

SUMMARY
The ability of Porphyromonas gingivalis to overcome oxidative stress in the inflammatory environment of the periodontal pocket is critical for its survival. We have previously demonstrated that the recA locus, which carries the bacterioferritin co-migratory protein (bcp) gene and has a unique genetic architecture, plays a role in virulence regulation and oxidative stress resistance in P. gingivalis. To further characterize the bcp gene, which was confirmed to be part of the bcp-recA-vimA-vimE-vimF operon, we created a P. gingivalis bcp-defective isogenic mutant (FLL302) by allelic exchange. Compared with the wild-type, FLL302 had a similar growth rate, black pigmentation, β-hemolysis and UV sensitivity. Although there was no change in the distribution of gingipain activity, there was a 30% reduction in both Arg-X and Lys-X activities in the mutant strain compared with the wild-type. When exposed to 0.25 mM hydrogen peroxide, P. gingivalis FLL302 was more sensitive than the wild-type. In addition, the cloned P. gingivalis bcp gene increased resistance to 0.25 mM hydrogen peroxide in a bcp-defective Escherichia coli mutant. The mutant also demonstrated decreased aerotolerance when compared with the wild-type. Porphyromonas gingivalis FLL302 and the wild-type strain had similar virulence profiles in a mouse model of virulence. These observations suggest that the bcp gene may play a role in oxidative stress resistance but has a decreased functional significance in the pathogenic potential of P. gingivalis.
Keywords: ahpC, alkyl hydroperoxide reductase, bacterioferritin co-migratory protein, oxidative stress, virulence
INTRODUCTION
Porphyromonas gingivalis is a black-pigmented, gram-negative bacillus commonly identified as a major etiological agent in adult periodontitis. This periodontal pathogen possesses several virulence factors (e.g. hydrolytic enzymes, fimbriae, hemagglutinin, capsule and lipopolysaccharide) that can directly affect the periodontium or elicit host functions that result in destruction typical of advanced periodontitis (Genco & Slots, 1984; Mayrand & Holt, 1988; Nakayama, 1994). Survival of this organism in the periodontal pocket would necessitate mechanisms to surmount oxidative stress caused by exposure to air or reactive oxygen intermediates such as hydrogen peroxide (H2O2), superoxide and the hydroxyl radical (OH−) generated by neutrophils and macrophages (Chapple, 1996; Salyers, 1994). Oxidative stress is defined as a perturbation in the balance between pro-oxidants and antioxidants in favor of the pro-oxidants. Two major mechanisms used by bacteria to overcome oxidative stress include the use of (i) endonuclease gene products encoded by nei, nth, fpg and mutY (Michaels & Miller, 1992; Grollman & Moriya, 1993; Nash et al., 1996; Radicella & Boiteux, 1997; Boiteux & Radicella, 1999) and (ii) antioxidant enzymes such as superoxide dismutase, alkyl hydroperoxidase subunits C and F (AhpC/F) and catalase (Amano et al., 1986, 1992; Nakayama, 1994; Baillon et al., 1999; Rocha & Smith, 1999; Springer et al., 2001). Toxic oxygen metabolites can be neutralized by superoxide dismutase, catalase and peroxidase, all of which are generally expressed by aerobic and many anaerobic bacteria (Beaman & Beaman, 1984; Rocha et al., 1996). Although P. gingivalis is oxygen tolerant (Amano et al., 1988) and expresses superoxide dismutase activity (Nakayama, 1990; Choi et al., 1991; Nakayama, 1994), it is missing a catalase gene (Amano et al., 1986; Nakayama, 1990, 1994; Choi et al., 1991; Nelson et al., 2003). However, protection against oxidative damage may also use another unique mechanism in P. gingivalis. Cell surface heme acquisition has been postulated to be a defense mechanism against reactive oxygen species in P. gingivalis (Smalley et al., 1998). The storage of the heme on the cell surface, which gives the organism its characteristic black pigmentation, can form μ-oxo dimers in the presence of reactive oxygen species and can give rise to the catalytic degradation of hydrogen peroxide (Smalley et al., 1998, 2000).
In addition to the aforementioned antioxidant enzymes, recent reports have demonstrated a role for the bacterioferritin co-migratory protein (Bcp) in oxidative stress resistance (Jeong et al., 2000; Kong et al., 2000; Wang et al., 2005; Limauro et al., 2008). The Bcp is part of the thiol-specific antioxidant/alkyl hydroperoxidase family and functions in a similar way to AhpC in detoxifying hydrogen peroxide (Jeong et al., 2000). The bcp gene in P. gingivalis has been identified upstream of the recA, vimA, vimE and vimF genes and could be part of the same operon (Nelson et al., 2003). Previous reports have attributed the function of DNA repair and protease activation and maturation to the genes in this operon (Abaibou et al., 2001; Fletcher et al., 1997; Vanterpool et al., 2004, 2005a,b). To determine a role for the P. gingivalis bcp gene in oxidative stress resistance, we cloned and expressed this gene in Escherichia coli, and created a P. gingivalis bcp-defective mutant by allelic exchange. We now report that the bcp homologue in P. gingivalis may play an important role in protecting this organism against toxic hydrogen peroxide but does not demonstrate a discernible difference in virulence in the mouse model tested.
METHODS
Bacterial strains and culture conditions
Porphyromonas gingivalis W83 was grown in brain–heart infusion (BHI) broth (Difco Laboratories, Detroit, MI) supplemented with hemin (5 μg ml−1), and cysteine (0.1%). The E. coli strains were grown in Luria–Bertani (LB) broth. Unless otherwise stated, all cultures were incubated at 37 °C. The P. gingivalis was maintained in an anaerobic chamber (Coy Manufacturing, Ann Arbor, MI) in 10% H2, 10% CO2 and 80% N2. Growth rates for P. gingivalis strains were determined spectrophotometrically (optical density at 600 nm). Hemolysis and pigmentation were determined by incubation of P. gingivalis on Brucella Blood agar (Anaerobe Systems, Morgan Hill, CA) for 7–10 days.
DNA isolation and analysis
The P. gingivalis chromosomal DNA was prepared by the method of Marmur (1961). For plasmid DNA analysis, DNA extraction was performed following the alkaline lysis procedure of Birnboim & Doly (1979). For large-scale preparation, plasmids were purified using the Qiagen plasmid midi kit as per the manufacturer’s instructions (Qiagen, Valencia, CA). DNA was digested by restriction enzymes as specified by the manufacturer (Boehringer Mannheim Corporation, Indianapolis, IN). DNA fragments were separated by electrophoresis [1% agarose; TAE buffer (0.4 M Tris–acetate, 0.001 M EDTA, pH 8.0)] and purified using the Geneclean II kit according to the manufacturer’s recommendations (Bio 101 Inc., La Jolla, CA). Porphyromonas gingivalis chromosomal DNA and the plasmid pVA2198 were digested with BamHI and PstI (Roche, Indianapolis, IN), respectively, as specified by the manufacturer. Southern blot alkaline transfer was performed according to the method of Chomczynski (1992). The polymerase chain reaction (PCR)-amplified 0.5-kilobase (kb) bcp (P1 and P2, Table 1) and 2.1-kb ermF-ermAM (P3 and P4) genes were labeled with digoxigenin and used as probes in hybridization experiments. DNA labeling, hybridization and detection were performed using the digoxigenin High prime Labeling and Detection Starter kit II (Roche) according to the manufacturer’s instructions.
Table 1.
