

ABSTRACT
Cell junctions are critical for cell adhesion and communication in epithelial tissues. It is evident that the cellular distribution, size, and architecture of cell junctions play a vital role in regulating function. These details of junction architecture have been challenging to elucidate in part due to the complexity and size of cell junctions. A major challenge in understanding these features is attaining high resolution spatial information with molecular specificity. Fluorescence microscopy allows localization of specific proteins to junctions, but with a resolution on the same scale as junction size, rendering internal protein organization unobtainable. Super-resolution microscopy provides a bridge between fluorescence microscopy and nanoscale approaches, utilizing fluorescent tags to reveal protein organization below the resolution limit. Here we provide a brief introduction to super-resolution microscopy and discuss novel findings into the organization, structure and function of epithelial cell junctions.
KEYWORDS: adherens junction, desmosome, gap junction, hemidesmosome, PALM, SIM, STORM, STED, tight junction
Introduction
Cell junctions are complex macromolecular structures with critical roles in many diverse processes including embryonic development, immune function, and wound healing. Epithelial tissues can have several types of cell junctions including tight junctions, adherens junctions, desmosomes, and gap junctions on the lateral membrane physically linking neighboring cells; and hemidesmosomes on the basal membrane interacting with the extracellular matrix (Fig. 1). Each of these junctions have highly specialized structures and cellular organization specific to their unique roles in signaling and adhesion,1 mechanobiology,2 and barrier formation,3 to name a few. Investigating the structure/function of cell junctions is challenging due to their membrane localization, molecular complexity, and size, which ranges from ∼10 nm (gap junctions) to ∼500 nm (desmosomes).4,5
Figure 1.
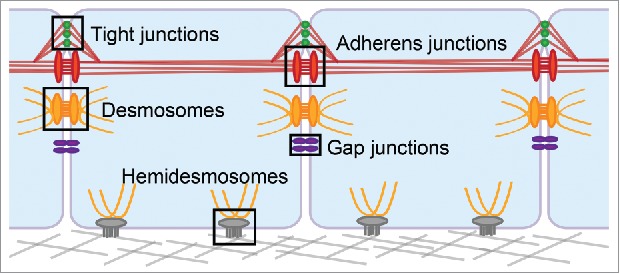
Epithelial cell junctions. Tight junctions are localized apically on the lateral membrane of epithelial cells. They interact with the neighboring adherens junctions, and both junctions integrate with the actin cytoskeleton, shown in red. Desmosomes are located throughout the lateral membrane, below the adherens junctions, and associate with the intermediate filament cytoskeleton, shown in yellow. Gap junctions are also distributed throughout the lateral cell membrane where they form channels that allows passage of ions and small molecules between the two cells. In stratified epithelia, hemidesmosomes are localized to the basal membrane where they promote adhesion to the underlying substrate and integrate with intermediate filaments.
Fluorescence microscopy is a critical tool in the study of cell junctions. The relatively straightforward sample preparation, ease of use, availability, and compatibility with live cell imaging distinguishes fluorescence microscopy. A central feature of fluorescence microscopy is the ability to specifically label and image proteins of interest, providing molecular specificity. Consequently, the spatial organization and dynamics of subcellular assemblies, proteins, or genomic sequences of interest in fixed or living samples can be determined using highly specific fluorescent labeling techniques such as immunocytochemistry, in situ hybridization, or chimeric fluorescent protein tags. However, a major limitation of conventional fluorescence microscopy for studying junction organization is the resolution limit. Resolution defines the smallest distance between two objects at which they can be individually identified. Based on relations described by Abbe and Rayleigh, the resolution of a conventional widefield or confocal microscope is limited to approximately 250 nm in the x-y imaging plane and 600 nm in the axial, or z dimension.6,7 Practically, this means a single protein with a physical size of 3 nm3 will appear as a 250 × 250 × 600 nm diffraction limited spot in x-y-z when imaged by fluorescence microscopy. A consequence of the resolution limit is that proteins can appear co-localized when in reality their organization is distinct. The sizes of epithelial junctions are close to or below the resolution limit, restricting the utility of conventional widefield or confocal fluorescence microscopy in assessing their structure.
