated with 100 mg/kg free naringin (III, P<0.05). However, there was insignificant difference in the mean ulcers number between groups received micelle (V) or free drug (IV) at respective doses of 100 and 200 mg/kg. Much less ulceration and hemorrhage were experienced in groups IV and V (Figure 6D and E).
Figure 6.

Gross appearance of stomach tissues.
Notes: (A) Normal control with normal gastric mucosa, (B) positive control showing multiple gastric ulcers and intensely hemorrhagic mucosa, (C) rats pretreated with naringin (100 mg/kg) displaying some gastric ulcers and moderately hemorrhagic mucosa, and rats pretreated with either (D) naringin (200 mg/kg) or (E) naringin–PF68 micelles (100 mg/kg) showing minimal gastric ulcers and mildly hemorrhagic mucosa. Arrows indicate gastric ulcers and hemorrhagic mucosa.
Abbreviation: PF68, pluronic F68.
Table 4.
Parameters of macroscopical evaluation of ethanol-induced ulcers in rats
| Animal group | Number of ulcers Mean ± SE n=6 |
Percentage incidence of animals | Paul’s index | AA | Severity of inflammation |
|---|---|---|---|---|---|
| I (normal control) | – | 0 | 0 | – | − (no inflammation) |
| II (positive control) | 26.50±2.32 | 100 | 26.50 | – | +++ (severe) |
| III (100 mg/kg free naringin) | 18.00±1.81# | 83.33 | 14.99 | 1.77 | ++ (moderate) |
| IV (200 mg/kg free naringin) | 7.50±1.04###,$$ | 66.67 | 5.00 | 5.30 | + (mild) |
| V (100 mg/kg naringin–PF68 micelles) | 8.75±0.85###,$ | 66.67 | 5.83 | 4.55 | + (mild) |
Notes:
P<0.05 and
P<0.001 vs positive control group (II).
P<0.05 and
P<0.01 vs 100 mg/kg free naringin pretreated group (III).
Abbreviations: AA, antiulcer activity; SE, standard error; PF68, pluronic F68.
Accordingly, the AA of naringin either free at a dose of 200 mg/kg (IV, AA=5.30) or micelle with PF68 at an amount equivalent to 100 mg/kg (V, AA=4.55) was highly increased relative to that produced on oral pretreatment with free drug at a dose of 100 mg/kg (III, AA=1.77). The AA was recognized if AA was at least of two units.51–53 Hence, the cytoprotective activity (AA>2) of naringin against ethanol-induced ulcer in rat model has been enhanced on micellization with PF68 allowing lowering in dose compared with free drug. This enhancement can be referred to more prolonged residence time and selective accumulation of nanomicelles in ulcerative tissues.35 Two principal mechanisms have been identified to account for the accumulation of nanoparticles in ulcerated tissue.77 Adhesion of nano-sized particles to the mucus layer in the stomach is favored due to their small mass. Compared to microparticles, uptake of nanoparticles by immune cells, such as macrophages, in the area of active inflammation is much easier. This enhancement can be referred to more prolonged residence time and selective accumulation of nanomicelles in ulcerative tissues.35
Histopathological examination
Gastric tissues of normal control group (I) showed normal gastric mucosa, gastric pits, lining epithelium, gastric glands, and submucosa (Figure 7A). In accordance with macroscopic findings, stomach of positive control rats (II) exhibited coagulative necrosis of entire mucosal thickness with intense hemorrhage and desquamation of necrotic glandular epithelium, as well as congestion of blood vessels, severe edema, and neutrophilic infiltrations in the submucosa (Figure 7B). Coagulative necrosis of luminal half of the gastric mucosa with hemorrhage and desquamation of necrotic glandular epithelium as well as severe edema and neutrophilic infiltrations in the submucosa were observed in gastric tissues of rats orally pretreated with 100 mg/kg naringin (III) (Figure 7C). Coagulative necrosis of only superficial layer of gastric mucosa and mild desquamation of necrotic glandular epithelium, besides mild edema and neutrophilic infiltrations in the submucosa, were noticed in gastric specimens of rats orally pretreated with either 200 mg/kg free naringin (IV) or naringin–PF68 micelles at a dose equivalent to 100 mg/kg of the drug (group V) (Figure 7D and E, respectively).
Figure 7.

Histological examination (HE, 100×) of rats stomach.
Notes: (A) Normal control showing normal gastric mucosa (arrow) and submucosa (arrow head), (B) positive control with coagulative necrosis of entire mucosal thickness with intense hemorrhage and desquamation of necrotic glandular epithelium (arrow) as well as congestion of blood vessels, severe edema, and neutrophilic infiltrations in the submucosa (arrow head), (C) orally pretreated with naringin (100 mg/kg) showing coagulative necrosis of luminal half of the gastric mucosa with hemorrhage and desquamation of necrotic glandular epithelium (arrow) as well as severe edema (arrow head) and neutrophilic infiltrations in the submucosa, and orally pretreated with either (D) naringin (200 mg/kg) or (E) naringin–PF68 micelles (100 mg/kg) displaying coagulative necrosis of only superficial layer of gastric mucosa (arrow) and mild desquamation of necrotic glandular epithelium, besides mild edema and neutrophilic infiltrations in the submucosa (arrow head).
Abbreviations: HE, hematoxylin and eosin; PF68, pluronic F68.