Oligonucleotide primers used in this study
| Primers | Oligonucleotide sequence | Characteristics | References |
|---|---|---|---|
| P1 | 5′ ATGACCTTTTTGTTACCTTTG 3′ | bcp 0.5 LEFT | This study |
| P2 | 5′ TTATCCGTTTAGTATCTGATC 3′ | bcp 0.5 RIGHT | This study |
| P3 | 5′ TATTAGGCCTATAGCTTCCGCTATT 3′ | ermStu LEFT | This study |
| P4 | 5′ AATTAGGCCTTAGTAACGTGTAACTTT 3′ | ermStu RIGHT | This study |
| P5 | 5′ CGTTCCCGTCTTGGTCTATC3′ | bcpPrimEx2 | This study |
| P6 | 5′GCUGAUGGCGAUGAAUGAACACUGCGUUUGCUGGCUUUGAUGAAA 3′ | 5′ RACE Adapter | FirstChoice® RLM-RACE Kit Manual |
| P7 | 5′GCTGATGGCGAATGAACACTG 3′ | 5′ RACE Outer primer | FirstChoice® RLM-RACE Kit Manual |
| P8 | 5′CGCGGATCCGAACACTGCGTTTGCTGGCTTTGATG 3′ | 5′ RACE Inner primer | FirstChoice® RLM-RACE Kit Manual |
| P9 | 5′ ATGATACAAATAGGAGACCGT 3′ | bcp ORF L | This study |
| P10 | 5′ TATGTTTATCCTCCGGAATAAGGACTCTAAGAACCCTAT 3′ | bcp M39L | This study |
| P11 | 5′ ATACAAATAGGAGGCCTTATTCCTGAGATTCTTGGGATA 3′ | bcp M39R | This study |
| P12 | 5′ CTCTAAGGCAAATTTAGGAAG 3′ | bcp LEFT | This study |
| P13 | 5′ GATGAAGATACAGGTTGTATTG 3′ | bcp RIGHT | This study |
| P14 | 5′ TACCTGTTTTTGCTGACCGG 3′ | vimA reverse | Abaibou et al., (2001) |
| P15 | 5′ ATGCCCATCCCTCTATACCTG 3′ | vimA forward | Abaibou et al., (2001) |
| P16 | 5′ ATGGCAGAAGAAAAGATACCC 3′ | recA forward | Fletcher et al., (1997) |
| P17 | 5′ TGAATGTTTGTTGCGAATGG 3′ | recA reverse | Fletcher et al., (1997) |
DNA sequencing
Nucleotide sequences were determined by the dideoxy-chain termination method (Devereux et al., 1984) at the DNA core facility of Loma Linda University (Loma Linda, CA) or at Retrogen (Retrogen, Inc., San Diego, CA). Oligonucleotide primers used in sequencing reactions were obtained from Invitrogen (Invitrogen Corp., Carlsbad, CA) and IDT (Integrated DNA Technologies, Coralville, IA). Nucleotide sequences were analysed using the SEQUENCER software package (Gene Codes Corporation, Ann Arbor, MI).
PCR analysis of RNase-treated chromosomal DNA from P. gingivalis
The PCR amplification was performed with the Perkin Elmer Cetus DNA Thermal Cycler (Perkin-Elmer Corporation, Norwalk, CT). The primers used in this study (Table 1) were synthesized at the Nucleic Acid Core Facility at Loma Linda University. The reaction mixture (50 μl), containing 1 μl template DNA (0.5 ng), 1 μM each primer, 0.2 mM dNTPs in 1× Expand High Fidelity System buffer, was denatured for 2 min at 94 °C, then 1.73 U Expand High Fidelity System enzyme was added (Roche). The PCR consisted of 30 cycles with a temperature profile of 94 °C for 30 s, 55 °C for 1 min and 72 °C for 2 min. The final extension was performed at 72 °C for 7 min. The PCR-amplified DNA was then identified by 1% agarose gel electrophoresis.
Reverse transcriptase–polymerase chain reaction analysis of DNase-treated RNA extracted from P. gingivalis
Total RNA was extracted from P. gingivalis W83 and P. gingivalis FLL302 grown to mid-log phase [optical density at 600 nm (OD600) of 0.7] using the RNAwiz RNA kit (Ambion, Austin, TX) as previously described (Johnson et al., 2004). Extracted RNA was subjected to DNase treatment using the DNA-free™ Kit (Ambion) according to the manufacturer’s protocol. Reverse transcription and PCR amplification were performed with a Perkin-Elmer Cetus DNA thermal Cycler (Perkin Elmer Corporation) also as previously described (Abaibou et al., 2001). The final products were analysed by 1% agarose gel electrophoresis.
Transcription start site (TSS) mapping of the bcp gene
To map the 5′ region of the bcp transcript, the First-Choice® RLM-RACE kit (Ambion) was used according to the defined protocols. Total RNA harvested from P. gingivalis grown to mid-log (OD600 ~0.6) was sequentially treated with calf intestinal phosphatase and tobacco acid phosphatase. A 5′ adapter sequence (P6) was ligated to the treated RNA by T4 RNA ligase before the sequence was reverse transcribed. An outer PCR using the 5′RACE outer primer (P7) and the bcp reverse primer (P2) was carried out. A nested PCR using the product from the outer PCR as template was performed using the 5′RACE inner primer (P8) and the same bcp reverse primer (P2). Minus template and minus tobacco acid phosphatase reactions were run as controls. The resulting inner PCR products were sequenced to reveal the TSS, which would be the nucleotide immediately upstream of the ligated adapter sequence.
Cloning and expression of the P. gingivalis bcp gene
Two possible open reading frames (ORF) for the bcp gene were amplified using primer pairs P1 and P2 or P9 and P2 by PCR. The amplified fragments for each ORF were TOPO® TA cloned into the pTrcHis2 expression vector (Invitrogen) and transformed into E. coli TOP10 cells. The transformed E. coli cells were incubated with shaking at 37 °C for 1 h and 50-μl aliquots were plated on LB agar plates containing ampicillin (50 μg ml−1) and incubated overnight at 37°C. Plasmid DNA was extracted from ampicillin-resistant colonies and digested with EcoRI (New England Biolabs, Beverly, MA) to confirm the orientation of the bcp gene. Two plasmids with the bcp gene cloned in opposite orientations relative to each other for each bcp ORF were chosen and designated pFLL301.3 or pFLL301.4 (forward orientation) and pFLL301.2 or pFLL301.5 (reverse orientation). The expression of the bcp gene was then induced for up to 5 h using 1 mM isopropyl-β-D-galactopyranoside (IPTG). Sodium dodecyl sulfate–polyacrylamide gel electrophoresis was then performed with a 4–12% Bis–Tris separating gel (Invitrogen). The samples were electrophoresed at a constant voltage of 200 V for 45 min then stained with SimplyBlue SafeStain (Invitrogen) for 1 h, destained in distilled water for 30 min and visualized.
Complementation of E. coli bcp-defective mutant
The E. coli KD2301 (Jeong et al., 2000), a bcp-defective mutant, was transformed by electroporation with the plasmid pFLL301.3 or pFLL301.4 (carrying the bcp gene in the forward orientation) and pFLL301.2 or pFLL301.5 (carrying the bcp gene in the reverse orientation). Additionally, the KD2301 strain was transformed with pFLL26, a plasmid containing a cloned 2.1-kb fragment encoding the bcp-recA-vimA genes (Fletcher et al., 1997). Transformed E. coli cells were plated on LB agar plates with ampicillin (50 μg ml−1) and grown overnight at 37°C. One ampicillin-resistant transformant, each carrying the appropriate plasmid, was compared with E. coli KD2301 (Jeong et al., 2000). All strains were grown overnight in LB broth; 100 μl of each of the overnight cultures was subcultured into 100 ml LB broth and grown to mid log-phase (OD600 0.4). Then, 100 μl aliquots of each of the cultures were then plated onto LB agar with and without 1 mM IPTG. Ten microliters of a 3% hydrogen peroxide solution was then placed on blank 6-mm antibiotic disks and placed in the center of the plate. Blank disks with distilled water were used as controls. The plates were then incubated at 37°C overnight and the zones of inhibition were measured. All experiments were performed three times, each in triplicate.
Mutagenesis of the cloned bcp gene
To facilitate the subsequent mutagenesis of the bcp gene with the ermF-ermAM antibiotic cassette, a unique StuI restriction site was created in the bcp gene by the PCR-based overlap extension method described previously (Horton et al., 1990, 1993; Horton, 1995). Primers 10 and 11 were engineered with restriction sequences for StuI at the Nucleic Acid Core facility at Loma Linda University. In two separate PCR using primer pairs P11 and P12 and P10 and P13 (Table 1), fragments of the bcp gene with flanking regions (1.0 and 0.8 kb, respectively) were amplified from P. gingivalis W83 chromosomal DNA. The amplified DNA fragments were electrophoresed on a 1% agarose gel at 110 V for 45 min, purified by the Gene Clean II kit (Bio 101 Inc.) and then used as a template for the third PCR using primers P12 and P13 (Table 1). The final 1.8-kb PCR amplified fragment with the bcp gene and flanking regions was identified by 1% agarose gel electrophoresis and purified. The 1.8-kb fragment containing the bcp gene and flanking regions constructed by the PCR-based overlap extension method (Horton et al., 1990, 1993; Horton, 1995) was cloned into the PCR2.1 TOPO® TA cloning vector (Invitrogen). The recombinant plasmid carrying the cloned bcp gene and flanking DNA (pFLL301.1) was digested with StuI. The pVA2198 containing the 2.1-kb ermF-ermAM cassette was digested with SacI and BamHI and treated with Klenow (Roche). The blunt-ended ermF-ermAM antibiotic cassette was ligated into the StuI site of pFLL301.1. The recombinant plasmid pFLL301.1 was used as a donor to electroporate P. gingivalis W83. Electroporation of P. gingivalis was performed as previously reported (Fletcher et al., 1995).
