

Abstract
Purpose of Review
Polycystic ovary syndrome (PCOS) is diagnosed by its characteristic reproductive features. However, PCOS is also associated with metabolic abnormalities, including insulin resistance and β-cell dysfunction. The severity of these abnormalities varies according to the reproductive phenotype, with the so-called NIH or classic phenotype conferring the greatest metabolic risk. The increased risk for type 2 diabetes (T2D) is well-established among affected women with the NIH phenotype, but whether PCOS also confers an increased risk for cardiovascular events remains unknown.
Recent Findings
Recent studies in daughters of affected women have found evidence for pancreatic β-cell dysfunction prior to menarche. Further, genetic analyses have provided evidence that metabolic abnormalities such as obesity and insulin resistance contribute to the pathogenesis of PCOS.
Summary
PCOS increases the risk for T2D. However, the risk for cardiovascular disease has not been quantified, and prospective, longitudinal studies are still critically needed.
Keywords: Polycystic ovary syndrome, insulin resistance, obesity, type 2 diabetes, cardiovascular disease
Introduction
Polycystic ovary syndrome (PCOS) is among the most common endocrine disorders in women of reproductive age with prevalence of 5–15%, depending on the diagnostic criteria applied [1]. PCOS is defined by its reproductive features of hyperandrogenism, chronic oligo-anovulation, and/or polycystic ovarian morphology (PCOM) [2]. However, it has become increasingly evident over the past 35 years that PCOS is also a major metabolic disorder associated with substantial defects in insulin action and pancreatic β-cell dysfunction [1, 2]. While these metabolic defects are independent of obesity [2, 3], obesity is a common feature of PCOS, and it substantially worsens these metabolic defects [1, 2].
PCOS is a leading risk factor for dysglycemia and type 2 diabetes (T2D) in adolescent [4, 5] as well as in young adult women [6]. These metabolic abnormalities may also confer increased risk for cardiovascular disease in affected women. However, prospective, longitudinal studies of long-term health outcomes in women with PCOS are lacking. Recent important contributions to the field include genetic evidence implicating metabolic factors such as obesity and insulin resistance in the causality of PCOS ●●[7]. Studies in daughters of affected women, who have increased risk for PCOS, suggest metabolic phenotypes may be present prior to menarche when the syndrome can be diagnosed ● [8, 9]. Accordingly, early identification and implementation of strategies to prevent T2D may be feasible. In this review, we will summarize the literature on metabolic risk in PCOS, highlight recent contributions to the field, and identify important gaps in our existing knowledge to be addressed in future studies.
Distinct Diagnostic Criteria PCOS
The diagnostic criteria for PCOS are all based on expert opinion, the lowest level of evidence ([10], Table 1). The first set of diagnostic criteria were established at the 1990 NICHD Conference on PCOS [11]. The NIH criteria require both hyperandrogenism (clinical or biochemical) and chronic anovulation, after exclusion of alternative diagnoses which could result in these phenotypes, including hyperprolactinemia, non-classical congenital adrenal hyperplasia, and androgen-secreting neoplasms [11]. This phenotype is referred to as NIH or “classic” PCOS [11]. In 2003, the diagnostic criteria were broadened with the addition of ovarian morphology on ultrasound at the ESHRE/ASRM Meeting in Rotterdam, Netherlands [12]. Although this meeting has been designated as a consensus meeting, there was no formal consensus process [10]. Rather, recommendations were based on expert opinion rather than on scientific evidence. The resulting Rotterdam criteria require 2 of the following 3 findings: 1) hyperandrogenism (clinical or biochemical); 2) chronic anovulation; and 3) PCOM [12]. These criteria added two additional PCOS phenotypes: women with PCOM and chronic anovulation but lacking hyperandrogenism, and women with PCOM and hyperandrogenism, but lacking anovulation.
Table 1.
Diagnostic Criteria for PCOS
| NIH [11] | Hyperandrogenism + chronic anovulation |
| Rotterdam [12] | Two of the following: hyperandrogenism, chronic anovulation, PCOM |
| Androgen Excess Society [13] | Hyperandrogenism + ovarian dysfunction, indicated by oligoanovulation and/or PCOM |
All criteria require exclusion of other disorders which can cause a similar phenotype, including hyperprolactinemia, non-classical congenital adrenal hyperplasia, and androgen-secreting neoplasms. Adapted from [2], used with permission.
In 2006, an expert panel of the Androgen Excess Society recommended that hyperandrogenemia be a required criterion for the diagnosis of PCOS [13], eliminating the phenotype of chronic anovulation and PCOM in the absence of hyperandrogenism. The significance of PCOM in the diagnosis of PCOS continues to be a matter of debate. The presence or absence of PCOM appears to be age-related [14] but not a marker for features of the syndrome [15]. In women with the NIH PCOS phenotype, PCOM does not confer additional metabolic risk [15]. PCOM does identify women at increased risk for ovarian hyperstimulation during assisted reproductive treatments [16].
There is now an extensive literature comparing the metabolic features of the major PCOS phenotypes: anovulation and hyperandrogenemia +/− PCOM (NIH criteria), PCOM with hyperandrogenemia but no anovulation, and anovulation and PCOM but no hyperandrogenemia [17–19]. There is expert agreement that the NIH phenotype confers the greatest metabolic risk [20]. Therefore, studies which include the Rotterdam phenotypes of anovulation + PCOM and/or hyperandrogenemia + PCOM may underestimate metabolic phenotypes observed in NIH phenotype PCOS.
Pathogenesis of PCOS: Roles of Hyperandrogenemia & Insulin Resistance
The pathophysiology of PCOS is complex, influenced by genetic and metabolic factors, and involves a multitude of features, including disordered gonadotropin secretion, hyperandrogenemia, insulin resistance and hyperinsulinemia, ovarian dysfunction, and alteration in follicular development and selection [1]. The pathophysiology of the reproductive features of PCOS has been comprehensively reviewed elsewhere [1]. However, the interaction between hyperandrogenemia and insulin resistance likely plays a central role in development of the key phenotypic features of PCOS [1, 2]. Hyperandrogenemia is the biochemical hallmark of PCOS [2]. While the ovaries are the main source of increased androgens in PCOS [21], adrenal androgen excess is also observed in 20–30% of affected women [13]. Evidence in animal models suggest that androgen exposure during early critical developmental periods constitutes one potential mechanism for later development of the reproductive and metabolic features of PCOS [22]. Epigenetic changes represent a likely mechanism for the persistent reproductive and metabolic consequences of intrauterine androgen exposure in these animals [23].
However, insulin resistance and resulting hyperinsulinemia may also play a central role in the pathogenesis of androgen excess in PCOS [1, 2]. Insulin acts as a co-gonadotropin to increase LH-induced androgen synthesis in theca cells [24] and can enhance GnRH-mediated gonadotropin secretion [25]. Insulin also reduces hepatic SHBG synthesis, thereby increasing the levels of bioavailable androgens [26]. Therefore, interventions aimed at targeting insulin sensitivity or androgen concentrations and/or activity may improve both the reproductive and metabolic features of the disorder [1, 2].
Metabolic Phenotype in PCOS
Whole Body Insulin Action
Hyperinsulinemia suggesting the presence of insulin resistance in PCOS was first reported in 1980, with the observation of increased insulin responses during the oral glucose tolerance test (OGTT), which were independent of obesity in affected women [27]. The defects in total body insulin action were quantified in PCOS using the gold-standard technique, the euglycemic clamp [28]. Insulin-mediated glucose disposal (IMGD), which reflects primarily skeletal muscle insulin action [29], was reduced by 35–40% in both lean and obese women with PCOS compared to control women of similar age and body composition [3]. The magnitude of the decrease was similar to that seen in substantially older patients with T2D [29]. IMGD was reduced by a similar extent by obesity per se; therefore, PCOS and obesity had independent and additive effects on IMGD [3]. Endogenous glucose production was increased only in obese women with PCOS [30] indicating a synergistic deleterious effect of adiposity and PCOS on hepatic insulin action [30].
Insulin resistance is a consistent finding in obese women with PCOS, whereas some studies in lean affected women have failed to detect defects in in vivo insulin action [31]. The reason for this inconsistency is unclear, and ethnic/racial differences in insulin sensitivity may be one factor contributing to these inconsistent findings [32].. Nevertheless, a recent meta-analysis of clamp assessments of insulin action in PCOS found decreased mean IMGD in the majority of studies of both lean and obese affected women ●[33].
Many studies of the prevalence of insulin resistance in affected women have been limited by the use of surrogate markers of insulin action [34, 35]. These surrogate markers, such as fasting and postchallenge insulin and glucose levels, are confounded by differences in pancreatic β-cell function, insulin clearance, and glucose absorption [9, 36, 37]. Even when euglycemic clamp was used, normative data has been problematic, and studies have often relied upon historical rather than concurrently-studied control women [18]. Further, there is considerable overlap in measures of insulin action between PCOS and control groups [2]. These data suggest that insulin resistance is common among women with PCOS, but not a universal feature of the syndrome.