Immunohistochemical localization of TNF-α and caspase-3
Figure 8A illustrates immunohistochemical evaluation of gastric TNF-α expression. Normal control rats (group I) displayed minimal immunoreactivity for TNF-α as indicated by minimal brown-stained gastric glands. Intragastric administration of ethanol (70%, 10 mL/kg, group II) without naringin pretreatment suggested strong immunoreactivity as clarified by intense brown staining in gastric mucosal epithelium, gastric glands, and submucosa. Moderate brown staining of the gastric mucosal epithelium, gastric glands, and submucosa on oral pretreatment with 100 mg/kg naringin (group III) indicated a reduction in the gastric expression of TNF-α. Doubling the dose of naringin (IV) or use of an equivalent amount of micelle drug (V) resulted in mild immunoreactivity indicated by mild brown staining of superficial gastric mucosal epithelium and gastric glands and submucosa.
Figure 8.

Microphotographs of gastric tissue of rats using immunohistochemical staining for (A) TNF-α and (B) caspase-3 in comparison with normal and positive control groups (IHC, 100×).
Notes: (I) Normal control displaying minimal brown-stained gastric mucosal epithelium (arrow), (II) positive control showing intense brown staining in gastric mucosal epithelium (arrow), gastric glands, and submucosa (arrow head), (III) rats orally pretreated with naringin (100 mg/kg) showing moderate brown staining of the gastric mucosal epithelium (arrow), gastric glands, and submucosa (arrow head), and orally pretreated with either (IV) naringin (200 mg/kg) or (V) naringin–PF68 micelles (100 mg/kg) displaying mild brown staining of superficial gastric mucosal epithelium (arrow) and gastric glands and submucosa (arrow head).
Abbreviations: IHC, immunohistochemical; PF68, pluronic F68; TNF-α, tumor necrosis factor-alpha.
Similar results have been recorded for caspase-3 (Figure 8B). In comparison with rats pretreated with 100 mg/kg free drug, significantly lower gastric expression of TNF-α and caspase-3 has been resulted from dose doubling or use of equivalent micelle drug (Figure 9). Both later treatments produced statistically indifferent gastric expression of TNF-α and caspase-3. The drug accumulation and longer residence time at inflamed tissues on micellization with PF68 may explain the reduction of gastric expression of TNF-α and caspase-3.35,77
Figure 9.

Effects of naringin oral pretreatment on ethanol-induced increase of gastric expression of TNF-α and caspase-3 in rats in comparison with normal and positive control groups (IHC, 100×).
Notes: (A) TNF-α and (B) caspase-3. Data are mean ± SD, n=6. ***P<0.001 vs normal control group. #P<0.05 and ###P<0.001 vs positive control group. $$P<0.01, and $$$P<0.001 vs 100 mg/kg naringin pretreated group.
Abbreviations: IHC, immunohistochemical; PF68, pluronic F68; TNF-α, tumor necrosis factor-alpha.
Gastric oxidative stress
Gastric tissue levels of MDA, GSH, and SOD in different studied rat groups are depicted in Figure 10. Ethanol (70%, 10 mL/kg) intragastric administration (II) induced a highly significant (P<0.001) increase in MDA content concomitant with a highly significant (P<0.001) depression in GSH and SOD activities when compared with normal control rats (I). Oral pretreatment with naringin (100 mg/kg, group III) resulted in a significant decrease in MDA content and a significant (P<0.001) elevation in GSH and SOD activities when compared with positive control rats (II). Compared to rats orally pretreated with naringin (100 mg/kg), significantly greater suppression of MDA release and elevation of antioxidant markers was produced on pretreatment with naringin either free at a higher dose of 200 mg/kg (IV) or micelle with PF68 (V) at a dose equivalent to 100 mg/kg of the drug. There was an insignificant difference between the two later groups (IV and V) regarding their effects on tissue levels of MDA, GSH, and SOD as well as when compared with normal control (I).
Figure 10.

Effects of naringin oral pretreatment on ethanol-induced changes in oxidative stress markers.
Notes: (A) MDA, (B) GSH, and (C) SOD in comparison with normal and positive control groups. Data are mean ± SD, n=6. ***P<0.001 vs normal control group. ##P<0.01, and ###P<0.001 vs positive control group. $P<0.05 and $$P<0.01 vs 100 mg/kg naringin pretreated group.
Abbreviations: GSH, reduced glutathione; MDA, malondialdehyde; PF68, pluronic F68; SOD, superoxide dismutase.
Consequently, it can be said that polymeric micelles of naringin with PF68 enhanced its cytoprotective activity against ulcer induced in rats by ethanol allowing the reduction of naringin dose to the half possibly due to the high uptake of these nanoparticles by macrophages at the ulcerative areas.35,77
ELISA of NF-κB and IL-6 in gastric tissue homogenate
Figure 11 represents the effects of naringin oral pretreatment on NF-κB and IL-6 expressions in comparison with normal and positive control groups using ELISA. As shown in this figure, NF-κB and IL-6 were significantly (P<0.001) expressed in positive control rats (II) when compared with those of normal control group (I). In contrast, naringin pretreated groups (III, IV, and V) showed a significant reduction in NF-κB and IL-6 expressions as compared to positive control group (II). Compared to rats pretreated with 100 mg/kg naringin (I), such reduction was statistically significant (P<0.05) when the dose of free drug was increased to 200 mg/kg or encapsulated naringin within PF68 micelles at a dose of 100 mg/kg was administered. There was an insignificant difference in gastric NF-κB and IL-6 expressions on the dose increase or micellization of the drug, and hence PF68 micelles can be still suggested as a promising drug delivery system of this drug for potentiated cytoprotective action against ethanol-induced ulcer. Lowering of gastric NF-κB and IL-6 expressions encountered with naringin–PF68 micelles could be explained by the excessive accumulation of these nanoscopic particles in the ulcerative tissues.35,77
Figure 11.

Effects of naringin oral pretreatment on ethanol-induced cytokines expression in comparison with normal and positive control groups using ELISA.