Hydrogen peroxide sensitivity testing and determination of tolerance to air
Porphyromonas gingivalis W83 and P. gingivalis FLL302 the bcp-defective isogenic mutant strain were tested for sensitivity to hydrogen peroxide as previously described (Johnson et al., 2004). Both strains were also tested for resistance to killing by atmospheric oxygen using a method similar to one previously described (Diaz et al., 2006). Both P. gingivalis W83 and FLL302 were grown to mid-log (OD600 ~0.8). A 10−6 dilution of each culture was made with pre-warmed BHI medium and 20 μl of the dilution was plated on each BHI agar plate. Test plates were immediately incubated at 37°C in air for 30 min or 1 h before being returned to anaerobic incubation for 4–6 days. Control plates were only incubated under anaerobic conditions. Percentage survival was determined as: (colony-forming units on plates exposed to air/colony-forming units on control plates) × 100. All experiments were performed three times, each in triplicate.
Bacterial cell fraction preparation and protease activity assay
Preparations of whole-cell culture media were performed as previously reported (Abaibou et al., 2000). Briefly, bacterial cultures were centrifuged at 6000 g for 30 min at 4°C. The supernatant (cell-free) was removed from the cell pellet and further clarified by ultracentrifugation (100,000 g, 60 min, 4°C) to yield vesicle-free (supernatant) and vesicle (pellet) fractions. The cell pellet from the first centrifugation was washed three times and resuspended in 0.1 M Tris–HCl buffer. The presence of Arg-X and Lys-X activity in each fraction (cell-free, cell suspension, vesicle-free and vesicles) was determined using a microplate reader (Bio-Rad, Hercules, CA) according to the methods of Potempa et al. (1998).
Ultraviolet sensitivity measurements
The UV sensitivity experiments were performed as previously described (Fletcher et al., 1997). All experiments were performed in triplicate.
Virulence testing
Both P. gingivalis strain W83 and the isogenic mutant strain FLL302 were tested for invasiveness in a murine model as previously described (Fletcher et al., 1995). Briefly, actively growing overnight cultures of P. gingivalis W83 and FLL302, each grown in BHI were centrifuged and washed in sterile phosphate-buffered saline. Each strain was then adjusted to 1 × 1010 bacterial ml−1 dose and 3 × 109 bacteria ml−1 dose in phosphate-buffered saline. Female BALB/c mice were challenged with 100 μl of each bacterial culture from each strain and at each concentration at two sites on the dorsal surface (200 μl total). The mice were examined daily to assess their general health status as well as the presence and location of lesions. Weights were determined for all surviving mice. These experiments were performed under the authorization of an institutionally approved protocol (Loma Linda University Animal Research Committee, 2010).
Statistical analysis
Statistical analysis was performed using Student’s t-test.
RESULTS
The bcp gene is part of a multigene transcriptional unit
The recA-vimA-vimE-vimF was previously shown to be part of the same multigene transcriptional unit (Abaibou et al., 2001; Vanterpool et al., 2004, 2005a). To further confirm if the bcp gene was part of the same transcriptional unit, reverse transcription (RT-) PCR analysis of the DNase-treated RNA extracted from P. gingivalis W83 was amplified using bcp forward (P1) and vimA reverse (P14) oligonucleotide primers. A 2.4-kb fragment [bcp (0.5 kb), recA (1.0 kb) and vimA (0.9 kb)] was amplified only in the presence of reverse transcriptase.
TSS mapping of the bcp gene
RLM-RACE was performed to determine the start site for this transcriptional unit. As shown in Fig. 1, an adenine residue was identified as the TSS. It is noteworthy that this residue is located 25 bases downstream from the start of the current predicted ORF of the bcp gene (http://www.oralgen.lanl.gov). An ATG start codon is observed 23 nucleotide bases downstream of the TSS (Fig. 1B). This ORF would encode for a 151-amino-acid protein in contrast to the previous 167-amino-acid protein (http://www.oralgen.lanl.gov). A comparison of the Bcp proteins from P. gingivalis W83 and P. gingivalis ATCC 33277 shows 98% homology. Together they are similar to the Bcp proteins of other oral anaerobes and of E. coli (Fig. 1C).
Figure 1.
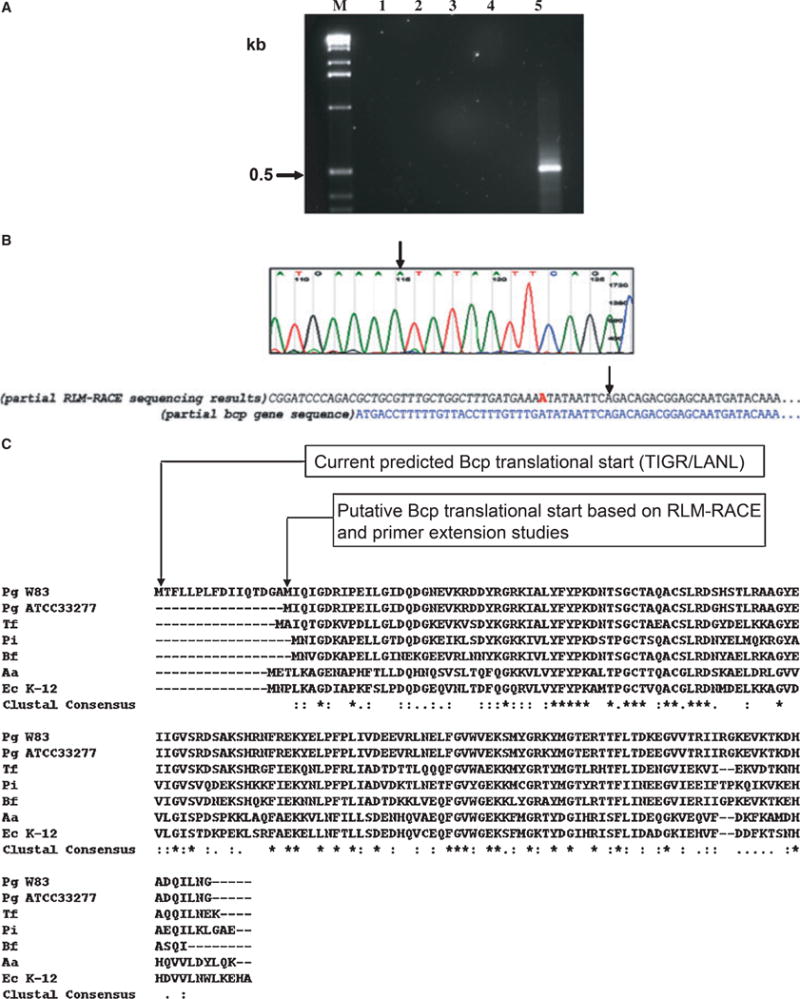
Identification of the transcriptional start site (TSS) of the bcp gene of Porphyromonas gingivalis by RLM-RACE. (A) Total RNA was harvested from a mid log-phase culture (OD600 ~0.6) of P. gingivalis W83 and sequentially treated with calf intestinal phosphatase and tobacco acid phosphatase (TAP). After ligation of a 5′ RACE adapter sequence (P6, Table 1) the RNA was reverse transcribed. An outer polymerase chain reaction (PCR) using primers P7 and P2 was performed (data not shown) and the product from this outer primer reaction was used as the template for the nested PCR using primers P8 and P2. No-template –TAP reactions (lane 1) and –TAP (lane 2) reactions were run as controls. The inner PCR revealed no PCR product in the absence of template DNA (lane 3) but yielded a fragment slightly larger than 0.5 kb (lane 4) with template DNA. The size of observed PCR products was based on the 1 kb DNA marker (M) used. (B) Sequencing analysis of the RLM-RACE inner PCR product revealed the transcription start site (TSS) to be an ‘A’ (indicated by arrows) located immediately at the junction of the ligated 5′ adapter sequence (in italics). A partial sequence of the bcp gene is shown for reference. This TSS is at the −23 position upstream of an ATG. (C) Sequence alignment of P. gingivalis Bcp proteins and Bcp homologues in common oral anaerobes and Escherichia coli (CLUSTAL W version 2.0). Pg, Porphyromonas gingivalis; Tf, Tannerella forsythensis; Pi, Prevotella intermedia; Bf, Bacteroides fragilis; Aa, Aggregatibacter actinomycetemcomitans; Ec, Escherichia coli. Asterisks indicate positions that have a fully conserved residue; whereas conservative replacements are indicated by a full stop or a colon.