Many junctional protein interactions and modifications have been investigated biochemically. While biochemical methods provide molecular specificity, they do not reveal cellular or tissue localizations and report population averages, obscuring heterogeneity. To access structural information unavailable through biochemical studies, immunogold and tomographic electron microscopy (EM) have been used to study junctions.8-10 An advantage of EM is nanometer scale resolution; however it comes with a tradeoff of limited molecular specificity and requirement of sample fixation. Further, the sample preparation can be time-consuming, labor-intensive, and requires a high level of skill. High-resolution structural techniques such as X-ray crystallography have elucidated the structure of individual proteins or smaller sub-complexes, but have not determined the structure of whole junctions. Between widefield microscopy, biochemical analysis, electron microscopy, and crystallography, no single technique can access dynamics, molecular specificity, and high resolution information simultaneously. This resolution gap makes integrating data obtained from different techniques at different scales a formidable challenge.
Advancements in microscopy have bridged this gap by combining the advantages of fluorescence techniques with nanoscale resolution. Super-resolution microscopy overcomes the diffraction barrier, allowing imaging of fluorescently labeled samples at resolutions from 120 nm, double the resolution of conventional microscopy, down to 20 nm in the x-y plane.11-14 These revolutionary techniques allow for the study of nanoscale organization of protein arrangement, order, and dynamics in macromolecular complexes.
The complex structure of epithelial junctions is readily accessible to investigation with super-resolution microscopy. Sub-junctional protein arrangement, as well as how junctions are organized relative to each other, play critical roles in function. On the sub-junctional level, proteins can be distributed homogeneously or clustered heterogeneously (Fig. 2A). By widefield, these types of organization are indistinguishable and both appear co-localized. In contrast, super-resolution can reveal co-localization on the nanoscale and distinguish between homogenous mixing and sub-junctional clustering. In some junctions, proteins are arranged in multi-layer assemblies parallel to the plasma membrane (Fig. 2B). By widefield, proteins in these layers appear to co-localize and the entire structure is encompassed by a diffraction limited puncta. Super-resolution can reveal the relative position of the proteins within the junction, proximal or distal to the membrane. Finally, junctions can associate with each other or the cytoskeleton to form sub-micron clusters (Fig. 2C). These clusters all appear the same by widefield, but super resolution allows more precise identification and measurement of the size, distribution, and composition of junctional clusters.
Figure 2.
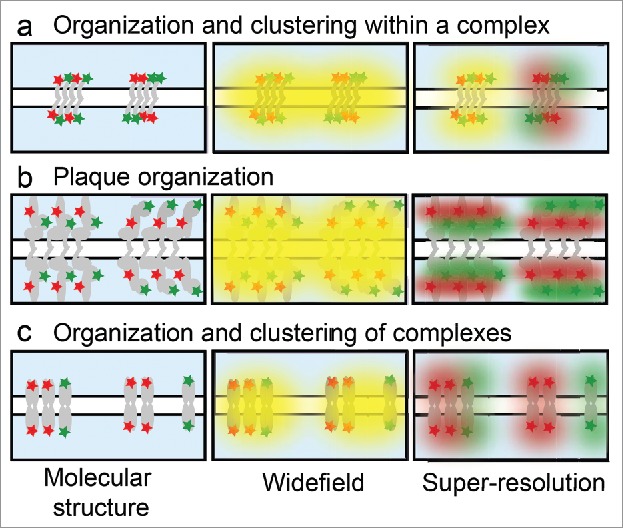
Protein organization in epithelial junctions. Illustration of different junctional molecular structures within the diffraction limit that cannot be distinguished by widefield microscopy, but are distinct by super-resolution. (a) Junctions can contain a homogenous mix of proteins (left) or distinct clusters (right). (b) Junctional plaque proteins are organized in layers parallel to the plasma membrane, with the red protein more distal (left) or proximal (right) to the membrane. (c) Junctions can have higher-order organization within the cell membrane with clusters of mixed junctions (left) or segregation of like junctions into distinct structures (right).