Notes: (A) NF-κB and (B) IL-6. Data are mean ± SD, n=6. **P<0.01, and ***P<0.001 vs normal control group. #P<0.05 and ###P<0.001 vs positive control group. $P<0.05 vs 100 mg/kg naringin pretreated group.
Abbreviations: ELISA, enzyme-linked immunosorbent assay; IL-6, interleukin-6; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PF68, pluronic F68.
Evaluation of naringin antitumor activity
In vitro cytotoxicity
Effects of PF68 micelles on naringin cytotoxicity against HepG2, Caco-2, and MCF-7 were assessed via MTT assay (Table 5). Values of IC50 were estimated for naringin–PF68 micelles, free naringin, plain micelles, and cisplatin as a reference anticancer. Naringin and its micelles showed different sensitivities to the cell lines used. Caco-2 cells were the most sensitive to naringin showing the IC50 of 7.67±0.14 µM followed by HepG2 that exhibited the IC50 of 25±0.17 µM. The highest IC50 (30.40±1.34 µM) has been recorded for MCF-7 that is very much consistent with published data, revealing IC50 between 20 and 40 µM as estimated by MTT assay.78 These results reflect the lower cytotoxicity of naringin against the breast cancer cell line compared to the other two cell lines used. This can be referred to the dose-dependent estrogenic and antiestrogenic activities of naringin.79 At low concentration, this drug showed estrogenic agonist activity. In contrast, it acted as estrogenic antagonist at high concentrations and, hence, it cannot be considered as an efficient estrogen antagonist and high drug concentration can be expected to significantly affect the viability and proliferation of estrogen-sensitive MCF-7 cells. Generally, encapsulation of naringin into PF68 pluronic micelles significantly (P<0.001) potentiated its cytotoxicity against all cell lines particularly Caco-2 cells as indicated by the lowest IC50 of 0.19±0.01 µM. The results may represent the micellar formulation of 1:50 naringin–PF68 as a clinically superior delivery system as suggested by significantly improved in vitro cytotoxicity than free drug and the reference anticancer cisplatin.
Table 5.
The in vitro cytotoxicity estimated as the IC50 values after 72 h
| Treatment | IC50 (μM)
|
||
|---|---|---|---|
| HepG2 | Caco-2 | MCF-7 | |
| Naringin | 25±0.17 | 7.67±0.14 | 30.40±1.34 |
| Naringin–PF68 micelles | 14.00±0.17@@@,*** | 0.19±0.01@@@,*** | 23.00±1.32@@@,* |
| PF68 plain micelles | 98.00±0.08 | 25.00±0.04 | 34.00±0.01 |
| Cisplatin | 30.75±0.22 | 9.80±1.13 | 20.51±0.57 |
Notes: Data are expressed as mean ± SD (n=3).
P<0.001 vs free naringin.
P<0.05, and
P<0.001 vs cisplatin.
Abbreviations: Caco-2, colorectal carcinoma; HepG2, human hepatocellular carcinoma; IC50, half maximal inhibitory concentration; MCF-7, human breast cancer; PF68, pluronic F68.
The lower in vitro cytotoxicity of free naringin relative to its micelles may be explained on the basis that drug aggregates could hinder drug access into cells or be eliminated from tumors by efflux pumps. The increased permeation and retention of micelles in cancer cells37,66 as well as the inhibition of drug efflux pump or P-glycoproteins by PF68 may explain the improvement of naringin in vitro cytotoxicity by micellization with pluronics. The direct inhibition of drug efflux pump by pluronics can be explained on the basis that they can incorporate into cellular membranes and translocate into cells hindering ATP synthesis, mitochondrial respiration, apoptotic signal transduction, gene expression, and drug efflux transporters’ activity.80 It has been reported that pluronics reduced ATP formation in drug-resistant cells rendering (MDR) tumors sensitive to multiple chemotherapeutics.34,35 These effects of pluronics could explain their successful use in developing drug delivery systems to overcome MDR of tumors.39,40,81
Mixed micelles of pluronic L61 and F127 containing doxorubicin (SP1049C) was the first clinically studied micellar formulation of anticancer.82 The micelle drug was effectively accumulated in the tumors with higher drug residence time and more delayed peak level than free doxorubicin. Improved cytotoxicity against MCF-7 cells and different intracellular distributions have been noticed for doxorubicin–copolymer conjugate compared with free drug.83 Endocytosis was the main cell transport mechanism of doxorubicin conjugate with pluronics, while free drug transported by transmembrane diffusion, and hence conjugates could overcome MDR in tumor cells.
In vivo antitumor activity
A solid palpable tumor mass was observed 5 days after inoculation of EAC cells (day 0). A progressive growth of the formed tumor to almost 10-fold its initial mass was observed after additional 14 days (day 14) and recognized as 100% tumor growth.
The mean values of tumor growth (ΔT/ΔC,%) and tumor growth inhibition are shown in Table 6. All treatment groups showed significantly (P<0.05) lower tumor size than untreated EAC-bearing group. The estimated values of percentage tumor growth were 20%, 42%, and 28% for groups receiving cisplatin, free naringin, and naringin–PF68 micelles, respectively, with the respective percentage tumor growth inhibition of 80, 58, and 72. Antitumor activity of naringin was significantly (P<0.05) lower than that of the reference anticancer cisplatin. Encapsulation of naringin within PF68 micelles significantly (P<0.05) improved its antitumor activity as indicated by the higher percentage of tumor inhibition 72 vs 58 for free naringin. Interestingly, the antitumor activity of naringin micelle with PF68 insignificantly differed from that of cisplatin. At the end of the study, treatment groups of naringin either free or encapsulated with PF68 showed the survival percentage of 100 vs 83.3 for cisplatin-treated group suggesting naringin and its micelles as safer alternative than the currently available chemotherapy (Figure 12).