Complementation of E. coli bcp-defective mutant with P. gingivalis bcp homologue
Bcp is known to play a functional role in oxidative stress resistance in E. coli (Jeong et al., 2000) To determine any functional similarity between the E. coli and P. gingivalis Bcp proteins chromosomal DNA from P. gingivalis W83 was subjected to PCR analysis using primers (P9 and P2; Table 1), which would amplify the 0.5-kb bcp gene. This purified DNA fragment was inserted into pTrcHis2 TOPO® TA vector (Invitrogen) under the control of the trc promoter (Mulligan et al., 1985; Brosius et al., 1985). The inserts in the resulting recombinant plasmids designated pFLL301.4 (correct orientation) and pFLL301.5 (opposite orientation) were confirmed by nucleotide sequencing analysis. In the presence of IPTG E. coli KD2301, a bcp-defective mutant transformed with pFLL301.4 (bcp homologue), showed increased resistance when exposed to hydrogen peroxide (Table 2). A recombinant plasmid carrying the bcp ORF from the previously predicted ORF had no effect on hydrogen peroxide sensitivity of the bcp-defective E. coli strain, either in the presence or absence of IPTG (Table 2). None of the chimeric constructs with the bcp gene in the reverse orientation had any effect on the level of resistance of the bcp-defective E. coli strain to hydrogen peroxide even in the presence of IPTG (Table 2). To further confirm if the P. gingivalis Bcp was expressed in E. coli, cell lysates of E. coli transformants carrying pFLL301.3, pFLL301.2, pFLL301.4 or pFLL301.5 were analysed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The predicted protein was observed to be expressed in the presence of IPTG in strains carrying the plasmids (pFLL301.3 and pFLL301.4) with bcp gene in the forward orientation (data not shown). In contrast, there was no expression of the expected 19-kDa Bcp protein in the absence of IPTG in E. coli lysates carrying those constructs or those with the bcp gene in the reverse orientation (pFLL301.2 and pFLL301.5) (data not shown).
Table 2.
Complementation of Escherichia coli bcp-defective mutant
| Background strain | Plasmid1 | Zone of inhibition (cm)2 mean (SD)
|
|
|---|---|---|---|
| −IPTG | +IPTG | ||
| E. coli BL21(DE3) | – | 1.58 (0.10) | 1.35 (0.28) |
| E. coli KD2301 | – | 1.25 (0.08) | 1.25 (0.10) |
| E. coli KD2301 | pFLL26 (Fletcher et al., 1997) | 1.23 (0.05) | 1.20 (0.09) |
| E. coli KD2301 | pFLL301.2 (bcp gene in reverse orientation) | 1.08 (0.12) | 1.10 (0.11) |
| E. coli KD2301 | pFLL301.3 (bcp gene in forward orientation) | 1.08 (0.08) | 1.12 (0.08) |
| E. coli KD2301 | pFLL301.4 (bcp gene in forward orientation) | 1.03 (0.05) | 0.87 (0.08)* |
| E. coli KD2301 | pFLL301.5 (bcp gene in reverse orientation) | 1.08 (0.08) | 1.07 (0.08) |
Plasmids containing various constructs of the bcp gene from Porphyromonas gingivalis W83 were used to transform the E. coli bcp-defective mutant, KD2301.
Sensitivity to hydrogen peroxide was determined by disk inhibition assays. The data shown are an average of three independent experiments. Complementation was determined as a statistically significant difference (P < 0.05) in resistance to hydrogen peroxide between the KD2301 mutant and the complemented strain and is indicated by an asterisk (*).
Inactivation of the bcp gene in P. gingivalis W83 by allelic exchange mutagenesis
An isogenic P. gingivalis W83 mutant defective in the bcp gene was constructed by allelic exchange mutagenesis. The recombinant plasmid pFLL301, which carries the ermF-ermAM cassette in the unique StuI site of the bcp gene, was used as a donor to electroporate P. gingivalis W83. Because the plasmid cannot replicate in P. gingivalis, we predicted that two double crossover events between the regions flanking the erm marker and the wild-type gene on the chromosome would result in replacement of a segment of the wild-type gene with the fragment conferring clindamycin resistance. Following electroporation and plating on selective media we detected 12 clindamycin-resistant colonies after 6 days of incubation. Randomly chosen colonies were further plated on Brucella blood agar to determine any pleiotropic phenotypic effects of the clindamycin-resistant mutants. Similar to the wild-type strain, all of the clindamycin-resistant mutants displayed a β-hemolytic and black-pigmented phenotype. Chromosomal DNA from two randomly chosen colonies and the wild-type W83 strain was digested with BamHI and subjected to Southern blot analysis. If the bcp gene was interrupted by the ermF-ermAM cassette, a 4.2-kb fragment should be observed when probed with the digoxigenin-labeled bcp DNA. The expected 4.2- and 2.1-kb fragments were observed in the two clindamycin-resistant strains and the wild-type, respectively (data not shown). In addition, when hybridization was performed with the digoxigenin-labeled erm DNA no hybridization was seen in wild-type DNA. However, the expected 4.2- and 9.2-kb fragments were observed in the two clindamycin-resistant colonies and the pVA2198 control plasmid, respectively (data not shown). Further confirmation of the clindamycin-resistant colonies was performed by PCR using primers P1 and P2 (Table 1) for the 0.5-kb bcp gene fragment. If the bcp gene was interrupted by the ermF-ermAM cassette, a 2.6-kb fragment was expected to be amplified. The expected 2.6- and 0.5-kb fragments were observed in the two clindamycin-resistant strains and the wild-type W83, respectively (data not shown). Taken together, these results suggest the insertional inactivation of the chromosomal bcp gene with the 2.1-kb ermF-ermAM antibiotic cassette. One strain designated P. gingivalis FLL302 was randomly chosen from the two bcp-defective mutants for further studies.
RT-PCR analysis of P. gingivalis FLL302 and W83
To further confirm the inactivation of the bcp gene in P. gingivalis FLL302 and to evaluate any polar effects on the recA and vimA genes, total RNA was isolated from the wild-type W83 and the bcp-defective mutant FLL302 grown to mid log-phase. Specific oligonucleotide primers for the bcp, recA and vimA genes (Table 1) were used in RT-PCR analysis. As primers specific for the bcp, recA and vimA genes would yield 0.5-, 1.0- and 0.9-kb fragments, respectively, a 0.5-kb fragment for the bcp gene should be amplified in the wild-type strain but should be missing in P. gingivalis FLL302. Furthermore, as recA and vimA are a part of the bcp-recA-vimA transcriptional unit, 1.0- and 0.9-kb fragments should be amplified in the wild-type strain, which should be absent in the mutant strain. As shown in Fig. 2A (lanes 4–6), fragments of 0.5, 1.0 and 0.9 kb in size were amplified in the wild-type strain using bcp, recA and vimA specific primers (Fig. 2B), respectively. In contrast, only 1.0-and 0.9-kb fragments using the same primer pairs were amplified in the bcp-defective mutant FLL302 (Fig. 2A, lanes 1–3). There were no amplified fragments observed for either the wild-type strain or the mutant when reverse transcriptase was absent in the reaction mix (Fig. 2A, lanes 7–12). To test if a read-through from the erm cassette could result in the expression of the downstream genes (recA and vimA), primers P11 (5′ oligonucleotide primer for the erm cassette) and P2 (3′ primer for the bcp gene) were used in RT-PCR analysis of total RNA isolated from P. gingivalis FLL302. There was no amplified product observed in RNA from P. gingivalis FLL302 (data not shown). Taken together, these data confirm the inactivation of the bcp gene in P. gingivalis FLL302 and may suggest that the recA and vimA genes can be independently expressed in P. gingivalis W83.
Figure 2.