Here we introduce three families of super-resolution methods capable of providing this increased resolution, all of which are commercially available and accessible to many researchers. We will not discuss super-resolution theory and principles in depth and instead refer the reader to the many excellent reviews 15-18 and practical comparisons between techniques.19-22 Finally we examine the use of super-resolution microscopy for studying epithelial cell junctions, including a discussion of novel findings from these cutting edge approaches.
Structured illumination microscopy
Structured illumination microscopy (SIM) employs a widefield configuration in which the specimen is illuminated by an excitation pattern of parallel lines which interact with the fluorescent structures in the sample. The illumination pattern is phase-shifted, rotated, and a series of images are collected. High frequency information from this image series is reconstructed computationally resulting in a super-resolution image.14,23 Reconstruction reveals finer structures in the sample with up to twice the resolution in all three dimensions, 120 nm in the x-y plane and 300 nm in z. Variations of SIM using non-linear excitation have achieved resolutions down to 50 nm.24-27
SIM offers many advantages, although it can suffer from artifacts with improper implementation.28 SIM is compatible with most widely used fluorescent labels including both fluorescent proteins and antibodies. Multiplexing multiple channel acquisition is straightforward and SIM can easily address protein co-localization. These features make SIM widely accessible. Images can be obtained at speeds that are limited only by exposure time and camera sensitivities. This grants compatibility with live cell imaging, assuming the dynamics are slower than the acquisition time.29-32 Additionally, SIM provides optical sectioning, can resolve structures many microns deep into a sample, and can be combined with z-stacks to achieve super-resolution in 3D.33-35
Single molecule localization microscopy
Single molecule localization microscopy (SMLM) encompasses several methods including photoactivated localization microscopy (PALM),12 stochastic optical reconstruction microscopy (STORM),11,36 and ground state depletion individual molecule return (GSDIM),37 among others.38-41 Each of these methods depends on the use of photoswitchable fluorophores to achieve stochastic activation, such that only a small percentage of fluorophores are in a fluorescent “on” state at a given time. Thousands of images are acquired over time in which fluorophores are stochastically turning “on”. By temporally separating the emission, individual fluorophores can be resolved in each image. A reconstruction algorithm determines the localization of individual molecules by Gaussian fitting. The super-resolution image is created when the localizations from all the images in the series are combined. SMLM achieves a 10-fold improvement in resolution over the diffraction limit with an x-y resolution of ∼20 nm.11,12,36,37,42 Improvements in axial resolution are provided by total internal reflection fluorescence (TIRF) illumination or point spread function engineering to achieve z-resolutions of 50–100 nm.
SMLM depends on the photophysical properties of the fluorescent label. STORM and direct STORM (dSTORM) utilize photoswitchable probes typically consisting of chemical dyes conjugated to antibodies in special imaging buffer, which provide multiple localizations per fluorophore. For example, Alexa 647 has optimal photophysical properties including a high photon yield and low duty cycle.43 Alternatively, irreversibly activatable fluorophores provide a single off-on cycle. The typical photoactivatable probes used in PALM are genetically encoded, offer a 1:1 labeling stoichiometry, and can provide density information with each localization representing a single protein.44 SMLM can accommodate multiplexed imaging with multiple probes, live cell imaging,45-48 and 3D imaging with ∼50 nm axial resolution.49 SMLM has proven to be a powerful tool in studying nanoscale structures and organization in cells and tissues, and its capabilities continue to evolve.
Stimulated emission depletion microscopy
Reversible saturable optical fluorescence transitions (RESOLFT) microscopy is a category of methods encompassing stimulated emission depletion microscopy (STED) and ground state depletion (GSD) microscopy. Like all super-resolution techniques, STED relies on measuring only a subset of fluorophores to overcome the diffraction limit.13,50,51 In most STED microscopes, two overlapping lasers are synchronized, one to excite the sample and the other to drive excited fluorophores to the ground state without emission of a photon, thereby depleting the fluorescence.52 Because of the “doughnut” shape of the depletion beam, only fluorophores in the center of the excitation emit fluorescence. The excitation and depletion beams are raster-scanned over the sample, as in a laser scanning confocal, to acquire a super-resolution image. An advantage of RESOLFT microscopy is that a super-resolution image is directly acquired, with no reconstruction necessary.