Table 6.
Effects of micelled naringin (100 mg/kg) on tumor growth in mice-bearing EAC cells in comparison with cisplatin (1 mg/kg) and free naringin (100 mg/kg)
| Treatment group | Tumor size (mm3)
|
|||
|---|---|---|---|---|
| Day 0 | Day 14 | ΔT/ΔC (%) | Percentage of inhibition | |
| EAC-bearing mice | 72.40±4.27 | 722.4±41.91 | 100 | 0 |
| Cisplatin | 71.33±3.97 | 206.2±12.70‡ | 20 | 80 |
| Naringin | 70.00±3.86 | 346.0±19.26‡,* | 42 | 58 |
| Naringin–PF68 micelles | 74.33±4.44 | 258.0±13.35‡,@ | 28 | 72 |
Notes: Data are expressed as mean ± SE (n=6). ΔT, changes of tumor size in the treatment group; ΔC, changes of tumor size in the EAC-bearing mice; ΔT/ΔC (%), percentage of tumor growth.
P<0.05 vs EAC-bearing mice group,
P<0.05 vs free naringin group,
P<0.05 vs cisplatin group.
Abbreviations: EAC, Ehrlich ascites carcinoma; PF68, pluronic F68.
Figure 12.
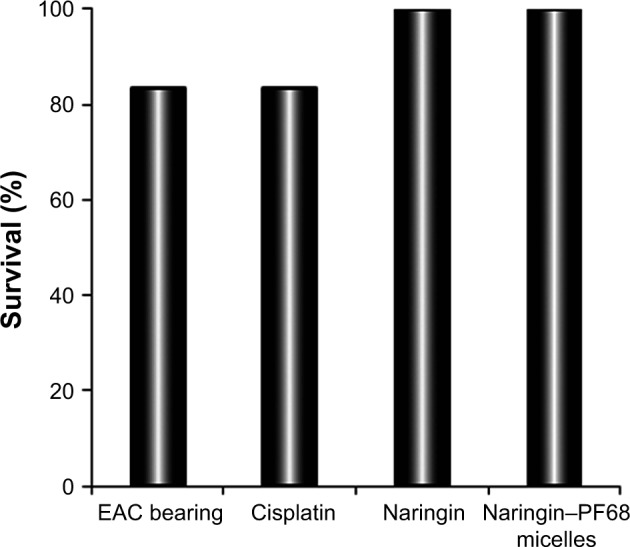
Survival (%) of mice of different groups at the end of tumor growth study.
Abbreviations: EAC, Ehrlich ascites carcinoma; PF68, pluronic F68.
The superiority of PF68 micelles of naringin over the free drug indicated by the significantly improved percentage inhibition of tumor growth may suggest it as a novel promising drug delivery system of naringin for the treatment of tumor. Potentiation of in vivo anticancer effect of many drugs as paclitaxel and doxorubicin in different tumor models has been recorded for formulations with pluronics.82,84–86
Conclusion
Naringin–PF68 micelles were dispersed spherical particles with nanoscopic diameter <100 nm and narrow size distribution suggesting prolonged circulation times and facilitated access to cells and tissues. A total of 1:50 naringin–PF68 showed the highest drug entrapment. The encapsulation of drug within these micelles was indicated by results of FT-IR, DSC, and XRD. The micelles provided extended release up to 48 vs 10 h for free naringin in different pH release media. These nanomicelles potentiated naringin cytoprotection against ethanol-induced ulcer in rats with dose reduction as reflected by minimized mucosal damage, oxidative stress, and gastric levels of TNF-α, caspase-3, NF-κB, and IL-6. As well, improved antitumor activity has been recorded by enhanced in vitro cytotoxicity against HepG2, Caco-2, and MCF-7 cell lines and tumor inhibition in EAC-bearing mice. Therefore, 1:50 polymeric micelles with PF68 might be represented as a promising nanocarrier of the phytopharmaceutical naringin with prolonged release as well as enhanced antiulcer and antitumor activities encouraging their clinical investigation as alternative of the currently available treatment regimens of ulcer and cancer that exhibited some side effects.