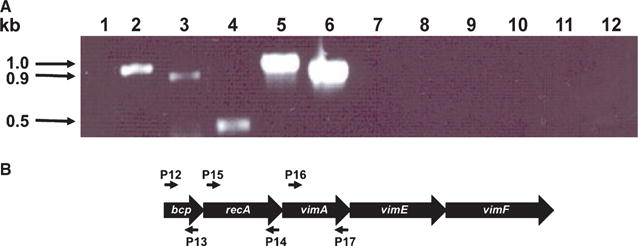
(A) Reverse transcription–polymerase chain reaction (RT-PCR) analysis of RNA extracted from Porphyromonas gingivalis W83 and FLL302. Total RNA was extracted from FLL302 (lanes 1–3 and 7–9) and W83 (lanes 4–6 and 10–12) grown to mid log-phase (OD600, 0.6) and subjected to RT-PCR. Porphyromonas gingivalis FLL302: lane 1, primers (P1 and P2, Table 1) plus reverse transcriptase; lane 2, primers (P16 and P17, Table 1) plus reverse transcriptase; lane 3, primers (P14 and P15, Table 1) plus reverse transcriptase; P. gingivalis W83: lane 4, primers (P1 and P2, Table 1) plus reverse transcriptase; lane 5, primers (P16 and P17, Table 1) plus reverse transcriptase; lane 6, primers (P14 and P15, Table 1) plus reverse transcriptase; lanes 7–12, corresponding primer sets for the bcp, recA and vimA genes minus reverse transcriptase (negative controls). All lanes contain 5 μl of the amplified mixture. (B) Location of the primers used in RT-PCR analysis.
UV sensitivity of P. gingivalis FLL302
The recA gene plays a role in DNA repair in P. gingivalis (Fletcher et al., 1997). The RT-PCR showed that the recA gene was expressed in P. gingivalis FLL302 so we expected that the sensitivity to UV should be similar to that of the wild-type. The P. gingivalis FLL302, FLL32 (recA-defective isogenic mutant) and the wild-type W83 were exposed to UV radiation at 2000 and 4000 μJ. After 2000 μJ UV irradiation there was 15 and 11% survival of the wild-type W83 and the isogenic bcp-defective mutant FLL302, respectively (data not shown). In contrast, there was no survival of the recA-defective mutant FLL32 at 2000 μJ UV irradiation (data not shown). No survival of any of the P. gingivalis strains was observed after exposure to 4000 μJ of UV irradiation. Taken together these data indicate that under the same physiological conditions the UV sensitivity of the wild-type P. gingivalis W83 and the bcp-defective mutant FLL302 is similar.
Sensitivity of P. gingivalis FLL302 to hydrogen peroxide and tolerance to air
Because the P. gingivalis Bcp can increase the resistance of E. coli to peroxides, P. gingivalis W83 and the isogenic mutant P. gingivalis FLL302 were assessed for sensitivity to hydrogen peroxide. Hydrogen peroxide was prepared at a concentration of 0.25 mM in BHI. In contrast to the parent strain, P. gingivalis FLL302 demonstrated greater sensitivity to hydrogen peroxide at a concentration of 0.25 mM (3A). Sensitivity of both bacterial stains was similar when a higher concentration was used (0.5 mM) (data not shown). Taken together, these data suggest that P. gingivalis FLL302 has an increased sensitivity to hydrogen peroxide compared with the wild-type W83. In addition, when exposed to air, the bcp mutant, FLL302, demonstrated decreased aerotolerance when compared with the wild-type (Fig. 3B).
Figure 3.
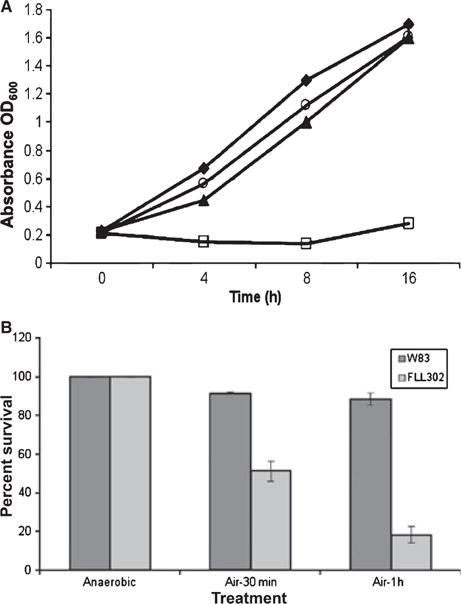
Sensitivity of the Porphyromonas gingivalis bcp mutant to hydrogen peroxide. The P. gingivalis was grown to early log-phase (OD600 0.2) in brain–heart infusion broth and 0.25 mM hydrogen peroxide (▲ W83, □ FLL302) was then added to the cell cultures and incubated for a further 16 h. Cell cultures without hydrogen peroxide (♦ W83, ○ FLL302) were used as controls. The results shown are representative of three independent experiments performed in triplicate.
Protease distribution and activity of P. gingivalis FLL302
We have previously shown that the vimA gene is involved in protease activation and distribution in P. gingivalis W83 (Abaibou et al., 2001). Because the vimA transcript was expressed in the bcp-defective mutant FLL302 we expected that the proteolytic activity and distribution would be similar to that in the wild-type. Strains of P. gingivalis W83 and P. gingivalis FLL302 were assayed for proteolytic activity using N-α-benzoyl-DL-arginine p-nitroanilide and Ac-Lys-p-nitroanilide HCl. In late exponential growth phase cultures, both the arginine-X and lysine-X proteolytic activities of P. gingivalis FLL302 were 70% of the activity compared with the wild-type W83 (Fig. 4).
Figure 4.
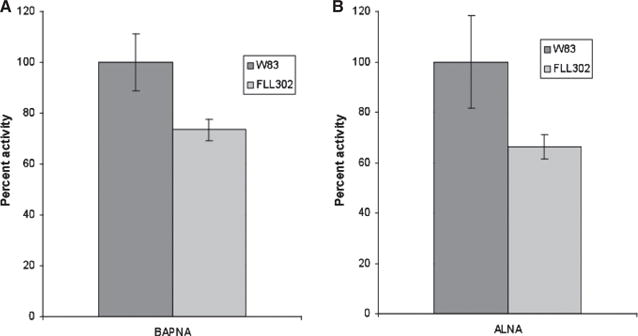
Arg-X and Lys-X cysteine protease activity in Porphyromonas gingivalis strains W83 and FLL302. The P. gingivalis was grown to late log-phase OD600 (1.2) in 50 ml brain–heart infusion broth supplemented with hemin and vitamin K. Activity against N-α-benzoyl-DL-arginine p-nitroanilide (BAPNA) and Ac-Lys-p-nitroanilide HCl (ALNA) was tested in whole cell culture according to the method of Potempa et al. (1998). The Arg-X (A) and Lys-X (B) activities of W83 were assumed to be 100%; this activity measured in P. gingivalis FLL302 represents 70% of the activity of the wild-type strain W83 for both cysteine proteases. The results shown are representative of three independent experiments performed in triplicate.
Taken together, these data suggest that under the same physiological conditions, there is a 30% reduction in total proteolytic activity of P. gingivalis FLL302 when compared with the wild-type W83 whereas the distribution remained similar in both strains.
Virulence testing of P. gingivalis FLL302
Protection against peroxide damage during the course of an infection plays a major role in pathogenesis (Nathan & Shiloh, 2000). As P. gingivalis FLL302 was more sensitive to hydrogen peroxide and had decreased proteolytic activity when compared with W83, its virulence potential in the mouse model was assessed. Our previous reports showed that the 50% lethal dose of P. gingivalis W83 in the BALB/c mouse model is approximately 3 × 109 bacteria per animal (Fletcher et al., 1995). Within 6 days, four of ten animals challenged with P. gingivalis (wild-type) at a dose of 3 × 109 bacteria per animal had developed spreading, ulcerative abdominal lesions and died (data not shown). The surviving animals appeared cachectic and hunched with ruffled hair. However, all six recovered before the end of the 14-day observation period. Similarly, within 4 days, four of ten animals challenged with P. gingivalis FLL302 (bcp-defective mutant) at a dose of 3 × 109 bacteria per animal had developed spreading, ulcerative abdominal lesions and died (data not shown). All of the surviving animals appeared cachectic and hunched with ruffled hair and also recovered during the 14-day observation period. In both groups, the lesions began healing by day 6 after challenge. All animals challenged with a dose of 1 × 1010 bacteria per animal of the wild-type (10 of 10) or the P. gingivalis FLL302 (10 of 10) died by 48 h (data not shown). Although the mice did not display lesions at the dorsal surface site of injection, they had developed spreading, ulcerative abdominal lesions. These data suggest that there is no detectable difference in the virulence potential of the bcp-defective mutant P. gingivalis FLL302 compared with the parent strain.