Insulin Secretion
There is now considerable evidence that defects in insulin secretion are required for the development of T2D [38, 39]. Normal pancreatic β-cells are able to increase insulin secretion to compensate for reduced insulin sensitivity [38], such that the product of insulin secretion and insulin sensitivity is constant [40]. This hyperbolic relationship is known as the disposition index (DI) (Figure 1, [40]). It is only when the β-cell is unable to increase insulin secretion sufficiently to compensate for peripheral insulin resistance that dysglycemia and T2D develop [38, 39]. Decreased DI is the most powerful predictor of T2D risk [41]. DI can be reduced despite fasting and postprandial hyperinsulinemia [42]. Studies that failed to correct insulin secretion for insulin sensitivity have concluded that insulin secretion was increased in women with PCOS [43, 44]. However, direct assessments of insulin secretion found evidence for β-cell dysfunction in affected women [2, 45, 46]. DI was decreased in both lean and obese women with PCOS and this decrease could precede decompensations in glucose tolerance [45, 47]. These defects were even more pronounced in PCOS women with a family history of T2D [45]. It is notable that β-cell defects seem to be an early finding in women with PCOS, as adolescent girls with PCOS and impaired glucose tolerance (IGT) had decreased DI [48].
Figure 1. Defects in Disposition Index (DI) Observed in Premenarchal PCOS Daughters.
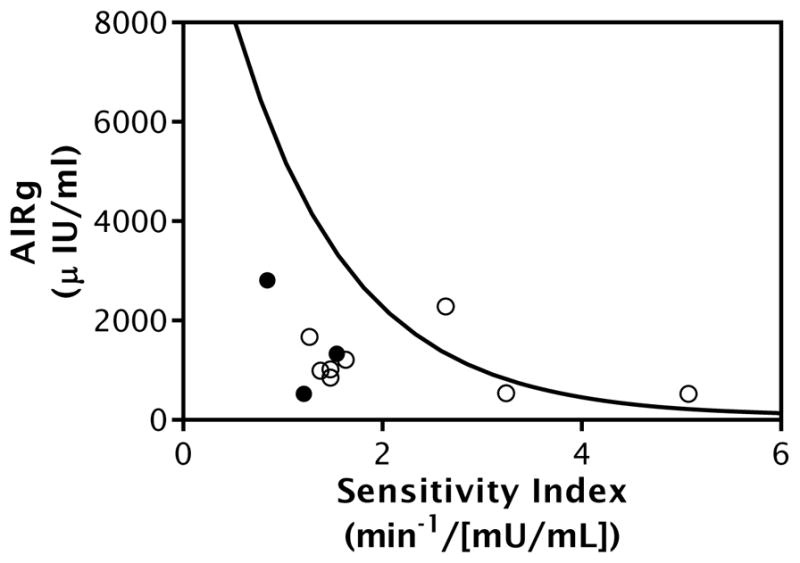
Pictured data from PCOS daughters and control girls of comparable age, BMI, and pubertal stage. All girls were aged 8 to 12 years of age and breast Tanner Stage I-III. Frequently-sampled IV glucose tolerance test (FSIGT)-derived measure of insulin sensitivity (Sensitivity Index, SI) plotted on the X axis, measure of insulin secretion (Acute Insulin Response to Glucose, AIRg) plotted on the Y axis. The product of SI and AIRg is termed the disposition index (DI) and is a measure of β-cell function. In the normally functioning β-cell, the relationship between insulin sensitivity and secretion follows a hyperbolic pattern. Hyperbolic line fit for the control girl data, the PCOS daughter data are plotted individually (open circles). PCOS daughters with dysglycemia denoted by black circles. Most PCOS daughters had a DI below the control population average indicating that they are at high risk to progress to type 2 diabetes [41]. Figure originally published in [9], used with permission.
Insulin Clearance
Reductions in insulin clearance may also contribute to hyperinsulinemia in PCOS [37]. Indeed, C-peptide model analysis for hepatic insulin extraction [46] suggested insulin clearance is decreased in affected women [46]. Studies have shown that differences in insulin clearance are also influenced by genetic factors and associated with insulin resistance [49]. Therefore, fasting hyperinsulinemia observed in PCOS likely results from decreased hepatic insulin clearance, in addition to increased basal insulin secretion in the setting of decreased insulin sensitivity [2].
Cellular & Molecular Mechanisms for Defects in Insulin Action
The most striking defect in adipocyte insulin action in PCOS was a significant increase in the ED50 of insulin for stimulation of glucose uptake, indicating decreased insulin sensitivity, in subcutaneous adipocytes from lean and obese PCOS women compared to those from weight-comparable, reproductively normal women [30, 50]. There were also modest but significant decreases in maximal rates of insulin mediated glucose disposal in PCOS subcutaneous adipocytes [30, 51], while the number and affinity of insulin receptors did not differ [30]. Therefore, in PCOS there is a post-binding defect in insulin sensitivity resulting in reduced insulin-mediated receptor signaling [30, 51].
Studies in skin fibroblasts and skeletal muscle have provided further detail regarding the mechanism for this post-binding defect in insulin-mediated receptor signaling in PCOS [52]. As observed in isolated adipocytes, there was no change in insulin binding or receptor affinity in vitro in skin fibroblasts [52]. However, basal and insulin-stimulated autophosphorylation of insulin receptors was reduced in approximately 50% of PCOS fibroblasts [52]. Constitutive serine phosphorylation was also increased in fibroblast insulin receptors, resulting in inhibition of post-receptor signaling [52]. Similarly, in vivo studies have confirmed decreases in post-receptor insulin signaling, including insulin-mediated IRS-1-associated PI3-K activation, in serial skeletal muscle biopsies obtained during euglycemic clamp studies in women with PCOS, which were associated with reduced IMGD in the affected women [53].
In addition to these metabolic actions, insulin also has mitogenic actions on cell growth and differentiation. Previous studies have shown that the metabolic actions of insulin can be disrupted while the mitogenic actions are preserved [54], as has been observed in cultured skin fibroblasts in patients with severe insulin resistance syndromes [55]. Indeed, a similar selective defect in insulin action has been found in skin fibroblasts [56] and ovarian granulosa-lutein cells [57] in women with PCOS.
Adipose depot-specific alterations in lipolysis have also been reported in women with PCOS. While subcutaneous adipocytes had decreased sensitivity to catecholamine-stimulated lipolysis [58], adipocytes from the visceral fat depot had increased catecholamine-stimulated lipolysis [59]. This increase in visceral adipose lipolytic sensitivity to catecholamines may result in increased portal delivery of free fatty acids, causing increased hepatic lipid accumulation and ultimately, worsening of hepatic insulin resistance [2, 59].
In summary, in vitro and in vivo evidence in adipocytes, fibroblasts, and skeletal muscle has confirmed a post-binding defect in insulin signal transduction characterized by constitutive serine phosphorylation, resulting in inhibition of post-receptor signaling in PCOS [51–53]. These defects in insulin signaling may affect the metabolic but not the mitogenic actions of insulin in PCOS women [56]. Alterations in visceral adipose tissue lipolysis may represent an additional contributing mechanism for the insulin resistance observed in PCOS women [59].
Obesity and PCOS
Obesity is common in women with PCOS [1], leading some to question whether obesity may play a central role in the pathogenesis of the reproductive and metabolic features of PCOS. In the United States, the prevalence of obesity in women with PCOS is as high as 80% [60, 61], while outside the United States, the prevalence of obesity in women with PCOS is only around 50% [62]. The consistent prevalence rate for PCOS among populations with very disparate obesity prevalence provides one piece of evidence that obesity is not a critical factor in the pathogenesis of the syndrome [63]. In addition, there are many lean women with PCOS [3], and the majority of obese women are reproductively normal [64]. Further, the prevalence of PCOS has remained fairly stable over time despite marked increases in the prevalence of obesity [63]. Therefore, while obesity may impact the severity of the reproductive and metabolic features of the disorder, the pathogenesis of PCOS is independent of obesity.
The question of whether women with PCOS are predisposed to obesity has not been adequately studied. Two early investigations of basal metabolic rate in PCOS using indirect calorimetry, a precise measure of metabolic rate, had conflicting results [65, 66]. A recent well-powered study in women with NIH phenotype PCOS found no difference in metabolic rate compared with reproductively normal control women of comparable age and BMI, but used a less accurate measure of basal metabolic rate [67]. There is no study investigating caloric intake in women with PCOS. While several studies have reported decreased meal-stimulated and/or basal ghrelin levels in women with PCOS [68–70], there is no convincing evidence for differences in satiety among affected women [68, 69].
Some studies have found women with PCOS have increased waist circumference [71] suggesting that they have increased abdominal fat. However, studies that have quantified visceral adipose tissue volume by accurate modalities such as CT or MRI have failed to show differences in visceral adipose tissue volume or distribution compared with reproductively normal women of comparable BMI [72, 73]. Regardless, as catecholamine-stimulated lipolytic activity is reduced in PCOS visceral adipose tissue [2, 59], this adipose depot likely plays a role in the insulin resistance in the disorder irrespective of differences in visceral adipose volume. Indeed, the insulin resistance observed in PCOS has a direct impact not only on metabolic features of the syndrome but even on androgen action in affected women through insulin-mediated reduction in circulating sex hormone binding globulin levels, resulting in increased free, biochemically active, circulating androgens [26].