Acknowledgments
The authors are grateful for the technical support from MERC “Medical Experimental Research Center”, Mansoura University, and would like to thank Professor Dr M Sobh, Dr H Sheash, and Mr Husam Eid for their technical support during cytotoxicity assays.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Bharti S, Rani N, Krishnamurthy B, Arya DS. Preclinical evidence for the pharmacological actions of naringin: a review. Planta Med. 2014;80(6):437–451. doi: 10.1055/s-0034-1368351. [DOI] [PubMed] [Google Scholar]
- 2.Rouseff RL, Martin SF, Youtsey CO. Quantitative survey of narirutin, naringin, hesperidin, and neohesperidin in citrus. J Agric Food Chem. 1987;35(6):1027–1030. [Google Scholar]
- 3.Benavente-García O, Castillo J. Update on uses and properties of citrus flavonoids: new findings in anticancer, cardiovascular, and anti-inflammatory activity. J Agric Food Chem. 2008;56(15):6185–6205. doi: 10.1021/jf8006568. [DOI] [PubMed] [Google Scholar]
- 4.Chanet A, Milenkovic D, Manach C, Mazur A, Morand C. Citrus flavanones: what is their role in cardiovascular protection? J Agric Food Chem. 2012;60(36):8809–8822. doi: 10.1021/jf300669s. [DOI] [PubMed] [Google Scholar]
- 5.Choi BS, Sapkota K, Kim S, Lee HJ, Choi HS, Kim SJ. Antioxidant activity and protective effects of Tripterygium regelii extract on hydrogen peroxideinduced injury in human dopaminergic cells, SH-SY5Y. Neurochem Res. 2010;35(8):1269–1280. doi: 10.1007/s11064-010-0185-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li P, Wang S, Guan X, et al. Acute and 13 weeks subchronic toxicological evaluation of naringin in Sprague-Dawley rats. Food Chem Toxicol. 2013;60:1–9. doi: 10.1016/j.fct.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 7.Wallace JL, Granger DN. The cellular and molecular basis of gastric mucosal defense. FASEB J. 1996;10(7):731–740. doi: 10.1096/fasebj.10.7.8635690. [DOI] [PubMed] [Google Scholar]
- 8.Konturek SJ, Konturek PC, Pawlik T, Sliwowski Z, Ochmański W, Hahn EG. Duodenal mucosal protection by bicarbonate secretion and its mechanisms. J Physiol Pharmacol. 2004;55(Suppl 2):5–17. [PubMed] [Google Scholar]
- 9.Bruntan LL, Lazo JS, Parker KL. Goodman and Gilman’s: The Pharmacological Basis of Therapeutics. 11th ed. New York: McGraw Hill Companies; 2006. [Google Scholar]
- 10.Martin MJ, Marhuenda E, Pérez-Guerrero C, Franco JM. Antiulcer effect of naringin on gastric lesions induced by ethanol in rats. Pharmacology. 1994;49(3):144–150. doi: 10.1159/000139228. [DOI] [PubMed] [Google Scholar]
- 11.Borrelli F, Izzo AA. The plant kingdom as a source of anti-ulcer remedies. Phytother Res. 2000;14(8):581–591. doi: 10.1002/1099-1573(200012)14:8<581::aid-ptr776>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 12.Galati EM, Monforte MT, d’Aquino A, Miceli N, Di Mauro D, Sanogo R. Effects of naringin on experimental ulcer in rats. Phytomedicine. 1998;5(5):361–366. doi: 10.1016/S0944-7113(98)80018-4. [DOI] [PubMed] [Google Scholar]
- 13.Kanno S, Shouji A, Tomizawa A, et al. Inhibitory effect of naringin on lipopolysaccharide (LPS)-induced endotoxin shock in mice and nitric oxide production in RAW 264.7 macrophages. Life Sci. 2006;78(7):673–681. doi: 10.1016/j.lfs.2005.04.051. [DOI] [PubMed] [Google Scholar]
- 14.Yokoyama M. Polymeric micelles drug carriers for tumor targeting. In: Svenson S, editor. Polymeric Drug Delivery. Particulate Drug Carriers. Washington, DC: ACS Publications; 2006. pp. 27–36. [Google Scholar]
- 15.Vanamala J, Leonardi T, Patil BS, et al. Suppression of colon carcinogenesis by bioactive compounds in grapefruit. Carcinogenesis. 2006;27(6):1257–1265. doi: 10.1093/carcin/bgi318. [DOI] [PubMed] [Google Scholar]
- 16.Li H, Yang B, Huang J, et al. Naringin inhibits growth potential of human triple-negative breast cancer cells by targeting β-catenin signaling pathway. Toxicol Lett. 2013;220(3):219–228. doi: 10.1016/j.toxlet.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 17.Schindler R, Mentlein R. Flavonoids and vitamin E reduce the release of the angiogenic peptide vascular endothelial growth factor from human tumor cells. J Nutr. 2006;136(6):1477–1482. doi: 10.1093/jn/136.6.1477. [DOI] [PubMed] [Google Scholar]
- 18.Thangavel P, Muthu R, Vaiyapuri M. Antioxidant potential of naringin–a dietary flavonoid–in N-nitrosodiethylamine induced rat liver carcinogenesis. Biomed Prev Nutr. 2012;2(3):193–202. [Google Scholar]
- 19.D’souza P, Deepak M, Rani P, et al. Brine shrimp lethality assay of Bacopa monnieri. Phytother Res. 2002;16(2):197–198. doi: 10.1002/ptr.1001. [DOI] [PubMed] [Google Scholar]
- 20.Waris G, Ahsan H. Reactive oxygen species: role in the development of cancer and various chronic conditions. J Carcinog. 2006;5:14–21. doi: 10.1186/1477-3163-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsiu SL, Huang TY, Han YC, Ching DH, Lee Chao PD. Comparison of metabolic pharmacokinetics of naringin and naringenin in rabbits. Life Sci. 2002;70(13):1481–1489. doi: 10.1016/s0024-3205(01)01491-6. [DOI] [PubMed] [Google Scholar]