DISCUSSION
Periodontal disease is a chronic infection marked by massive inflammation (Seymour et al., 1993). This inflammatory process is marked by the recruitment of neutrophils and macrophages to the site of infection to effect the clearance of the offending organism(s) through the oxidative burst (Van Dyke et al., 1985). As a result, the inflammatory microenvironment of the periodontal pocket is also a highly oxidatively stressful environment characterized by several reactive oxygen intermediates (Beaman & Beaman, 1984; Genco & Slots, 1984). Bacterial exposure to these reactive oxygen intermediates can result in damage to bacterial membranes and nucleic acids (Imlay & Linn, 1988). Bacteria have therefore developed several mechanisms to subvert the damage caused under oxidative stress through the use of antioxidant enzymes such as superoxide dismutase, AhpC and Bcp (Nakayama, 1994; Rocha & Smith, 1999; Jeong et al., 2000). Bcp has been reported to detoxify hydrogen peroxide in E. coli and so plays a role in oxidative stress resistance (Jeong et al., 2000). In this study we examined the role of the bcp gene in the oxidative stress resistance and pathogenesis. It was our hypothesis that the bcp gene in P. gingivalis W83 would play a significant role in oxidative stress and virulence.
In a previous communication we hypothesized that the bcp gene may be part of a unique transcriptional unit together with the recA, vimA, vimE and vimF genes in P. gingivalis (Abaibou et al., 2001). In this study we have confirmed bcp as part of the multigene transcriptional unit. The recA and vim genes in P. gingivalis are involved in DNA repair and protease activation/distribution, respectively (Fletcher et al., 1997; Abaibou et al., 2001). This bcp-recA-vimA-vimE-vimF genetic architecture would be advantageous to P. gingivalis because DNA repair activity, protease activation and oxidative stress resistance could be co-ordinately regulated. The significance of this observation would be consistent with the ability of P. gingivalis to use the heme on its surface as an oxidative sink to neutralize reactive oxygen intermediates (Smalley et al., 1998, 2000). Because P. gingivalis proteases are major virulence factors involved in heme acquisition and binding to the bacterial membrane (Okamoto et al., 1998; Genco & Dixon, 2001; Sroka et al., 2001), this could be an important strategy for survival in the periodontal pocket.
In our analysis of the bcp transcript from cells in the exponential growth phase we identified the TSS to be an adenine residue. Interestingly, this was located within the current predicted ORF for the bcp gene. Identification of another in-frame start codon 23 bases downstream of that predicted for the ORF of Bcp suggests that the Bcp protein may be smaller than predicted. When the current predicted ORF is aligned with other Bcp proteins, including that of another P. gingivalis strain, ATCC 33277, the P. gingivalis Bcp appears to have additional amino acid residues in its N-terminal region that are not conserved with the other proteins. Based on the transcript observed in our experiments, a smaller Bcp protein would be consistent with the observed size of Bcp that is seen in other bacterial species. This presents two interesting scenarios: either in silico prediction of the Bcp product is incorrect or there is possibly an alternative bcp gene product depending on differential regulation based on growth phase, stress or other environmental factors. These hypotheses would need to be further evaluated and validated.
The inactivation of bcp in E. coli does not significantly change its sensitivity to hydrogen peroxide (Kong et al., 2000). However, complementation using the smaller Bcp homologue from P. gingivalis significantly increases its resistance to hydrogen peroxide. It is possible that the Bcp protein in P. gingivalis may use hydrogen peroxide as a substrate. Bioinformatic analysis of the P. gingivalis Bcp (http://tigr-blast.tigr.org/cmr-blast) revealed that it was 60% similar to the E. coli Bcp. Although both proteins share sequence similarity, the differences in their sequences could account for differences in their specificity for or efficiency in degrading hydrogen peroxide. Collectively, these data suggest that the Bcp homologue in P. gingivalis can detoxify hydrogen peroxide.
Inactivation of the P. gingivalis bcp gene allowed us to determine a role for the bcp gene in oxidative stress. Comparison of the sensitivities of the wild-type W83 and the bcp-defective mutant FLL302 to hydrogen peroxide and exposure to air demonstrated that the Bcp plays a significant role in oxidative stress resistance. These findings are consistent with other reports that suggest similar roles for Bcp (Jeong et al., 2000; Atack et al., 2008).
RT-PCR analysis of the DNase-treated RNA from P. gingivalis FLL302 (bcp-defective mutant) revealed that both the recA and vimA genes can be independently expressed. Because the ermF-ermAM antibiotic cassette was constructed with transcriptional stop signals (Fletcher et al., 1995) it is unlikely that a read-through of this cassette would facilitate the expression of the down stream recA and vimA genes. This would also be consistent with the failure to RT-PCR amplify any product from the bcp-defective mutant using a specific 5′ oligonucleotide primer for the erm cassette and a 3′ primer from the bcp or recA genes. From this study however, our observations of the independent expression of the recA and vimA genes could further help to explain the multiple hybridizing bands previously observed when RNA from the wild-type strain was probed with the recA gene (Abaibou et al., 2001). Taken together, these data suggest that there may be some differential expression of the genes in the bcp-recA-vimA transcriptional unit. Preliminary analysis of the nucleotide sequence of this locus indicates putative promoter regions for each gene (data not shown). The significance of this expression pattern in P. gingivalis is under further investigation in the laboratory. Because the gingipains are downregulated at elevated temperature (Percival et al., 1999), typical of the inflammatory microenvironment of the periodontal pocket, it is tempting to speculate that under conditions of oxidative stress and where a decrease in proteolytic activity is desirable, an ability to differentially express genes in the bcp-recA-vimA-vimE-vimF operon may be an important strategy for its adaptation to conditions of oxidative stress.
Consistent with previous reports on P. gingivalis FLL33, a recA-defective mutant (Fletcher et al., 1997) and P. gingivalis FLL92, a vimA-defective mutant (Abaibou et al., 2001), the independent expression of the recA and vimA gene in P. gingivalis FLL302 should not alter UV sensitivity and gingipain activity/distribution in this mutant. The bcp-defective mutant was black-pigmented, and had a similar growth rate, β-hemolysis and UV sensitivity as the parent strain. There was a 30% reduction in the Arg-X and Lys-X cysteine protease activities in P. gingivalis FLL302 though the distribution was similar to the wild-type P. gingivalis W83 strain (data not shown). The source of the reduction in proteolytic activity is currently unknown but it is likely that the reduced expression of the vimA gene, as demonstrated in the FLL302 mutant, may play a role.
Reduced gingipain activity is correlated with reduced virulence in P. gingivalis (Fletcher et al., 1997; Abaibou et al., 2001). Further, protection against peroxide damage during the course of an infection is important for survival of the invading microorganism (Miller & Britigan, 1997). Results of our virulence studies in the mouse model suggest that there is no discernible difference in the virulence potential of P. gingivalis FLL302, the bcp-defective mutant, compared with the wild-type strain. It is likely that the level of gingipain activity of P. gingivalis FLL302 is not significant enough to alter its virulence potential. Furthermore, the bcp defect by itself might not be enough to alter the virulence potential of P. gingivalis. These data are consistent with similar experiments conducted with a P. gingivalis ahpC mutant (Johnson et al., 2004) and a Salmonella typhimurium ahpC-defective mutant (Taylor et al., 1998). Results from these studies confirmed increased sensitivity of the ahpC mutant to peroxides in vitro but no change in its virulence in a mouse model (Taylor et al., 1998). However, we cannot rule out the possibility that the similarity in virulence potential observed between the wild-type and the bcp-defective mutant may be partly the result of the mouse abscess model of virulence used in these experiments. It is possible that in other mouse models of virulence, we may be able to mimic more closely the conditions of the periodontal cavity, possibly allowing for subtle differences in the attenuation of virulence to be observed. A second likely explanation for the inability of a single oxidative stress resistance gene to affect virulence in this mouse model, could be the presence of other redundant mechanisms of oxidative stress resistance upregulated during the infectious process. Recent reports on mechanisms of oxidative stress resistance in P. gingivalis indicate a role for several genes including sod, dps, oxyR (Diaz et al., 2006), FeoB2 (He et al., 2006) and rubrerythrin in this process (Amano et al., 1992; Sztukowska et al., 2002; Ueshima et al., 2003). In addition, a survey of the P. gingivalis genome [http://www.oralgen.lanl.gov/] has identified other genes (NADH oxidase, oxyR, thiol peroxidase and thioredoxin) that may play a role in oxidative stress defense. Although many of the genes may play some role in oxidative stress, their role in virulence has not been investigated and remains unknown.