If the size and intensity of the depletion beam is precisely tuned, the resolution of STED is theoretically limitless.53 Practically, factors including sample type, laser intensity, and photobleaching result in a lateral resolution limit of ∼10 to 70 nm. Resolutions below 10 nm have been reported in a non-biological sample,54 while 30–40 nm resolution in the x-y plane is generally achieved in biological samples.55 Without a 3D depletion beam, the axial resolution of STED remains comparable to widefield microscopy, ∼ 600 nm. STED is compatible with multi-color imaging, with fluorophores matched to specific imaging lasers. While STED can be used in live cell experiments, concerns centered on phototoxicity due to high laser powers are raised. STED principles have been combined with atomic force microscopy (AFM)56 and fluorescence correlation spectroscopy (FCS)57 revealing additional dimensions of molecular organization and dynamics.
General imaging considerations for super-resolution microscopy
Each super-resolution technique involves tradeoffs between spatial resolution (Fig. 3), temporal resolution, and ease of sample preparation. Sample preparation is always critical in fluorescence microscopy, and improper handling can result in decreased resolution and obscured data interpretation.20,58,59 When the target resolution is higher and distances being measured are smaller, the negative effects of poor sample preparation are exacerbated. Disruption to the ultrastructure from fixation and processing need to be minimized and the density of fluorescent labels should be high. In microscopy, the sample itself becomes part of the optical system and thus must be prepared with coverslips of optimal thickness (#1.5) and appropriate mounting medium. The sample coverslip and objective should both be clean, refractive index-matched immersion oil used, and the correction collar on the objective properly set. Any non-specific background or refractive index variations in the sample will be detrimental to the image quality. The calibration and function of the microscope is also critical, and should be verified routinely with standardized samples.
Figure 3.
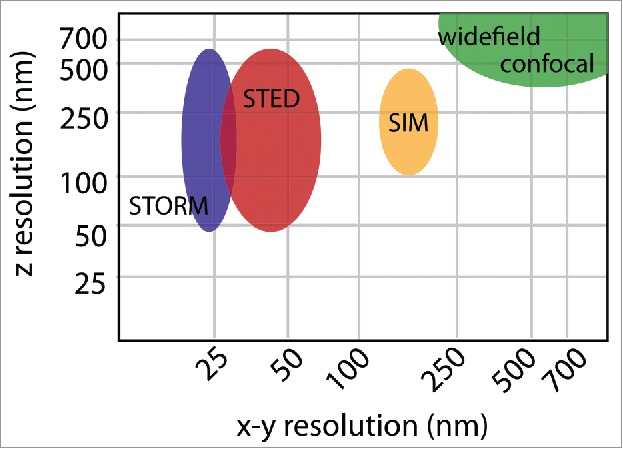
Spatial resolution. The resolution of widefield or confocal microscopy is determined by the numerical aperture of the objective and wavelength of the excitation light and is ∼250 nm in x-y and 600 nm in z. SIM doubles the resolution in all three dimensions achieving ∼125 nm lateral resolution and ∼300 nm axial resolution. STED and STORM can achieve a range of axial resolutions, depending on the use of TIRF or 3D imaging modalities. In general STORM achieves a slightly better x-y resolution of ∼20 nm, as opposed to ∼40 nm by STED. These resolutions are approximations based on commercially available systems. Variations in the resolution of each method depend on the properties and quality of the sample, as well as the particular optical setup.
A daunting challenge is the imaging of thick biological specimens. Distortion from light scattering and optical aberrations decreases image resolution when the structure of interest is located further from the coverslip. In this case it may be beneficial to reorient the sample closer to coverslip to improve imaging resolution, for example by cutting thinner tissue sections, preparing sections directly on coverslips to avoid mounting media between coverslip and sample, or by artificially manipulating protein localization within cells. In particular, SMLM techniques are often used in conjunction with TIRF for improved z-resolution, though that limits the excitation field to ∼140 nm adjacent to the coverslip. If the junction of interest is not located within the TIRF excitation field, a sub-critical incidence angle (also called inclined plane illumination) can be used to reduce out-of-plane fluorescence. While both cell culture models and thin tissue sections (∼5 µm) are suitable for localization-based imaging, the resolution of the resulting image may depend on the proximity of the junction to the coverslip. SIM can typically image up to 20 µm into a sample, with resolution decreasing with depth. STED can image as deep as the objective working distance, but resolution will be best within 20 µm of the coverslip.