- 22.Spencer JP, Chowrimootoo G, Choudhury R, Debnam ES, Srai SK, Rice Evans C. The small intestine can both absorb and glucoronidate luminal flavonoids. FEBS Lett. 1999;458(2):224–230. doi: 10.1016/s0014-5793(99)01160-6. [DOI] [PubMed] [Google Scholar]
- 23.Rechner AR, Smith MA, Kuhnle G, et al. Colonic metabolism of dietary polyphenols: influence of structure on microbial fermentation products. Free Radic Biol Med. 2004;36(2):212–225. doi: 10.1016/j.freeradbiomed.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 24.Lauro MR, De Simone F, Sansone F, Iannelli P, Aquino RP. Preparations and release characteristics of naringin and naringenin gastro-resistant microparticles by spray-drying. J Drug Del Sci Tech. 2007;17(2):119–124. [Google Scholar]
- 25.Chakraborty S, Stalin S, Das N, Choudhury ST, Ghosh S, Swarnakar S. The use of nano-quercetin to arrest mitochondrial damage and MMP-9 upregulation during prevention of gastric inflammation induced by ethanol in rat. Biomaterials. 2012;33(10):2991–3001. doi: 10.1016/j.biomaterials.2011.12.037. [DOI] [PubMed] [Google Scholar]
- 26.Krishnakumara N, Sulfikkarali N, RajendraPrasad N, Karthikeyan S. Enhanced anticancer activity of naringenin-loaded nanoparticles in human cervical (HeLa) cancer cells. Biomed Prev Nutr. 2011;1(4):223–231. [Google Scholar]
- 27.Li N, Zhao L, Qi L, Li Z, Luan Y. Polymer assembly: promising carriers as co-delivery systems for cancer therapy. Prog Polym Sci. 2016;58:1–26. [Google Scholar]
- 28.Zhao L, Li N, Wang K, Shi C, Zhang L, Luan Y. A review of polypeptide-based polymersomes. Biomaterials. 2014;35(4):1284–1301. doi: 10.1016/j.biomaterials.2013.10.063. [DOI] [PubMed] [Google Scholar]
- 29.Shi C, Zhang Z, Shi J, Wang F, Luan Y. Co-delivery of docetaxel and chloroquine via PEO-PPO-PCL/TPGS micelles for overcoming multidrug resistance. Int J Pharm. 2015;495(2):932–939. doi: 10.1016/j.ijpharm.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 30.Guo Y, He W, Yang S, Zhao D, Li Z, Luan Y. Co-delivery of docetaxel and verapamil by reduction-sensitive PEG-PLGA-SS-DTX conjugate micelles to reverse the multi-drug resistance of breast cancer. Colloids Surf B Biointerfaces. 2017;151:119–127. doi: 10.1016/j.colsurfb.2016.12.012. [DOI] [PubMed] [Google Scholar]
- 31.Feng X, Ding J, Gref R, Chen X. Poly(β-cyclodextrin)-mediated polylactide-cholesterol stereocomplex micelles for controlled drug delivery. Chin J Polymer Sci. 2017;35(6):693–699. [Google Scholar]
- 32.Li D, Ding J, Zhuang X, Chen L, Chen X. Drug binding rate regulates the properties of polysaccharide prodrugs. J Mater Chem B. 2016;4(30):5167–5177. doi: 10.1039/c6tb00991c. [DOI] [PubMed] [Google Scholar]
- 33.Xu W, Ding J, Chen X. Reduction-responsive polypeptide micelles for intracellular delivery of antineoplastic agent. Biomacromolecules. 2017;18(10):3291–3301. doi: 10.1021/acs.biomac.7b00950. [DOI] [PubMed] [Google Scholar]
- 34.Batrakova EV, Kabanov AV. Pluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J Control Release. 2008;130(2):98–106. doi: 10.1016/j.jconrel.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamprecht A, Ubrich N, Yamamoto H, et al. Design of rolipram loaded nanoparticles: comparison of two preparation methods. J Control Release. 2001;71(3):297–306. doi: 10.1016/s0168-3659(01)00230-9. [DOI] [PubMed] [Google Scholar]
- 36.Batrakova EV, Li S, Brynskikh AM, et al. Effects of pluronic and doxorubicin on drug uptake, cellular metabolism, apoptosis and tumor inhibitionin animal models of MDR cancers. J Control Release. 2010;143(3):290–301. doi: 10.1016/j.jconrel.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Croy SR, Kwon GS. Polymeric micelles for drug delivery. Curr Pharm Des. 2006;12(36):4669–4684. doi: 10.2174/138161206779026245. [DOI] [PubMed] [Google Scholar]
- 38.Kwon GS. Polymeric micelles for delivery of poorly water-soluble compounds. Crit Rev Ther Drug Carr Syst. 2003;20(5):357–403. doi: 10.1615/critrevtherdrugcarriersyst.v20.i5.20. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Tang L, Sun L, et al. A novel paclitaxel-loaded poly(epsilon-caprolactone)/Poloxamer 188 blend nanoparticle overcoming multidrug resistance for cancer treatment. Acta Biomater. 2010;6(6):2045–2052. doi: 10.1016/j.actbio.2009.11.035. [DOI] [PubMed] [Google Scholar]
- 40.Zhang W, Shi Y, Chen Y, Ye J, Sha X, Fang X. Multifunctional Pluronic P123/F127 mixed polymeric micelles loaded with paclitaxel for the treatment of multidrug resistant tumors. Biomaterials. 2011;32(11):2894–2906. doi: 10.1016/j.biomaterials.2010.12.039. [DOI] [PubMed] [Google Scholar]