We have constructed an isogenic mutant of P. gingivalis that is defective in the bcp gene, which is part of the bcp-recA-vimA transcriptional unit. Further, we have demonstrated that the genes in this operon can be differentially expressed. While P. gingivalis FLL302, the bcp-defective mutant, showed in vitro sensitivity to hydrogen peroxide, reduced aerotolerance and its virulence potential in a mouse model were unaltered when compared with the wild-type. This suggests that there may be other redundant mechanism(s) involved in oxidative stress defense in P. gingivalis. Further studies characterizing the relationship of the bcp gene to other oxidative stress genes may give insight into the global regulation of these genes in oxidative stress defense in P. gingivalis.
Acknowledgments
This work was supported by the Loma Linda University, School of Dentistry and Public Health Service grant DE13664 and DE019730 from the National Institute of Dental and Craniofacial Research (HMF).
References
- Abaibou H, Ma Q, Olango GJ, Potempa J, Travis J, Fletcher HM. Unaltered expression of the major protease genes in a non-virulent recA-defective mutant of Porphyromonas gingivalis W83. Oral Microbiol Immunol. 2000;15:40–47. doi: 10.1034/j.1399-302x.2000.150107.x. [DOI] [PubMed] [Google Scholar]
- Abaibou H, Chen Z, Olango GJ, Liu Y, Edwards J, Fletcher HM. vimA gene downstream of recA is involved in virulence modulation in Porphyromonas gingivalis W83. Infect Immun. 2001;69:325–335. doi: 10.1128/IAI.69.1.325-335.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano A, Tamagawa H, Shizukuishi S, Tsunemitsu A. Superoxide dismutase, catalase and peroxidases in oral anaerobic bacteria. J Osaka Univ Dent Sch. 1986;26:187–192. [PubMed] [Google Scholar]
- Amano A, Tamagawa H, Takagaki M, Murakami Y, Shizukuishi S, Tsunemitsu A. Relationship between enzyme activities involved in oxygen metabolism and oxygen tolerance in black-pigmented Bacteroides. J Dent Res. 1988;67:1196–1199. doi: 10.1177/00220345880670090901. [DOI] [PubMed] [Google Scholar]
- Amano A, Ishimoto T, Tamagawa H, Shizukuishi S. Role of superoxide dismutase in resistance of Porphyromonas gingivalis to killing by polymorphonuclear leukocytes. Infect Immun. 1992;60:712–714. doi: 10.1128/iai.60.2.712-714.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atack JM, Harvey P, Jones MA, Kelly DJ. The Campylobacter jejuni thiol peroxidases Tpx and Bcp both contribute to aerotolerance and peroxide-mediated stress resistance but have distinct substrate specificities. J Bacteriol. 2008;190:5279–5290. doi: 10.1128/JB.00100-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillon ML, van Vliet AH, Ketley JM, Constantinidou C, Penn CW. An iron-regulated alkyl hydroperoxide reductase (AhpC) confers aerotolerance and oxidative stress resistance to the microaerophilic pathogen Campylobacter jejuni. J Bacteriol. 1999;181:4798–4804. doi: 10.1128/jb.181.16.4798-4804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaman L, Beaman BL. The role of oxygen and its derivatives in microbial pathogenesis and host defense. Annu Rev Microbiol. 1984;38:27–48. doi: 10.1146/annurev.mi.38.100184.000331. [DOI] [PubMed] [Google Scholar]
- Birnboim HC, Doly J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979;7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiteux S, Radicella JP. Base excision repair of 8-hydroxyguanine protects DNA from endogenous oxidative stress. Biochimie. 1999;81:59–67. doi: 10.1016/s0300-9084(99)80039-x. [DOI] [PubMed] [Google Scholar]
- Brosius J, Erfle M, Storella J. Spacing of the -10 and -35 regions in the tac promoter. Effect on its in vivo activity. J Biol Chem. 1985;260:3539–3541. [PubMed] [Google Scholar]
- Chapple ILC. Role of free radicals and antioxidants in the pathogenesis of inflammatory periodontal disease. J Clin Pathol Mol Pathol. 1996;49:M247–M255. doi: 10.1136/mp.49.5.m247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JI, Takahashi N, Kato T, Kuramitsu HK. Isolation, expression, and nucleotide sequence of the sod gene from Porphyromonas gingivalis. Infect Immun. 1991;59:1564–1566. doi: 10.1128/iai.59.4.1564-1566.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P. One-hour downward alkaline capillary transfer for blotting of DNA and RNA. Anal Biochem. 1992;201:134–139. doi: 10.1016/0003-2697(92)90185-a. [DOI] [PubMed] [Google Scholar]
- Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz PI, Slakeski N, Reynolds EC, Morona R, Rogers AH, Kolenbrander PE. Role of oxyR in the oral anaerobe Porphyromonas gingivalis. J Bacteriol. 2006;188:2454–2462. doi: 10.1128/JB.188.7.2454-2462.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher HM, Schenkein HA, Morgan RM, Bailey KA, Berry CR, Macrina FL. Virulence of a Porphyromonas gingivalis W83 mutant defective in the prtH gene. Infect Immun. 1995;63:1521–1528. doi: 10.1128/iai.63.4.1521-1528.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher HM, Morgan RM, Macrina FL. Nucleotide sequence of the Porphyromonas gingivalis W83 recA homolog and construction of a recA-deficient mutant. Infect Immun. 1997;65:4592–4597. doi: 10.1128/iai.65.11.4592-4597.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genco CA, Dixon DW. Emerging strategies in microbial haem capture. Mol Microbiol. 2001;39:1–11. doi: 10.1046/j.1365-2958.2001.02231.x. [DOI] [PubMed] [Google Scholar]
- Genco RJ, Slots J. Host responses in periodontal diseases. J Dent Res. 1984;63:441–451. doi: 10.1177/00220345840630031601. [DOI] [PubMed] [Google Scholar]
- Grollman AP, Moriya M. Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet. 1993;9:246–249. doi: 10.1016/0168-9525(93)90089-z. [DOI] [PubMed] [Google Scholar]
- He J, Miyazaki H, Anaya C, Yu F, Yeudall WA, Lewis JP. Role of Porphyromonas gingivalis FeoB2 in metal uptake and oxidative stress protection. Infect Immun. 2006;74:4214–4223. doi: 10.1128/IAI.00014-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM. PCR-mediated recombination and mutagenesis. SOEing together tailor-made genes. Mol Biotechnol. 1995;3:93–99. doi: 10.1007/BF02789105. [DOI] [PubMed] [Google Scholar]
- Horton RM, Cai ZL, Ho SN, Pease LR. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. BioTechniques. 1990;8:528–535. [PubMed] [Google Scholar]
- Horton RM, Ho SN, Pullen JK, Hunt HD, Cai Z, Pease LR. Gene splicing by overlap extension. Methods Enzymol. 1993;217:270–279. doi: 10.1016/0076-6879(93)17067-f. [DOI] [PubMed] [Google Scholar]
- Imlay JA, Linn S. DNA damage and oxygen radical toxicity. Science. 1988;240:1302–1309. doi: 10.1126/science.3287616. [DOI] [PubMed] [Google Scholar]
- Jeong W, Cha MK, Kim IH. Thioredoxin-dependent hydroperoxide peroxidase activity of bacterioferritin comigratory protein (BCP) as a new member of the thiol-specific antioxidant protein (TSA)/Alkyl hydroperoxide peroxidase C (AhpC) family. J Biol Chem. 2000;275:2924–2930. doi: 10.1074/jbc.275.4.2924. [DOI] [PubMed] [Google Scholar]
- Johnson NA, Liu Y, Fletcher HM. Alkyl Hydroperoxide peroxidase subunit C(AhpC) protects against organic peroxides but does not affect the virulence of Porphyromonas gingivalis W83. Oral Microbiol Immunol. 2004;19:233–239. doi: 10.1111/j.1399-302X.2004.00145.x. [DOI] [PubMed] [Google Scholar]
- Kong W, Shiota S, Shi Y, Nakayama H, Nakayama K. A novel peroxiredoxin of the plant Sedum lineare is a homologue of Escherichia coli bacterioferritin co-migratory protein (Bcp) Biochem J. 2000;351:107–114. doi: 10.1042/0264-6021:3510107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limauro D, Pedone E, Galdi I, Bartolucci S. Peroxiredoxins as cellular guardians in Sulfolobus solfataricus: characterization of Bcp1, Bcp3 and Bcp4. FEBS J. 2008;275:2067–2077. doi: 10.1111/j.1742-4658.2008.06361.x. [DOI] [PubMed] [Google Scholar]