In addition to its impact on the metabolic phenotype in PCOS [3, 30], obesity also contributes to the reproductive abnormalities observed in affected women. Studies of morbidly obese women undergoing bariatric surgery suggest that dramatic weight loss can result in resolution of both the reproductive and metabolic features of the PCOS phenotype in this subset of patients [74, 75]. In addition, a longitudinal study found that treatment with hypocaloric diet and metformin resulting in even less dramatic weight loss resulted in improvements in measures of insulin sensitivity, androgen concentrations, hirsutism scores, and menstrual cyclicity [76]. Finally, a recent randomized controlled trial found that pre-conception lifestyle modifications resulting in weight loss was superior to pretreatment with oral contraceptive pills in improving ovulation rates in overweight and obese PCOS women undergoing ovulation induction, providing additional evidence for an impact of weight loss on improvement in the reproductive features of PCOS ●●[77].
Impaired Glucose Tolerance (IGT) and T2D
PCOS represents one of the leading risk factors for dysglycemia and T2D in women. Because the major defect in insulin action in PCOS involves primarily peripheral insulin-mediated glucose uptake rather than insulin-mediated hepatic glucose production [29, 30], dysglycemia typically presents with IGT rather than impaired fasting glucose in PCOS [61]. Indeed, IGT is very common and an early feature of the disorder, with prevalence estimates of ~30% in both adult women [60, 61, 78] and affected adolescents [4]. Data from the Nurses’ Health Study suggests PCOS is associated with a two times increased risk for T2D, using irregular menses as a proxy for PCOS diagnosis [79]. Obesity and family history of T2D are independent additive risk factors which increase the risk of IGT and T2D in affected women [61, 80]. Because dysglycemia is mainly postprandial in PCOS [61], fasting glucose levels and hemoglobin A1C levels are not sensitive for detecting glucose intolerance [81]. Accordingly, two hour post-glucose challenge glucose levels are necessary to assess glucose tolerance in affected women [82, 83].
There have been limited studies investigating the conversion rates from IGT to T2D in PCOS [84, 85]. While some studies [84, 85] have suggested that the annual conversion rate from IGT to T2D in women with PCOS is roughly half the general population conversion rate of 7% [86, 87], these studies were limited by small sample size [84, 85]. Larger, prospective longitudinal studies are needed to better assess the course of IGT and T2D in PCOS. There have been no prospective studies to assess unique diabetes prevention strategies in affected women. The current recommendation favoring metformin for PCOS women with IGT who fail lifestyle modification [82] is based on largely on extrapolation of data in the T2D literature [88]. While studies have demonstrated improvement in metabolic and reproductive features of the syndrome through treatment with metformin [76, 89], longitudinal data on the impact of metformin in reducing conversion to T2D in PCOS women are unavailable [86].
Metabolic Syndrome (MBS) and Dyslipidemia
MBS is a constellation of cardiometabolic risk factors associated with increased risk for T2D and cardiovascular disease [90]. There are a number of diagnostic criteria for MBS used in clinical practice, which are all characterized by the associations of central obesity, hypertriglyceridemia, low circulating high-density lipoprotein-cholesterol (HDL), elevated systolic and/or diastolic blood pressure, and elevated fasting glucose [90]. While women with PCOS have increased prevalence of MBS, the most common features observed in affected women are increased waist circumference, increased triglyceride levels, and low HDL levels [91–93]. The risk for MBS increases with adiposity; PCOS women in the highest BMI quartile have nearly 14 times greater MBS risk compared with lean women with PCOS [91]. The risk for MBS in PCOS also varies by national origin and ethnicity, with lower prevalence observed in Europe compared with the US [94, 95], suggesting possible environmental factors may compound the risk for MBS among women with PCOS in the US.
Women with PCOS also have elevated LDL levels independent of obesity [96], with prevalence of at-risk LDL (>130 mg/dL) of around 30% [97]. The dyslipidemia observed in PCOS women did not improve with improvements in insulin action after treatment with troglitazone [97], suggesting insulin resistance is not the primary driver of dyslipidemia observed in PCOS women. In contrast, the risk for MBS increased with increasing quartiles of bioavailable testosterone levels in adolescent girls with PCOS, after correcting for differences in insulin sensitivity [98]. In another study, treatment with flutamide, a pure androgen receptor antagonist, resulted in a significant decrease in the HDL/LDL ratio in women with PCOS [99]. These studies suggest that hyperandrogenemia may contribute to MBS risk and LDL increases in PCOS.
Cardiovascular Disease Risk Factors
Many studies have found evidence for increased cardiovascular disease risk in women with PCOS using surrogate endpoints [1, 83] such as increased carotid intima-media thickness [100, 101]. Studies have also reproducibly found evidence for endothelial dysfunction in affected women [102, 103]. While obesity had a major impact on carotid intima-media thickness, the endothelial dysfunction observed in PCOS women seemed to be largely independent of body weight [100–103]. Coronary artery calcification assessed by electron beam computed tomography, the most powerful predictor of coronary events [104], has been found to be significantly increased in some studies of women with PCOS compared with reproductively normal control women of comparable age and BMI, with odds radios in the range of 2.3–2.4 [105, 106]. However, another study found no difference in prevalence of coronary artery calcification or abdominal aortic plaque [107]. A subanalysis of a large a population-based multicenter cohort, the Coronary Artery Risk Development in Young Adults (CARDIA) Women’s study, found women with a history of both hyperandrogenemia and oligomenorrhea had an increased prevalence of coronary artery calcifications and increased carotid intima-media thickness, while women with isolated oligomenorrhea or hyperandrogenemia had no increased risk ●[108]. Therefore, risk for coronary artery disease may be increased only in NIH phenotype PCOS, while hyperandrogenemia alone may not be associated with increased risk.
Cardiovascular Events
There are no prospective, longitudinal studies investigating cardiovascular events in women with PCOS. Available studies investigating the prevalence of cardiovascular events in PCOS have been limited by retrospective or cross-sectional design, small sample size, and/or failure to study women during the late post-menopausal years, the age group in which cardiovascular events would be likely to occur. Wild et al found increased risk of nonfatal cerebrovascular disease in a retrospective analysis of PCOS defined primarily by histological evidence of polycystic ovaries following ovarian wedge resection [109]. In this study, there was also a 50% increase in cardiovascular disease deaths in women with PCOS, but as the mean age in the cohort was only 56.7 years, there were only 17 such events, and the increase did not achieve statistical significance [109]. The Nurse’s Health study also reported a 50% increased risk for coronary heart disease in over 80,000 women studied using irregular menstrual history as a proxy for PCOS diagnosis [110], providing further corroboration for the findings in the Wild study [109]. However, some studies in postmenopausal women with PCOS suggest the increased cardiovascular disease risk observed in younger affected women plateaus in later life, while unaffected women continue to develop more cardiovascular risk, ultimately resulting in similar cardiovascular risk in PCOS and reproductively normal women during the postmenopausal years [111, 112].
Obstructive Sleep Apnea (OSA)
Women with PCOS also have increased risk for OSA compared with reproductively normal control women of comparable BMI [113, 114], and OSA may be a risk factor for glucose intolerance in affected women. Indeed, multiple studies have demonstrated that sleep restriction is an independent risk factor for insulin resistance and decompensation in glucose tolerance in the general population [115]. Studies in PCOS women have found associations between the risk of OSA in PCOS and insulin resistance, obesity, and/or hyperandrogenemia [113, 116]. Treatment of OSA with continuous positive airway pressure may result in modest improvement in insulin sensitivity, diastolic blood pressure, and cardiac sympathovagal balance in these women [117].
Non-Alcoholic Fatty Liver Disease (NAFLD)
The association of PCOS and NAFLD remains somewhat unclear. Some studies have suggested that PCOS is associated with increased risk for hepatic steatosis, with prevalence estimates ranging broadly from 15–60% [118–120]. However, many of these studies have been limited by small sample size or failure to account for the impact of insulin resistance using accurate measures of insulin sensitivity [118–120]. There is some evidence that higher androgen concentrations increase risk for NAFLD in women with PCOS, as well as in reproductively normal women [119, 121]. Fewer studies have investigated the risk of non-alcoholic steatohepatitis (NASH; NAFLD associated with liver cell injury) in PCOS [120, 122], due to the need for invasive liver biopsy for confirmation of NASH. One study using hepatic apoptotic markers as evidence for NASH reported a prevalence of 27% in women with PCOS compared with only 1% of control women, but failed to account for the impact of differences in insulin resistance using accurate measures of insulin sensitivity [122].
Metabolic Phenotypes in First Degree Relatives of Women with PCOS
A number of observations have supported the hypothesis that there is a genetic susceptibility to PCOS and its associated metabolic features [2]. Twin studies have shown that PCOS is very heritable [123]. Male and female first degree relatives of women with PCOS have reproductive and metabolic abnormalities characteristic of the syndrome, consistent with a genetic contribution to these phenotypes in family members [8, 9, 124–127].