- 41.Dabholkar RD, Sawant RM, Mongayt DA, Devarajan PV, Torchilin VP. Polyethylene glycol-phosphatidylethanolamine conjugate (PEG-PE)-based mixed micelles: some properties, loading with paclitaxel, and modulation of P-glycoprotein-mediated efflux. Int J Pharm. 2006;315(1–2):148–157. doi: 10.1016/j.ijpharm.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 42.Wei Z, Hao J, Yuan S, et al. Paclitaxel-loaded Pluroic P123/F127 mixed polymeric micelles: formulation, optimization and in vitro characterization. Int J Pharm. 2009;376(1–2):176–185. doi: 10.1016/j.ijpharm.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 43.Zu Y, Wang D, Zhao X, et al. A novel preparation method for camptothecin (CPT) loaded folic acid conjugated dextran tumor-targeted nanoparticles. Int J Mol Sci. 2011;12(7):4237–4249. doi: 10.3390/ijms12074237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krishnaiah YS, Bhaskar Reddy PR, Satyanarayana V, Karthikeyan RS. Studies on the development of oral colon targeted drug delivery systems for metronidazole in the treatment of amoebiasis. Int J Pharm. 2002;236(1–2):43–55. doi: 10.1016/s0378-5173(02)00006-6. [DOI] [PubMed] [Google Scholar]
- 45.Li Y, Bi Y, Xi Y, Li L. Enhancement on oral absorption of paclitaxel by multifunctional pluronic micelles. J Drug Target. 2013;21(2):188–199. doi: 10.3109/1061186X.2012.737001. [DOI] [PubMed] [Google Scholar]
- 46.Higuchi T. Mechanism of sustained action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52:1145–1149. doi: 10.1002/jps.2600521210. [DOI] [PubMed] [Google Scholar]
- 47.Ritger PL, Peppas NA. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Control Release. 1987;5(1):23–36. [PubMed] [Google Scholar]
- 48.Huang CC, Chen YM, Wang DC, et al. Cytoprotective effect of American ginseng in a rat ethanol gastric ulcer model. Molecules. 2013;19(1):316–326. doi: 10.3390/molecules19010316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.El-Maraghy SA, Rizk SM, Shahin NN. Gastroprotective effect of crocin in ethanol-induced gastric injury in rats. Chem Biol Interact. 2015;229:26–35. doi: 10.1016/j.cbi.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 50.Arab HH, Salama SA, Omar HA, Ammar el-SM, Maghrabi IA. Diosmin protects against ethanol-induced gastric injury in rats: novel anti-ulcer actions. PLoS One. 2015;10(3):e0122417. doi: 10.1371/journal.pone.0122417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krylova SG, Khotimchenko YS, Zueva EP, et al. Gastroprotective effect of natural non-starch polysaccharides. Bull Exp Biol Med. 2006;142(4):454–457. doi: 10.1007/s10517-006-0390-y. [DOI] [PubMed] [Google Scholar]
- 52.Elmowafy EM, Awad GAS, Mansour S, El-Shamy AA. Ionotropically emulsion gelled polysaccharides beads: preparation, in vitro and in vivo evaluation. Carbohyd Polym. 2009;75(1):135–142. [Google Scholar]
- 53.Yusif RM, Abu Hashim II, Mohamed EA. Gastroretentive matrix tablets of boswellia oleogum resin: preparation, optimization, in vitro evaluation, and cytoprotective effect on indomethacin-induced gastric ulcer in rabbits. AAPS PharmSciTech. 2016;17(2):328–338. doi: 10.1208/s12249-015-0351-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laine L, Weinstein WM. Histology of alcoholic hemorrhagic “gastritis”: a prospective evaluation. Gastroenterology. 1988;94(6):1254–1262. doi: 10.1016/0016-5085(88)90661-0. [DOI] [PubMed] [Google Scholar]
- 55.Bancroft JD, Stevens A, Turner DR. Theory and Practice of Histological Techniques. 4th ed. New York: Churchill Livingstone; 1996. [Google Scholar]
- 56.El-Sheakh AR, Ghoniem HA, Suddek GM, Ammar el-SM. Antioxidant and anti-inflammatory effects of flavocoxid in high-cholesterol-fed rabbits. Naunyn Schmiedebergs Arch Pharmacol. 2015;388(12):1333–1344. doi: 10.1007/s00210-015-1168-4. [DOI] [PubMed] [Google Scholar]
- 57.Atmaca G. Antioxidant effects of sulfur-containing amino acids. Yonsei Med J. 2004;45(5):776–788. doi: 10.3349/ymj.2004.45.5.776. [DOI] [PubMed] [Google Scholar]
- 58.Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82(1):70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 59.Marklund SL. Superoxide dismutase isoenzymes in tissues and plasma from New Zealand black mice, nude mice and normal BALB/c mice. Mutat Res. 1985;148(1–2):129–134. doi: 10.1016/0027-5107(85)90216-7. [DOI] [PubMed] [Google Scholar]
- 60.Naresh RRA, Udupa N, Uma DP. Effect of macrophage activation on niosome encapsulated bleomycin in tumor bearing mice. Ind J Pharamcol. 1996;28(3):175–180. [Google Scholar]
- 61.Schirner M, Hoffmann J, Menrad A, Schneider MR. Antiangiogenic chemotherapeutic agents: characterization in comparison to their tumor growth inhibition in human renal cell carcinoma models. Clin Cancer Res. 1998;4(5):1331–1336. [PubMed] [Google Scholar]
- 62.Goel MK, Khanna P, Kishore J. Understanding survival analysis: Kaplan-Meier estimate. Int J Ayurveda Res. 2010;1(4):274–278. doi: 10.4103/0974-7788.76794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allen C, Maysinger D, Eisenberg A. Nano-engineering block copolymer aggregates for drug delivery. Colloids Surf B Biointerfaces. 1999;16(1–4):3–27. [Google Scholar]
- 64.Sezgin Z, Yüksel N, Baykara T. Preparation and characterization of polymeric micelles for solubilization of poorly soluble anticancer drugs. Eur J Pharm Biopharm. 2006;64(3):261–268. doi: 10.1016/j.ejpb.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 65.Liu Z, Liu D, Wang L, Zhang J, Zhang N. Docetaxel-loaded pluronic P123 polymeric micelles: in vitro and in vivo evaluation. Int J Mol Sci. 2011;12(3):1684–1696. doi: 10.3390/ijms12031684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41(1):189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 67.Fissan H, Ristig S, Kaminski H, Asbach C, Epple M. Comparison of different characterization methods for nanoparticle dispersions before and after aerosolization. Anal Methods. 2014;6(18):7324–7334. [Google Scholar]