- Loma Linda University Animal Research Committee. Approved Protocol IACUC # 89037. Loma Linda, CA: Loma Linda University; 2010. [Google Scholar]
- Marmur J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J Mol Biol. 1961;3:208–218. [Google Scholar]
- Mayrand D, Holt SC. Biology of asaccharolytic black-pigmented Bacteroides species. Microbiol Rev. 1988;52:134–152. doi: 10.1128/mr.52.1.134-152.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels ML, Miller JH. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine) J Bacteriol. 1992;174:6321–6325. doi: 10.1128/jb.174.20.6321-6325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Britigan BE. Role of oxidants in microbial pathophysiology. Clin Microbiol Rev. 1997;10:1–18. doi: 10.1128/cmr.10.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan ME, Brosius J, McClure WR. Characterization in vitro of the effect of spacer length on the activity of Escherichia coli RNA polymerase at the TAC promoter. J Biol Chem. 1985;260:3529–3538. [PubMed] [Google Scholar]
- Nakayama K. The superoxide dismutase-encoding gene of the obligately anaerobic bacterium Bacteroides gingivalis. Gene. 1990;96:149–150. doi: 10.1016/0378-1119(90)90357-w. [DOI] [PubMed] [Google Scholar]
- Nakayama K. Rapid viability loss on exposure to air in a superoxide dismutase-deficient mutant of Porphyromonas gingivalis. J Bacteriol. 1994;176:1939–1943. doi: 10.1128/jb.176.7.1939-1943.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash HM, Bruner SD, Scharer OD, et al. Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr Biol. 1996;6:968–980. doi: 10.1016/s0960-9822(02)00641-3. [DOI] [PubMed] [Google Scholar]
- Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U S A. 2000;97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KE, Fleischmann RD, DeBoy RT, et al. Complete genome sequence of the oral pathogenic bacterium Porphyromonas gingivalis strain W83. J Bacteriol. 2003;185:5591–5601. doi: 10.1128/JB.185.18.5591-5601.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Nakayama K, Kadowaki T, Abe N, Ratnayake DB, Yamamoto K. Involvement of a lysine-specific cysteine proteinase in hemoglobin adsorption and heme accumulation by Porphyromonas gingivalis. J Biol Chem. 1998;273:21225–21231. doi: 10.1074/jbc.273.33.21225. [DOI] [PubMed] [Google Scholar]
- Percival RS, Marsh PD, Devine DA, et al. Effect of temperature on growth, hemagglutination, and protease activity of Porphyromonas gingivalis. Infect Immun. 1999;67:1917–1921. doi: 10.1128/iai.67.4.1917-1921.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potempa J, Mikolajczyk-Pawlinska J, Brassell D, et al. Comparative properties of two cysteine proteinases (gingipains R), the products of two related but individual genes of Porphyromonas gingivalis. J Biol Chem. 1998;273:21648–21657. doi: 10.1074/jbc.273.34.21648. [DOI] [PubMed] [Google Scholar]
- Radicella JP, Boiteux S. Repair of oxidized guanine in mammals: OGG1 genes. C R Seances Soc Biol Fil. 1997;191:755–763. [PubMed] [Google Scholar]
- Rocha ER, Smith CJ. Role of the alkyl hydroperoxide reductase (ahpCF) gene in oxidative stress defense of the obligate anaerobe Bacteroides fragilis. J Bacteriol. 1999;181:5701–5710. doi: 10.1128/jb.181.18.5701-5710.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha ER, Selby T, Coleman JP, Smith CJ. Oxidative stress response in an anaerobe, Bacteroides fragilis: a role for catalase in protection against hydrogen peroxide. J Bacteriol. 1996;178:6895–6903. doi: 10.1128/jb.178.23.6895-6903.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salyers AA. Bacterial Pathogenesis: a Molecular Approach. Washington DC: ASM Press; 1994. [Google Scholar]
- Seymour GJ, Gemmell E, Reinhardt RA, Eastcott J, Taubman MA. Immunopathogenesis of chronic inflammatory periodontal disease: cellular and molecular mechanisms. J Periodontal Res. 1993;28:478–486. doi: 10.1111/j.1600-0765.1993.tb02108.x. [DOI] [PubMed] [Google Scholar]
- Smalley JW, Silver J, Marsh PJ, Birss AJ. The periodontopathogen Porphyromonas gingivalis binds iron protoporphyrin IX in the μ-oxo dimeric form: an oxidative buffer and possible pathogenic mechanism. Biochem J. 1998;331(Pt 3):681–685. doi: 10.1042/bj3310681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley JW, Birss AJ, Silver J. The periodontal pathogen Porphyromonas gingivalis harnesses the chemistry of the μ-oxo bishaem of iron protoporphyrin IX to protect against hydrogen peroxide. FEMS Microbiol Lett. 2000;183:159–164. doi: 10.1111/j.1574-6968.2000.tb08951.x. [DOI] [PubMed] [Google Scholar]
- Springer B, Master S, Sander P, et al. Silencing of oxidative stress response in Mycobacterium tuberculosis: expression patterns of ahpC in virulent and avirulent strains and effect of ahpC inactivation. Infect Immun. 2001;69:5967–5973. doi: 10.1128/IAI.69.10.5967-5973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sroka A, Sztukowska M, Potempa J, Travis J, Genco CA. Degradation of host heme proteins by lysine and arginine-specific cysteine proteinases(Gingipains) of Porphyromonas gingivalis. J Bacteriol. 2001;183:5609–5616. doi: 10.1128/JB.183.19.5609-5616.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztukowska M, Bugno M, Potempa J, Travis J, Kurtz DM., Jr Role of rubrerythrin in the oxidative stress response of Porphyromonas gingivalis. Mol Microbiol. 2002;44:479–488. doi: 10.1046/j.1365-2958.2002.02892.x. [DOI] [PubMed] [Google Scholar]
- Taylor PD, Inchley CJ, Gallagher MP. The Salmonella typhimurium AhpC polypeptide is not essential for virulence in BALB/c mice but is recognized as an antigen during infection. Infect Immun. 1998;66:3208–3217. doi: 10.1128/iai.66.7.3208-3217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueshima J, Shoji M, Ratnayake DB, et al. Purification, gene cloning, gene expression, and mutants of Dps from the obligate anaerobe Porphyromonas gingivalis. Infect Immun. 2003;71:1170–1178. doi: 10.1128/IAI.71.3.1170-1178.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke TE, Levine MJ, Genco RJ. Neutrophil function and oral disease. J Oral Pathol. 1985;14:95–120. doi: 10.1111/j.1600-0714.1985.tb00474.x. [DOI] [PubMed] [Google Scholar]
- Vanterpool E, Roy F, Fletcher HM. The vimE gene downstream of vimA is independently expressed and is involved in modulating proteolytic activity in Porphyromonas gingivalis W83. Infect Immun. 2004;72:5555–5564. doi: 10.1128/IAI.72.10.5555-5564.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanterpool E, Roy F, Fletcher HM. Inactivation of vimF, a putative glycosyltransferase gene downstream of vimE, alters glycosylation and activation of the gingipains in Porphyromonas gingivalis W83. Infect Immun. 2005a;73:3971–3982. doi: 10.1128/IAI.73.7.3971-3982.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanterpool E, Roy F, Sandberg L, Fletcher HM. Altered gingipain maturation in vimA- and vimE-defective isogenic mutants of Porphyromonas gingivalis. Infect Immun. 2005b;73:1357–1366. doi: 10.1128/IAI.73.3.1357-1366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Olczak AA, Walton JP, Maier RJ. Contribution of the Helicobacter pylori thiol peroxidase bacterioferritin comigratory protein to oxidative stress resistance and host colonization. Infect Immun. 2005;73:378–384. doi: 10.1128/IAI.73.1.378-384.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]