Both mothers [126] and brothers [125] of women with PCOS have defects in glucose homeostasis and circulating lipid levels. While brothers and fathers of affected women also have higher prevalence for MBS, this increased risk seems to be largely driven by higher rates of obesity among these men [124]. Increased risk of cardiovascular events has been observed in fathers but not mothers of women with PCOS [127].
Studies in daughters of affected women have identified distinct reproductive phenotypes early in development, even prior to the onset of puberty, suggesting PCOS may be recognized in affected girls at an early age [8, 9, 128]. These reproductive phenotypes include increased 5α-reductase activity starting during early childhood [128], and increased serum testosterone levels by the time of peripuberty ●[9].
Early metabolic phenotypes are also observed in PCOS daughters, suggesting metabolic defects may be early findings in the pathogenesis of the syndrome. Beginning between the ages of 4–9 years, PCOS daughters develop elevated glucose-stimulated insulin levels and decreased adiponectin levels [8], suggestive of insulin resistance. Starting around the time of peripuberty, defects in insulin secretion have been observed in PCOS daughters ●[9], evidenced by decreased DI compared to control girls of comparable age and BMI, measured during a frequently-sampled IV glucose tolerance test ●(Figure 1, [9]). Decreased DI persisted during 3 years of longitudinal follow up in these girls ●[9]. As decreased DI is a powerful predictor of development of T2D [41], this finding suggests PCOS daughters may have markedly increased risk for T2D. Further, β-cell dysfunction may be present in affected women even prior to development of the diagnostic reproductive features of the syndrome.
Genetic Factors in the Pathogenesis of PCOS
Genome-wide association studies (GWAS) have now reproducibly mapped over 20 PCOS susceptibility loci in PCOS cohorts of Han Chinese [129] and of European ancestry ●● [7, 130]. Recent GWAS studies have provided new evidence for the importance of neuroendocrine changes in disease pathogenesis, as a genetic locus in the region of the follicle-stimulating hormone B polypeptide (FSHB) gene was reproducibly associated with PCOS and also strongly associated with luteinizing hormone levels in affected women ●● [7, 130].
However, recent investigations of genetic causality using Mendelian randomization have also implicated metabolic factors in the pathogenesis of PCOS ●● [7]. Mendelian randomization is a technique employed in genetic epidemiologic studies to investigate for causal associations in observational data [131]. Using this technique, a recently-published PCOS genome-wide association study found significant associations between genetic susceptibility loci for obesity (P=2.5 × 10−9), insulin resistance (P=6 × 10−4), and low circulating SHBG levels (P=5 × 10−4) and PCOS diagnosis ●● [7]. The association of genetic loci which impact SHBG levels had also previously been implicated in the genetic causality of T2D using Mendelian randomization [132]. These findings imply that genetic mechanisms for obesity and insulin resistance may represent causal factors in the development of PCOS.
Conclusions and Remaining Questions
PCOS is a common metabolic disorder and a critical risk factor for glucose intolerance, MBS, and T2D in women [1, 83]. Indeed, obesity and insulin resistance contribute to disease pathogenesis [7], and studies in young girls suggest metabolic features of the syndrome may be present in girls at risk, even by the time of early puberty [8, 9]. Despite the attention to metabolic risk in PCOS, a number of unanswered questions remain. Prospective, longitudinal studies of progression of metabolic defects and development of cardiovascular outcomes in PCOS are critically needed. We also lack data on the impact of unique early prevention approaches for T2D and cardiovascular disease in PCOS, including whether responses to prevention or treatment approaches for T2D differ in women with PCOS compared with other causes for T2D. Continued efforts to study the early origins in PCOS are needed to progress toward development and implementation of such prevention measures.
Footnotes
Compliance with Ethical Standards
Conflict of Interest
Laura C. Torchen declares that she has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any original studies with human or animal subjects performed by the author.
References
Papers of particular interest, published recently, have been highlighted as:
● Of importance
●● Of major importance
- 1.Dumesic DA, Oberfield SE, Stener-Victorin E, Marshall JC, Laven JS, Legro RS. Scientific Statement on the Diagnostic Criteria, Epidemiology, Pathophysiology, and Molecular Genetics of Polycystic Ovary Syndrome. Endocr Rev. 2015;36(5):487–525. doi: 10.1210/er.2015-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev. 2012;33(6):981–1030. doi: 10.1210/er.2011-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes. 1989;38(9):1165–74. doi: 10.2337/diab.38.9.1165. [DOI] [PubMed] [Google Scholar]
- 4.Palmert MR, Gordon CM, Kartashov AI, Legro RS, Emans SJ, Dunaif A. Screening for abnormal glucose tolerance in adolescents with polycystic ovary syndrome. J Clin Endocrinol Metab. 2002;87(3):1017–23. doi: 10.1210/jcem.87.3.8305. [DOI] [PubMed] [Google Scholar]
- 5.Coviello AD, Legro RS, Dunaif A. Adolescent girls with polycystic ovary syndrome have an increased risk of the metabolic syndrome associated with increasing androgen levels independent of obesity and insulin resistance. J Clin Endocrinol Metab. 2006;91(2):492–7. doi: 10.1210/jc.2005-1666. [DOI] [PubMed] [Google Scholar]
- 6.Ehrmann DA, Barnes RB, Rosenfield RL, Cavaghan MK, Imperial J. Prevalence of impaired glucose tolerance and diabetes in women with polycystic ovary syndrome. Diabetes Care. 1999;22(1):141–6. doi: 10.2337/diacare.22.1.141. [DOI] [PubMed] [Google Scholar]
- 7●●.Day FR, Hinds DA, Tung JY, Stolk L, Styrkarsdottir U, Saxena R, et al. Causal mechanisms and balancing selection inferred from genetic associations with polycystic ovary syndrome. Nat Commun. 2015;6:8464. doi: 10.1038/ncomms9464. This recent genome wide association study identified both replicated and novel genetic susceptibility loci significantly associated with PCOS. This study also reported mendelian randomization evidence for association of genetic risk for obesity and insulin resistance with PCOS diagnosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sir-Petermann T, Maliqueo M, Codner E, Echiburu B, Crisosto N, Perez V, et al. Early metabolic derangements in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2007;92(12):4637–42. doi: 10.1210/jc.2007-1036. [DOI] [PubMed] [Google Scholar]
- 9●.Torchen LC, Fogel NR, Brickman WJ, Paparodis R, Dunaif A. Persistent apparent pancreatic beta-cell defects in premenarchal PCOS relatives. J Clin Endocrinol Metab. 2014;99(10):3855–62. doi: 10.1210/jc.2014-1474. This study found peripubertal daughters of women with PCOS have evidence for beta-cell dysfunction, which persists as puberty progresses. This finding suggests PCOS daughters have increased risk for type 2 diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atkins D, Best D, Briss PA, Eccles M, Falck-Ytter Y, Flottorp S, et al. Grading quality of evidence and strength of recommendations. Bmj. 2004;328(7454):1490. doi: 10.1136/bmj.328.7454.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunaif Andrea, et al., editors. Polycystic ovary syndrome. Boston: Blackwell Scientific Publications; St. Louis, Mo: Distributors, USA and Canada, Mosby-Year Book; 1992. [Google Scholar]
- 12.Rotterdam EA-SPcwg. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS) Hum Reprod. 2004;19(1):41–7. doi: 10.1093/humrep/deh098. [DOI] [PubMed] [Google Scholar]
- 13.Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W, et al. Positions statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J Clin Endocrinol Metab. 2006;91(11):4237–45. doi: 10.1210/jc.2006-0178. [DOI] [PubMed] [Google Scholar]