When interpreting super-resolution microscopy data, it is important to consider the distance between the fluorescent label and the protein of interest. A full-size antibody is approximately 10 nm from end-to-end, resulting in a 20 nm “cloud” of uncertainty around an object labeled with both primary and secondary antibodies. The impact of antibody size has been documented with imaging of microtubules by EM and STORM.60,61 Immunolabeling methods that reduce the distance between the protein and the fluorophore can be considered including directly labeling primary antibodies,62 Fab fragments,63 nanobodies,64 and chromobodies.65 Considerations for genetically encoded tags include the location (i.e. N- or C- terminus) and the size (for example, GFP is a 4.2 nm long cylinder with a diameter of 2.4 nm). Super-resolution microscopy will be most successful when attention is given to optimizing labeling and sample preparation methods to avoid artifacts.
Super-resolution microscopy of epithelial junctions
Understanding the complex molecular organization of epithelial junctions is an ideal challenge for nanoscale imaging. Now that we have given an overview of techniques and introduced sample preparation considerations, we turn to the application of super-resolution imaging for studying epithelial junctions. In the following sections, we highlight advances in the understanding of the structure and function of epithelial junctions with super-resolution microscopy.
Tight junctions
Tight junctions (TJs) are localized just below the apical surface of epithelial cells where they form a barrier between the basolateral and apical membranes, contributing to cell polarity and regulating paracellular flux.66,67 Two classes of TJ transmembrane proteins, claudins and occludins, tightly link neighboring cells.68 These transmembrane proteins, along with intracellular scaffold proteins, link TJs to the actin cytoskeleton and to neighboring adherens junctions (AJs).69,70 The size, shape, molecular composition, and distribution of TJs are thought to be important for their function.
The polarized apical location of TJs excludes the use of TIRF illumination, which presents a challenge to SMLM based approaches. One way this was overcome was by utilizing reconstituted tight junction strands, which form claudin-claudin interactions throughout regions of cell contact and can be imaged in TIRF.71 A variant of SMLM, spectral position determination microscopy, showed that claudin-3 and claudin-5 expressed in HEK293 cells formed mesh-like structures. The size and shape of the structures differed by protein type, implying differences in their barrier function. This was a breakthrough in visualizing TJs, however the native context and organization was lost in the reconstituted junctions.
TJ architecture, and changes induced by alcohol, were investigated with STORM.72,73 In primary alveolar epithelial cells, TJs were found to be discrete punctate structures rather than a complex meshwork. Claudin-5 expression was increased and TJ function was impaired with an increase in paracellular leak following alcohol treatment. STORM revealed a corresponding remodeling of TJ architecture where claudin-18 co-localization with the scaffolding protein zonula occludens 1 (ZO-1) decreased at the same time as claudin-18 and claudin-5 co-localization increased. This suggested a mechanism for the reduced barrier function, where claudin-18 associating with claudin-5 dissociates from ZO-1. The differences in cluster size (<500 nm2) and co-localization would not have been detected without super-resolution microscopy.
While SIM, STED, and SMLM have each contributed to an improved understanding of TJ structure, much remains to be studied.71-76 Future applications of super-resolution microscopy will allow exploration of TJ structures as they relate to paracellular flux, barrier deficits, and cell polarity in disease.
Adherens junctions
AJs serve as cell signaling platforms, sites for transcriptional regulation, and play a central role in cell adhesion and polarity.77,78 AJs comprise transmembrane cadherins associating with the actin cytoskeleton through cytoplasmic catenin proteins. The many functions of AJs are paralleled by the complexity of the molecular architecture of the junction. Understanding the organization of protein-protein interactions in AJs is critical for determining the relationship between structure and function in these junctions.79
SIM has been used to study sub-junctional protein organization in AJs. Two transmembrane proteins responsible for mediating adhesion, E-cadherin and nectin, were found to localize to distinct clusters within AJs.80 The size and distribution of clusters differed significantly between the two proteins, suggesting that cluster organization regulates adhesive function. Live-cell SIM revealed that size and distribution of E-cadherin and nectin clusters were independent of each other. To further study the interactions between clusters, SIM was coupled with a proximity ligation assay (PLA). While SIM alone resolved distinct clusters, SIM-PLA showed that clusters maintain peripheral points of contact. This combination of SIM with biochemical methods allowed precise measurements of structural changes impacting adhesion.