- 68.Eaton P, Quaresma P, Soares C, et al. A direct comparison of experimental methods to measure dimensions of synthetic nanoparticles. Ultramicroscopy. 2017;182:179–190. doi: 10.1016/j.ultramic.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 69.Sahu N, Soni D, Chandrashekhar B, et al. Synthesis of silver nanoparticles using flavonoids: hesperidin, naringin and diosmin, and their antibacterial effects and cytotoxicity. Int Nano Lett. 2016;6(3):173–181. [Google Scholar]
- 70.Sansone F, Aquino RP, Del Gaudio P, Colombo P, Russo P. Physical characteristics and aerosol performance of naringin dry powders for pulmonary delivery prepared by spray-drying. Eur J Pharm Biopharm. 2009;72(1):206–213. doi: 10.1016/j.ejpb.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 71.Gao Y, Li LB, Zhai G. Preparation and characterization of pluronic/TPGS mixed micelles for solubilization of camptothecin. Colloids Surf B Biointerfaces. 2008;64(2):194–199. doi: 10.1016/j.colsurfb.2008.01.021. [DOI] [PubMed] [Google Scholar]
- 72.Wang Y, Li Y, Wang Q, Wu J, Fang X. Pharmacokinetics and biodistribution of paclitaxel loaded-pluronic P105/L101 mixed polymeric micelles. Yakugaku Zasshi. 2008;128(6):941–950. doi: 10.1248/yakushi.128.941. [DOI] [PubMed] [Google Scholar]
- 73.Mohamed EA, Abu Hashim II, Yusif RM, Suddek GM, Shaaban AA, Badria FA. Enhanced in vitro cytotoxicity and anti-tumor activity of vorinostat-loaded pluronic micelles with prolonged release and reduced hepatic and renal toxicities. Eur J Pharm Sci. 2017;96:232–242. doi: 10.1016/j.ejps.2016.09.029. [DOI] [PubMed] [Google Scholar]
- 74.Teng Y, Morrison ME, Munk P, Webber SE, Procházka K. Release kinetics studies of aromatic molecules into water from block polymer micelles. Macromolecules. 1998;31(11):3578–3587. [Google Scholar]
- 75.Sahu A, Kasoju N, Goswami P, Bora U. Encapsulation of curcumin in pluronic block copolymer micelles for drug delivery applications. J Biomater Appl. 2011;25(6):619–639. doi: 10.1177/0885328209357110. [DOI] [PubMed] [Google Scholar]
- 76.Chowdhary RK, Chansarkar N, Sharif I, Hioka N, Dolphin D. Formulation of benzoporphyrin derivatives in pluronics. Photochem Photobiol. 2003;77(3):299–303. doi: 10.1562/0031-8655(2003)077<0299:fobdip>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 77.Cortivo R, Vindigni V, Iacobellis L, Abatangelo G, Pinton P, Barbara Zavan B. Nanoscale particle therapies for wounds and ulcers. Nanomedicine. 2010;5(4):641–656. doi: 10.2217/nnm.10.32. [DOI] [PubMed] [Google Scholar]
- 78.Selvaraj S, Krishnaswamy S, Devashya V, et al. Investigations on the membrane interactions of naringin and its complexes with copper and iron: implications for their cytotoxicity. RSC Adv. 2014;4:46407–46417. [Google Scholar]
- 79.Guo D, Wang J, Wang X, et al. Double directional adjusting estrogenic effect of naringin from Rhizoma drynariae (Gusuibu) J Ethnopharmacol. 2011;138(2):451–457. doi: 10.1016/j.jep.2011.09.034. [DOI] [PubMed] [Google Scholar]
- 80.Gao Z, Zhang L, Sun Y. Nanotechnology applied to overcome tumor drug resistance. J Control Release. 2012;162(1):45–55. doi: 10.1016/j.jconrel.2012.05.051. [DOI] [PubMed] [Google Scholar]
- 81.Ji X, Gao Y, Chen L, Zhang Z, Deng Y, Li Y. Nanohybrid systems of non-ionic surfactant inserting liposomes loading paclitaxel for reversal of multidrug resistance. Int J Pharm. 2012;422(1–2):390–397. doi: 10.1016/j.ijpharm.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 82.Alakhov V, Klinski E, Li S, et al. Block copolymer-based formulation of doxorubicin. From cell screen to clinical trials. Colloids Surf B Biointerfaces. 1999;16(1–4):113–134. [Google Scholar]
- 83.Lee Y, Park SY, Mok H, Park TG. Synthesis, characterization, antitumor activity of pluronic mimicking copolymer micelles conjugated with doxorubicin via acid-cleavable linkage. Bioconjug Chem. 2008;19(2):525–531. doi: 10.1021/bc700382z. [DOI] [PubMed] [Google Scholar]
- 84.Alakhov VY, Moskaleva EY, Batrakova EV, Kabanov AV. Hyper-sensitization of multidrug resistant human ovarian carcinoma cells by Pluronic P85 block copolymer. Bioconjug Chem. 1996;7(2):209–216. doi: 10.1021/bc950093n. [DOI] [PubMed] [Google Scholar]
- 85.Batrakova EV, Dorodnych TY, Klinskii EY, et al. Anthracycline antibiotics non-covalently incorporated into the block copolymer micelles: in vivo evaluation of anti-cancer activity. Br J Cancer. 1996;74(10):1545–1552. doi: 10.1038/bjc.1996.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Venne A, Li S, Mandeville R, Kabanov A, Alakhov V. Hypersensitizing effect of pluronic L61 on cytotoxic activity, transport, and subcellular distribution of doxorubicin in multiple drug-resistant cells. Cancer Res. 1996;56(16):3626–3629. [PubMed] [Google Scholar]