- 14.Johnstone EB, Rosen MP, Neril R, Trevithick D, Sternfeld B, Murphy R, et al. The polycystic ovary post-rotterdam: a common, age-dependent finding in ovulatory women without metabolic significance. J Clin Endocrinol Metab. 2010;95(11):4965–72. doi: 10.1210/jc.2010-0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Legro RS, Chiu P, Kunselman AR, Bentley CM, Dodson WC, Dunaif A. Polycystic ovaries are common in women with hyperandrogenic chronic anovulation but do not predict metabolic or reproductive phenotype. J Clin Endocrinol Metab. 2005;90(5):2571–9. doi: 10.1210/jc.2004-0219. [DOI] [PubMed] [Google Scholar]
- 16.Fauser BC, Diedrich K, Devroey P. Predictors of ovarian response: progress towards individualized treatment in ovulation induction and ovarian stimulation. Hum Reprod Update. 2008;14(1):1–14. doi: 10.1093/humupd/dmm034. [DOI] [PubMed] [Google Scholar]
- 17.Carmina E, Chu MC, Longo RA, Rini GB, Lobo RA. Phenotypic variation in hyperandrogenic women influences the findings of abnormal metabolic and cardiovascular risk parameters. J Clin Endocrinol Metab. 2005;90(5):2545–9. doi: 10.1210/jc.2004-2279. [DOI] [PubMed] [Google Scholar]
- 18.Moghetti P, Tosi F, Bonin C, Di Sarra D, Fiers T, Kaufman JM, et al. Divergences in insulin resistance between the different phenotypes of the polycystic ovary syndrome. J Clin Endocrinol Metab. 2013;98(4):E628–37. doi: 10.1210/jc.2012-3908. [DOI] [PubMed] [Google Scholar]
- 19.Welt CK, Gudmundsson JA, Arason G, Adams J, Palsdottir H, Gudlaugsdottir G, et al. Characterizing discrete subsets of polycystic ovary syndrome as defined by the Rotterdam criteria: the impact of weight on phenotype and metabolic features. J Clin Endocrinol Metab. 2006;91(12):4842–8. doi: 10.1210/jc.2006-1327. [DOI] [PubMed] [Google Scholar]
- 20.Fauser BC, Tarlatzis BC, Rebar RW, Legro RS, Balen AH, Lobo R, et al. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): the Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil Steril. 2012;97(1):28–38. e25. doi: 10.1016/j.fertnstert.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 21.Ehrmann DA, Barnes RB, Rosenfield RL. Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion. Endocr Rev. 1995;16(3):322–53. doi: 10.1210/edrv-16-3-322. [DOI] [PubMed] [Google Scholar]
- 22.Dumesic DA, Abbott DH, Padmanabhan V. Polycystic ovary syndrome and its developmental origins. Rev Endocr Metab Disord. 2007;8(2):127–41. doi: 10.1007/s11154-007-9046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dumesic DA, Goodarzi MO, Chazenbalk GD, Abbott DH. Intrauterine environment and polycystic ovary syndrome. Semin Reprod Med. 2014;32(3):159–65. doi: 10.1055/s-0034-1371087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nestler JE, Jakubowicz DJ, de Vargas AF, Brik C, Quintero N, Medina F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab. 1998;83(6):2001–5. doi: 10.1210/jcem.83.6.4886. [DOI] [PubMed] [Google Scholar]
- 25.Adashi EY, Hsueh AJ, Yen SS. Insulin enhancement of luteinizing hormone and follicle-stimulating hormone release by cultured pituitary cells. Endocrinology. 1981;108(4):1441–9. doi: 10.1210/endo-108-4-1441. [DOI] [PubMed] [Google Scholar]
- 26.Nestler JE, Powers LP, Matt DW, Steingold KA, Plymate SR, Rittmaster RS, et al. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syndrome. J Clin Endocrinol Metab. 1991;72(1):83–9. doi: 10.1210/jcem-72-1-83. [DOI] [PubMed] [Google Scholar]
- 27.Burghen GA, Givens JR, Kitabchi AE. Correlation of hyperandrogenism with hyperinsulinism in polycystic ovarian disease. J Clin Endocrinol Metab. 1980;50(1):113–6. doi: 10.1210/jcem-50-1-113. [DOI] [PubMed] [Google Scholar]
- 28.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237(3):E214–23. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 29.DeFronzo RA. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes. 1988;37(6):667–87. doi: 10.2337/diab.37.6.667. [DOI] [PubMed] [Google Scholar]
- 30.Dunaif A, Segal KR, Shelley DR, Green G, Dobrjansky A, Licholai T. Evidence for distinctive and intrinsic defects in insulin action in polycystic ovary syndrome. Diabetes. 1992;41(10):1257–66. doi: 10.2337/diab.41.10.1257. [DOI] [PubMed] [Google Scholar]
- 31.Ciampelli M, Fulghesu AM, Cucinelli F, Pavone V, Caruso A, Mancuso S, et al. Heterogeneity in beta cell activity, hepatic insulin clearance and peripheral insulin sensitivity in women with polycystic ovary syndrome. Hum Reprod. 1997;12(9):1897–901. doi: 10.1093/humrep/12.9.1897. [DOI] [PubMed] [Google Scholar]
- 32.Chiu KC, Cohan P, Lee NP, Chuang LM. Insulin sensitivity differs among ethnic groups with a compensatory response in beta-cell function. Diabetes Care. 2000;23(9):1353–8. doi: 10.2337/diacare.23.9.1353. [DOI] [PubMed] [Google Scholar]
- 33●.Cassar S, Misso ML, Hopkins WG, Shaw CS, Teede HJ, Stepto NK. Insulin resistance in polycystic ovary syndrome: a systematic review and meta-analysis of euglycaemic-hyperinsulinaemic clamp studies. Hum Reprod. 2016;31(11):2619–31. doi: 10.1093/humrep/dew243. This meta-analysis confirms earlier studies which find women with PCOS have decreased insulin sensitivity, independent of BMI, using only evidence from highly accurate measures of insulin action. [DOI] [PubMed] [Google Scholar]
- 34.Carmina E, Lobo RA. Use of fasting blood to assess the prevalence of insulin resistance in women with polycystic ovary syndrome. Fertil Steril. 2004;82(3):661–5. doi: 10.1016/j.fertnstert.2004.01.041. [DOI] [PubMed] [Google Scholar]
- 35.DeUgarte CM, Bartolucci AA, Azziz R. Prevalence of insulin resistance in the polycystic ovary syndrome using the homeostasis model assessment. Fertil Steril. 2005;83(5):1454–60. doi: 10.1016/j.fertnstert.2004.11.070. [DOI] [PubMed] [Google Scholar]
- 36.Diamanti-Kandarakis E, Kouli C, Alexandraki K, Spina G. Failure of mathematical indices to accurately assess insulin resistance in lean, overweight, or obese women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2004;89(3):1273–6. doi: 10.1210/jc.2003-031205. [DOI] [PubMed] [Google Scholar]
- 37.Hucking K, Watanabe RM, Stefanovski D, Bergman RN. OGTT-derived measures of insulin sensitivity are confounded by factors other than insulin sensitivity itself. Obesity (Silver Spring) 2008;16(8):1938–45. doi: 10.1038/oby.2008.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bergman RN, Finegood DT, Kahn SE. The evolution of beta-cell dysfunction and insulin resistance in type 2 diabetes. Eur J Clin Invest. 2002;32(Suppl 3):35–45. doi: 10.1046/j.1365-2362.32.s3.5.x. [DOI] [PubMed] [Google Scholar]
- 39.Kahn BB. Type 2 diabetes: when insulin secretion fails to compensate for insulin resistance. Cell. 1998;92(5):593–6. doi: 10.1016/s0092-8674(00)81125-3. [DOI] [PubMed] [Google Scholar]
- 40.Bergman RN, Phillips LS, Cobelli C. Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J Clin Invest. 1981;68(6):1456–67. doi: 10.1172/JCI110398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lorenzo C, Wagenknecht LE, Rewers MJ, Karter AJ, Bergman RN, Hanley AJ, et al. Disposition index, glucose effectiveness, and conversion to type 2 diabetes: the Insulin Resistance Atherosclerosis Study (IRAS) Diabetes Care. 2010;33(9):2098–103. doi: 10.2337/dc10-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kahn SE, Prigeon RL, McCulloch DK, Boyko EJ, Bergman RN, Schwartz MW, et al. Quantification of the Relationship Between Insulin Sensitivity and β-Cell Function in Human Subjects: Evidence for a Hyperbolic Function. Diabetes. 1993;42(11):1663–72. doi: 10.2337/diab.42.11.1663. [DOI] [PubMed] [Google Scholar]
- 43.Holte J, Bergh T, Berne C, Berglund L, Lithell H. Enhanced early insulin response to glucose in relation to insulin resistance in women with polycystic ovary syndrome and normal glucose tolerance. J Clin Endocrinol Metab. 1994;78(5):1052–8. doi: 10.1210/jcem.78.5.8175959. [DOI] [PubMed] [Google Scholar]
- 44.Goodarzi MO, Erickson S, Port SC, Jennrich RI, Korenman SG. beta-Cell function: a key pathological determinant in polycystic ovary syndrome. J Clin Endocrinol Metab. 2005;90(1):310–5. doi: 10.1210/jc.2004-1006. [DOI] [PubMed] [Google Scholar]
- 45.Ehrmann DA, Sturis J, Byrne MM, Karrison T, Rosenfield RL, Polonsky KS. Insulin secretory defects in polycystic ovary syndrome. Relationship to insulin sensitivity and family history of non-insulin-dependent diabetes mellitus. J Clin Invest. 1995;96(1):520–7. doi: 10.1172/jci118064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Meara NM, Blackman JD, Ehrmann DA, Barnes RB, Jaspan JB, Rosenfield RL, et al. Defects in beta-cell function in functional ovarian hyperandrogenism. J Clin Endocrinol Metab. 1993;76(5):1241–7. doi: 10.1210/jcem.76.5.8496316. [DOI] [PubMed] [Google Scholar]
- 47.Dunaif A, Finegood DT. Beta-cell dysfunction independent of obesity and glucose intolerance in the polycystic ovary syndrome. J Clin Endocrinol Metab. 1996;81(3):942–7. doi: 10.1210/jcem.81.3.8772555. [DOI] [PubMed] [Google Scholar]
- 48.Arslanian SA, Lewy VD, Danadian K. Glucose intolerance in obese adolescents with polycystic ovary syndrome: roles of insulin resistance and beta-cell dysfunction and risk of cardiovascular disease. J Clin Endocrinol Metab. 2001;86(1):66–71. doi: 10.1210/jcem.86.1.7123. [DOI] [PubMed] [Google Scholar]
- 49.Polidori DC, Bergman RN, Chung ST, Sumner AE. Hepatic and Extrahepatic Insulin Clearance Are Differentially Regulated: Results From a Novel Model-Based Analysis of Intravenous Glucose Tolerance Data. Diabetes. 2016;65(6):1556–64. doi: 10.2337/db15-1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ciaraldi TP, Aroda V, Mudaliar S, Chang RJ, Henry RR. Polycystic ovary syndrome is associated with tissue-specific differences in insulin resistance. J Clin Endocrinol Metab. 2009;94(1):157–63. doi: 10.1210/jc.2008-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ciaraldi TP, el-Roeiy A, Madar Z, Reichart D, Olefsky JM, Yen SS. Cellular mechanisms of insulin resistance in polycystic ovarian syndrome. J Clin Endocrinol Metab. 1992;75(2):577–83. doi: 10.1210/jcem.75.2.1322430. [DOI] [PubMed] [Google Scholar]
- 52.Dunaif A, Xia J, Book CB, Schenker E, Tang Z. Excessive insulin receptor serine phosphorylation in cultured fibroblasts and in skeletal muscle. A potential mechanism for insulin resistance in the polycystic ovary syndrome. J Clin Invest. 1995;96(2):801–10. doi: 10.1172/jci118126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dunaif A, Wu X, Lee A, Diamanti-Kandarakis E. Defects in insulin receptor signaling in vivo in the polycystic ovary syndrome (PCOS) Am J Physiol Endocrinol Metab. 2001;281(2):E392–9. doi: 10.1152/ajpendo.2001.281.2.E392. [DOI] [PubMed] [Google Scholar]
- 54.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 55.Flier JS, Moller DE, Moses AC, O’Rahilly S, Chaiken RL, Grigorescu F, et al. Insulin-mediated pseudoacromegaly: clinical and biochemical characterization of a syndrome of selective insulin resistance. J Clin Endocrinol Metab. 1993;76(6):1533–41. doi: 10.1210/jcem.76.6.8388881. [DOI] [PubMed] [Google Scholar]
- 56.Book CB, Dunaif A. Selective insulin resistance in the polycystic ovary syndrome. J Clin Endocrinol Metab. 1999;84(9):3110–6. doi: 10.1210/jcem.84.9.6010. [DOI] [PubMed] [Google Scholar]
- 57.Rice S, Christoforidis N, Gadd C, Nikolaou D, Seyani L, Donaldson A, et al. Impaired insulin-dependent glucose metabolism in granulosa-lutein cells from anovulatory women with polycystic ovaries. Hum Reprod. 2005;20(2):373–81. doi: 10.1093/humrep/deh609. [DOI] [PubMed] [Google Scholar]
- 58.Ek I, Arner P, Bergqvist A, Carlstrom K, Wahrenberg H. Impaired adipocyte lipolysis in nonobese women with the polycystic ovary syndrome: a possible link to insulin resistance? J Clin Endocrinol Metab. 1997;82(4):1147–53. doi: 10.1210/jcem.82.4.3899. [DOI] [PubMed] [Google Scholar]
- 59.Ek I, Arner P, Ryden M, Holm C, Thorne A, Hoffstedt J, et al. A unique defect in the regulation of visceral fat cell lipolysis in the polycystic ovary syndrome as an early link to insulin resistance. Diabetes. 2002;51(2):484–92. doi: 10.2337/diabetes.51.2.484. [DOI] [PubMed] [Google Scholar]
- 60.Ehrmann DA, Barnes RB, Rosenfield RL, Cavaghan MK, Imperial J. Prevalence of impaired glucose tolerance and diabetes in women with polycystic ovary syndrome. Diabetes Care. 1999;22(1):141–6. doi: 10.2337/diacare.22.1.141. [DOI] [PubMed] [Google Scholar]
- 61.Legro RS, Kunselman AR, Dodson WC, Dunaif A. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab. 1999;84(1):165–9. doi: 10.1210/jcem.84.1.5393. [DOI] [PubMed] [Google Scholar]
- 62.Balen AH, Conway GS, Kaltsas G, Techatrasak K, Manning PJ, West C, et al. Polycystic ovary syndrome: the spectrum of the disorder in 1741 patients. Hum Reprod. 1995;10(8):2107–11. doi: 10.1093/oxfordjournals.humrep.a136243. [DOI] [PubMed] [Google Scholar]
- 63.Yildiz BO, Knochenhauer ES, Azziz R. Impact of obesity on the risk for polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93(1):162–8. doi: 10.1210/jc.2007-1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Azziz R. Reproductive endocrinologic alterations in female asymptomatic obesity. Fertil Steril. 1989;52(5):703–25. doi: 10.1016/s0015-0282(16)61020-8. [DOI] [PubMed] [Google Scholar]
- 65.Georgopoulos NA, Saltamavros AD, Vervita V, Karkoulias K, Adonakis G, Decavalas G, et al. Basal metabolic rate is decreased in women with polycystic ovary syndrome and biochemical hyperandrogenemia and is associated with insulin resistance. Fertil Steril. 2009;92(1):250–5. doi: 10.1016/j.fertnstert.2008.04.067. [DOI] [PubMed] [Google Scholar]
- 66.Segal KR, Dunaif A. Resting metabolic rate and postprandial thermogenesis in polycystic ovarian syndrome. Int J Obes. 1990;14(7):559–67. [PubMed] [Google Scholar]
- 67.Churchill SJ, Wang ET, Bhasin G, Alexander C, Bresee C, Pall M, et al. Basal metabolic rate in women with PCOS compared to eumenorrheic controls. Clin Endocrinol (Oxf) 2015;83(3):384–8. doi: 10.1111/cen.12740. [DOI] [PubMed] [Google Scholar]
- 68.Arusoglu G, Koksal G, Cinar N, Tapan S, Aksoy DY, Yildiz BO. Basal and meal-stimulated ghrelin, PYY, CCK levels and satiety in lean women with polycystic ovary syndrome: effect of low-dose oral contraceptive. J Clin Endocrinol Metab. 2013;98(11):4475–82. doi: 10.1210/jc.2013-1526. [DOI] [PubMed] [Google Scholar]
- 69.Moran LJ, Noakes M, Clifton PM, Wittert GA, Le Roux CW, Ghatei MA, et al. Postprandial ghrelin, cholecystokinin, peptide YY, and appetite before and after weight loss in overweight women with and without polycystic ovary syndrome. Am J Clin Nutr. 2007;86(6):1603–10. doi: 10.1093/ajcn/86.5.1603. [DOI] [PubMed] [Google Scholar]
- 70.Moran LJ, Norman RJ, Teede HJ. Metabolic risk in PCOS: phenotype and adiposity impact. Trends Endocrinol Metab. 2015;26(3):136–43. doi: 10.1016/j.tem.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 71.Escobar-Morreale HF, San Millan JL. Abdominal adiposity and the polycystic ovary syndrome. Trends Endocrinol Metab. 2007;18(7):266–72. doi: 10.1016/j.tem.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 72.Barber TM, Golding SJ, Alvey C, Wass JA, Karpe F, Franks S, et al. Global adiposity rather than abnormal regional fat distribution characterizes women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93(3):999–1004. doi: 10.1210/jc.2007-2117. [DOI] [PubMed] [Google Scholar]
- 73.Manneras-Holm L, Leonhardt H, Kullberg J, Jennische E, Oden A, Holm G, et al. Adipose tissue has aberrant morphology and function in PCOS: enlarged adipocytes and low serum adiponectin, but not circulating sex steroids, are strongly associated with insulin resistance. J Clin Endocrinol Metab. 2011;96(2):E304–11. doi: 10.1210/jc.2010-1290. [DOI] [PubMed] [Google Scholar]
- 74.Escobar-Morreale HF, Botella-Carretero JI, Alvarez-Blasco F, Sancho J, San Millan JL. The polycystic ovary syndrome associated with morbid obesity may resolve after weight loss induced by bariatric surgery. J Clin Endocrinol Metab. 2005;90(12):6364–9. doi: 10.1210/jc.2005-1490. [DOI] [PubMed] [Google Scholar]
- 75.Skubleny D, Switzer NJ, Gill RS, Dykstra M, Shi X, Sagle MA, et al. The Impact of Bariatric Surgery on Polycystic Ovary Syndrome: a Systematic Review and Meta-analysis. Obes Surg. 2016;26(1):169–76. doi: 10.1007/s11695-015-1902-5. [DOI] [PubMed] [Google Scholar]
- 76.Pasquali R, Gambineri A, Biscotti D, Vicennati V, Gagliardi L, Colitta D, et al. Effect of long-term treatment with metformin added to hypocaloric diet on body composition, fat distribution, and androgen and insulin levels in abdominally obese women with and without the polycystic ovary syndrome. J Clin Endocrinol Metab. 2000;85(8):2767–74. doi: 10.1210/jcem.85.8.6738. [DOI] [PubMed] [Google Scholar]
- 77●●.Legro RS, Dodson WC, Kris-Etherton PM, Kunselman AR, Stetter CM, Williams NI, et al. Randomized Controlled Trial of Preconception Interventions in Infertile Women With Polycystic Ovary Syndrome. J Clin Endocrinol Metab. 2015;100(11):4048–58. doi: 10.1210/jc.2015-2778. This RCT provides compelling evidence for the effectiveness of lifestyle modifications in improving ovulation rates in subfertile women with PCOS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dunaif A, Graf M, Mandeli J, Laumas V, Dobrjansky A. Characterization of groups of hyperandrogenic women with acanthosis nigricans, impaired glucose tolerance, and/or hyperinsulinemia. J Clin Endocrinol Metab. 1987;65(3):499–507. doi: 10.1210/jcem-65-3-499. [DOI] [PubMed] [Google Scholar]
- 79.Solomon CG, Hu FB, Dunaif A, Rich-Edwards J, Willett WC, Hunter DJ, et al. Long or highly irregular menstrual cycles as a marker for risk of type 2 diabetes mellitus. Jama. 2001;286(19):2421–6. doi: 10.1001/jama.286.19.2421. [DOI] [PubMed] [Google Scholar]
- 80.Ehrmann DA, Kasza K, Azziz R, Legro RS, Ghazzi MN. Effects of race and family history of type 2 diabetes on metabolic status of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2005;90(1):66–71. doi: 10.1210/jc.2004-0229. [DOI] [PubMed] [Google Scholar]
- 81.Velling Magnussen L, Mumm H, Andersen M, Glintborg D. Hemoglobin A1c as a tool for the diagnosis of type 2 diabetes in 208 premenopausal women with polycystic ovary syndrome. Fertil Steril. 2011;96(5):1275–80. doi: 10.1016/j.fertnstert.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 82.Legro RS, Arslanian SA, Ehrmann DA, Hoeger KM, Murad MH, Pasquali R, et al. Diagnosis and treatment of polycystic ovary syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2013;98(12):4565–92. doi: 10.1210/jc.2013-2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wild RA, Carmina E, Diamanti-Kandarakis E, Dokras A, Escobar-Morreale HF, Futterweit W, et al. Assessment of cardiovascular risk and prevention of cardiovascular disease in women with the polycystic ovary syndrome: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome (AE-PCOS) Society. J Clin Endocrinol Metab. 2010;95(5):2038–49. doi: 10.1210/jc.2009-2724. [DOI] [PubMed] [Google Scholar]
- 84.Legro RS, Gnatuk CL, Kunselman AR, Dunaif A. Changes in glucose tolerance over time in women with polycystic ovary syndrome: a controlled study. J Clin Endocrinol Metab. 2005;90(6):3236–42. doi: 10.1210/jc.2004-1843. [DOI] [PubMed] [Google Scholar]
- 85.Norman RJ, Masters L, Milner CR, Wang JX, Davies MJ. Relative risk of conversion from normoglycaemia to impaired glucose tolerance or non-insulin dependent diabetes mellitus in polycystic ovarian syndrome. Hum Reprod. 2001;16(9):1995–8. doi: 10.1093/humrep/16.9.1995. [DOI] [PubMed] [Google Scholar]
- 86.Knowler WC, Hamman RF, Edelstein SL, Barrett-Connor E, Ehrmann DA, Walker EA, et al. Prevention of type 2 diabetes with troglitazone in the Diabetes Prevention Program. Diabetes. 2005;54(4):1150–6. doi: 10.2337/diabetes.54.4.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344(18):1343–50. doi: 10.1056/nejm200105033441801. [DOI] [PubMed] [Google Scholar]
- 88.Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nestler JE, Jakubowicz DJ, Evans WS, Pasquali R. Effects of metformin on spontaneous and clomiphene-induced ovulation in the polycystic ovary syndrome. N Engl J Med. 1998;338(26):1876–80. doi: 10.1056/nejm199806253382603. [DOI] [PubMed] [Google Scholar]
- 90.Alberti KGMM, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the Metabolic Syndrome. A Joint Interim Statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. 2009;120(16):1640–5. doi: 10.1161/circulationaha.109.192644. [DOI] [PubMed] [Google Scholar]
- 91.Ehrmann DA, Liljenquist DR, Kasza K, Azziz R, Legro RS, Ghazzi MN. Prevalence and predictors of the metabolic syndrome in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91(1):48–53. doi: 10.1210/jc.2005-1329. [DOI] [PubMed] [Google Scholar]
- 92.Valkenburg O, Steegers-Theunissen RP, Smedts HP, Dallinga-Thie GM, Fauser BC, Westerveld EH, et al. A more atherogenic serum lipoprotein profile is present in women with polycystic ovary syndrome: a case-control study. J Clin Endocrinol Metab. 2008;93(2):470–6. doi: 10.1210/jc.2007-1756. [DOI] [PubMed] [Google Scholar]
- 93.Wild RA, Painter PC, Coulson PB, Carruth KB, Ranney GB. Lipoprotein lipid concentrations and cardiovascular risk in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1985;61(5):946–51. doi: 10.1210/jcem-61-5-946. [DOI] [PubMed] [Google Scholar]
- 94.Carmina E, Napoli N, Longo RA, Rini GB, Lobo RA. Metabolic syndrome in polycystic ovary syndrome (PCOS): lower prevalence in southern Italy than in the USA and the influence of criteria for the diagnosis of PCOS. Eur J Endocrinol. 2006;154(1):141–5. doi: 10.1530/eje.1.02058. [DOI] [PubMed] [Google Scholar]
- 95.Taponen S, Martikainen H, Jarvelin MR, Sovio U, Laitinen J, Pouta A, et al. Metabolic cardiovascular disease risk factors in women with self-reported symptoms of oligomenorrhea and/or hirsutism: Northern Finland Birth Cohort 1966 Study. J Clin Endocrinol Metab. 2004;89(5):2114–8. doi: 10.1210/jc.2003-031720. [DOI] [PubMed] [Google Scholar]
- 96.Legro RS, Kunselman AR, Dunaif A. Prevalence and predictors of dyslipidemia in women with polycystic ovary syndrome. Am J Med. 2001;111(8):607–13. doi: 10.1016/s0002-9343(01)00948-2. [DOI] [PubMed] [Google Scholar]
- 97.Legro RS, Azziz R, Ehrmann D, Fereshetian AG, O’Keefe M, Ghazzi MN. Minimal response of circulating lipids in women with polycystic ovary syndrome to improvement in insulin sensitivity with troglitazone. J Clin Endocrinol Metab. 2003;88(11):5137–44. doi: 10.1210/jc.2003-030044. [DOI] [PubMed] [Google Scholar]
- 98.Coviello AD, Legro RS, Dunaif A. Adolescent girls with polycystic ovary syndrome have an increased risk of the metabolic syndrome associated with increasing androgen levels independent of obesity and insulin resistance. J Clin Endocrinol Metab. 2006;91(2):492–7. doi: 10.1210/jc.2005-1666. [DOI] [PubMed] [Google Scholar]
- 99.Diamanti-Kandarakis E, Mitrakou A, Raptis S, Tolis G, Duleba AJ. The effect of a pure antiandrogen receptor blocker, flutamide, on the lipid profile in the polycystic ovary syndrome. J Clin Endocrinol Metab. 1998;83(8):2699–705. doi: 10.1210/jcem.83.8.5041. [DOI] [PubMed] [Google Scholar]
- 100.Guzick DS, Talbott EO, Sutton-Tyrrell K, Herzog HC, Kuller LH, Wolfson SK., Jr Carotid atherosclerosis in women with polycystic ovary syndrome: initial results from a case-control study. Am J Obstet Gynecol. 1996;174(4):1224–9. doi: 10.1016/s0002-9378(96)70665-8. discussion 9–32. [DOI] [PubMed] [Google Scholar]
- 101.Talbott EO, Guzick DS, Sutton-Tyrrell K, McHugh-Pemu KP, Zborowski JV, Remsberg KE, et al. Evidence for association between polycystic ovary syndrome and premature carotid atherosclerosis in middle-aged women. Arterioscler Thromb Vasc Biol. 2000;20(11):2414–21. doi: 10.1161/01.atv.20.11.2414. [DOI] [PubMed] [Google Scholar]
- 102.Kravariti M, Naka KK, Kalantaridou SN, Kazakos N, Katsouras CS, Makrigiannakis A, et al. Predictors of endothelial dysfunction in young women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2005;90(9):5088–95. doi: 10.1210/jc.2005-0151. [DOI] [PubMed] [Google Scholar]
- 103.Paradisi G, Steinberg HO, Hempfling A, Cronin J, Hook G, Shepard MK, et al. Polycystic ovary syndrome is associated with endothelial dysfunction. Circulation. 2001;103(10):1410–5. doi: 10.1161/01.cir.103.10.1410. [DOI] [PubMed] [Google Scholar]
- 104.Folsom AR, Kronmal RA, Detrano RC, O’Leary DH, Bild DE, Bluemke DA, et al. Coronary artery calcification compared with carotid intima-media thickness in the prediction of cardiovascular disease incidence: the Multi-Ethnic Study of Atherosclerosis (MESA) Arch Intern Med. 2008;168(12):1333–9. doi: 10.1001/archinte.168.12.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Christian RC, Dumesic DA, Behrenbeck T, Oberg AL, Sheedy PF, 2nd, Fitzpatrick LA. Prevalence and predictors of coronary artery calcification in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88(6):2562–8. doi: 10.1210/jc.2003-030334. [DOI] [PubMed] [Google Scholar]
- 106.Talbott EO, Zborowski JV, Rager JR, Boudreaux MY, Edmundowicz DA, Guzick DS. Evidence for an association between metabolic cardiovascular syndrome and coronary and aortic calcification among women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2004;89(11):5454–61. doi: 10.1210/jc.2003-032237. [DOI] [PubMed] [Google Scholar]
- 107.Chang AY, Ayers C, Minhajuddin A, Jain T, Nurenberg P, de Lemos JA, et al. Polycystic ovarian syndrome and subclinical atherosclerosis among women of reproductive age in the Dallas heart study. Clin Endocrinol (Oxf) 2011;74(1):89–96. doi: 10.1111/j.1365-2265.2010.03907.x. [DOI] [PubMed] [Google Scholar]
- 108●.Calderon-Margalit R, Siscovick D, Merkin SS, Wang E, Daviglus ML, Schreiner PJ, et al. Prospective association of polycystic ovary syndrome with coronary artery calcification and carotid-intima-media thickness: the Coronary Artery Risk Development in Young Adults Women’s study. Arterioscler Thromb Vasc Biol. 2014;34(12):2688–94. doi: 10.1161/ATVBAHA.114.304136. This epidemiologic study reports increased risk for subclinical cardiovascular disease in women with both hyperandrogenemia and oligomenorrhea, who likely fulfill NIH criteria for PCOS. [DOI] [PubMed] [Google Scholar]
- 109.Wild S, Pierpoint T, McKeigue P, Jacobs H. Cardiovascular disease in women with polycystic ovary syndrome at long-term follow-up: a retrospective cohort study. Clin Endocrinol (Oxf) 2000;52(5):595–600. doi: 10.1046/j.1365-2265.2000.01000.x. [DOI] [PubMed] [Google Scholar]
- 110.Solomon CG, Hu FB, Dunaif A, Rich-Edwards JE, Stampfer MJ, Willett WC, et al. Menstrual cycle irregularity and risk for future cardiovascular disease. J Clin Endocrinol Metab. 2002;87(5):2013–7. doi: 10.1210/jcem.87.5.8471. [DOI] [PubMed] [Google Scholar]
- 111.Polotsky AJ, Allshouse AA, Crawford SL, Harlow SD, Khalil N, Kazlauskaite R, et al. Hyperandrogenic oligomenorrhea and metabolic risks across menopausal transition. J Clin Endocrinol Metab. 2014;99(6):2120–7. doi: 10.1210/jc.2013-4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Talbott E, Clerici A, Berga SL, Kuller L, Guzick D, Detre K, et al. Adverse lipid and coronary heart disease risk profiles in young women with polycystic ovary syndrome: results of a case-control study. J Clin Epidemiol. 1998;51(5):415–22. doi: 10.1016/s0895-4356(98)00010-9. [DOI] [PubMed] [Google Scholar]
- 113.Fogel RB, Malhotra A, Pillar G, Pittman SD, Dunaif A, White DP. Increased prevalence of obstructive sleep apnea syndrome in obese women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2001;86(3):1175–80. doi: 10.1210/jcem.86.3.7316. [DOI] [PubMed] [Google Scholar]
- 114.Vgontzas AN, Legro RS, Bixler EO, Grayev A, Kales A, Chrousos GP. Polycystic ovary syndrome is associated with obstructive sleep apnea and daytime sleepiness: role of insulin resistance. J Clin Endocrinol Metab. 2001;86(2):517–20. doi: 10.1210/jcem.86.2.7185. [DOI] [PubMed] [Google Scholar]
- 115.Aurora RN, Punjabi NM. Obstructive sleep apnoea and type 2 diabetes mellitus: a bidirectional association. Lancet Respir Med. 2013;1(4):329–38. doi: 10.1016/S2213-2600(13)70039-0. [DOI] [PubMed] [Google Scholar]
- 116.Tasali E, Van Cauter E, Hoffman L, Ehrmann DA. Impact of obstructive sleep apnea on insulin resistance and glucose tolerance in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93(10):3878–84. doi: 10.1210/jc.2008-0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tasali E, Chapotot F, Leproult R, Whitmore H, Ehrmann DA. Treatment of obstructive sleep apnea improves cardiometabolic function in young obese women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2011;96(2):365–74. doi: 10.1210/jc.2010-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gutierrez-Grobe Y, Ponciano-Rodriguez G, Ramos MH, Uribe M, Mendez-Sanchez N. Prevalence of non alcoholic fatty liver disease in premenopausal, posmenopausal and polycystic ovary syndrome women. The role of estrogens. Ann Hepatol. 2010;9(4):402–9. [PubMed] [Google Scholar]
- 119.Jones H, Sprung VS, Pugh CJ, Daousi C, Irwin A, Aziz N, et al. Polycystic ovary syndrome with hyperandrogenism is characterized by an increased risk of hepatic steatosis compared to nonhyperandrogenic PCOS phenotypes and healthy controls, independent of obesity and insulin resistance. J Clin Endocrinol Metab. 2012;97(10):3709–16. doi: 10.1210/jc.2012-1382. [DOI] [PubMed] [Google Scholar]
- 120.Setji TL, Holland ND, Sanders LL, Pereira KC, Diehl AM, Brown AJ. Nonalcoholic steatohepatitis and nonalcoholic Fatty liver disease in young women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91(5):1741–7. doi: 10.1210/jc.2005-2774. [DOI] [PubMed] [Google Scholar]
- 121.Sarkar M, Wellons M, Cedars MI, VanWagner L, Gunderson EP, Ajmera V, et al. Testosterone Levels in Pre-Menopausal Women are Associated With Nonalcoholic Fatty Liver Disease in Midlife. Am J Gastroenterol. 2017;112(5):755–62. doi: 10.1038/ajg.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tan S, Bechmann LP, Benson S, Dietz T, Eichner S, Hahn S, et al. Apoptotic markers indicate nonalcoholic steatohepatitis in polycystic ovary syndrome. J Clin Endocrinol Metab. 2010;95(1):343–8. doi: 10.1210/jc.2009-1834. [DOI] [PubMed] [Google Scholar]
- 123.Vink JM, Sadrzadeh S, Lambalk CB, Boomsma DI. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J Clin Endocrinol Metab. 2006;91(6):2100–4. doi: 10.1210/jc.2005-1494. [DOI] [PubMed] [Google Scholar]
- 124.Coviello AD, Sam S, Legro RS, Dunaif A. High prevalence of metabolic syndrome in first-degree male relatives of women with polycystic ovary syndrome is related to high rates of obesity. J Clin Endocrinol Metab. 2009;94(11):4361–6. doi: 10.1210/jc.2009-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sam S, Coviello AD, Sung YA, Legro RS, Dunaif A. Metabolic phenotype in the brothers of women with polycystic ovary syndrome. Diabetes Care. 2008;31(6):1237–41. doi: 10.2337/dc07-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sam S, Legro RS, Essah PA, Apridonidze T, Dunaif A. Evidence for metabolic and reproductive phenotypes in mothers of women with polycystic ovary syndrome. Proc Natl Acad Sci U S A. 2006;103(18):7030–5. doi: 10.1073/pnas.0602025103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Taylor MC, Reema Kar A, Kunselman AR, Stetter CM, Dunaif A, Legro RS. Evidence for increased cardiovascular events in the fathers but not mothers of women with polycystic ovary syndrome. Hum Reprod. 2011;26(8):2226–31. doi: 10.1093/humrep/der101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Torchen LC, Idkowiak J, Fogel NR, O’Neil DM, Shackleton CH, Arlt W, et al. Evidence for Increased 5alpha-Reductase Activity During Early Childhood in Daughters of Women With Polycystic Ovary Syndrome. J Clin Endocrinol Metab. 2016;101(5):2069–75. doi: 10.1210/jc.2015-3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shi Y, Zhao H, Shi Y, Cao Y, Yang D, Li Z, et al. Genome-wide association study identifies eight new risk loci for polycystic ovary syndrome. Nat Genet. 2012;44(9):1020–5. doi: 10.1038/ng.2384. [DOI] [PubMed] [Google Scholar]
- 130●●.Hayes MG, Urbanek M, Ehrmann DA, Armstrong LL, Lee JY, Sisk R, et al. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat Commun. 2015;6:7502. doi: 10.1038/ncomms8502. This recent genome wide association study identified both replicated and novel genetic susceptibility loci significantly associated with PCOS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Burgess S, Butterworth A, Malarstig A, Thompson SG. Use of Mendelian randomisation to assess potential benefit of clinical intervention. BMJ. 2012;345:e7325. doi: 10.1136/bmj.e7325. [DOI] [PubMed] [Google Scholar]
- 132.Ding EL, Song Y, Manson JE, Hunter DJ, Lee CC, Rifai N, et al. Sex hormone-binding globulin and risk of type 2 diabetes in women and men. N Engl J Med. 2009;361(12):1152–63. doi: 10.1056/NEJMoa0804381. [DOI] [PMC free article] [PubMed] [Google Scholar]