3D SMLM has been used to examine E-cadherin clustering in cell culture and in in vivo models at higher resolution.81,82 The apical “band” of AJs observed by conventional microscopy was found to consist of distinct E-cadherin clusters.82 The size and shape of these clusters was similar to those in more lateral junctions. However, the surface distribution varied based on localization, with apical clusters more closely spaced. Interestingly, while size was unchanged, protein density was decreased in non-adherent versus adherent clusters. Furthermore, both F-actin and E-cadherin extracellular domain interactions were shown to regulate cluster size and density. In another study, E-cadherin cluster size was measured in Drosophila embryos.81 Both endocytosis and association of E-cadherin with actin were found to regulate cluster size, which followed a power law distribution with a maximum size limited to under 60 nm. These two studies highlight the power of 3D SMLM for investigating cluster size and organization in cells and tissue.
The architecture of AJs was mapped using iPALM, a specialized PALM microscope with <20 nm axial resolution.83,84 MDCK cells were plated on coverslips micropatterned with cadherins, which allowed AJs to form at the coverslip-cell interface.85 This unique system oriented the AJs so the axial position of proteins in the half-junction corresponded to the distance from the plasma membrane. Protein locations were mapped, identifying two compartments in the AJ molecular organization. A conformational change in vinculin following activation was shown to bridge the compartments while impacting the localization of several actin regulating proteins. This analysis identified a modular architecture and is a critical step toward our understanding of the relationship between structure and mechanical integration in AJs.
Gap junctions
Gap junctions form channels to transport ions and small molecules through hexameric assemblies of connexins, thereby providing direct communication between neighboring cells.86 Compared to the other epithelial junctions, gap junctions have a relatively simple molecular architecture. It was in part this simplicity that drove the use of connexin (Cx) 43 in seminal live-cell STED experiments.55 STED revealed many small Cx43 clusters previously not reported by confocal microscopy. Questions remain about the organization of connexin isoforms within gap junctions and the distribution of gap junctions along cell membranes.
Given that mutations in Cx26 are the most common genetic cause of childhood hearing loss,87 the structure and organization of gap junctions is of significant interest. The expression of Cx26 and Cx30 was examined by SIM to determine if junctions in the human cochlea were hetero- or homotypic.88 SIM showed that connexins form homotypic rather than heterotypic channels, as previously thought based on co-localization by widefield microscopy.89,90 The differential distribution of gap junctions based on connexin isoform in the sensory epithelium of the human cochlea was studied by SIM.91 Cx26 was found to be weakly expressed and present in small plaques, while Cx30 plaques dominated. Variations in the size of Cx30 plaques were found to be cell type dependent. In some areas of the cochlea, Cx30 formed a large network thought to be important for K+ signaling. SIM also revealed areas where Cx26 formed small puncta closely associated with the larger Cx30 network, which could not have been resolved by widefield or confocal microscopy. This work suggests that the distribution of connexins in the cochlea is important for function and indicates that disruption of connexin networks may be a mechanism for hearing loss.
Although not an epithelial tissue, super-resolution microscopy has proven a powerful tool in studying gap junctions in the cardiac intercalated disc. The molecular arrangement of these complex junctions relates to proper electrical and mechanical function and is disrupted in heart diseases including arrhythmogenic cardiomyopathy (AC).92,93 SMLM revealed that Cx43 and the “desmosomal” protein plakophilin-2 (Pkp2) form discrete punctate junctions that physically interact, with Pkp2 partially localized to the edge of Cx43 plaques.94 This result is in contrast to prior understanding of the molecular arrangement of these junctions as separate. The identification of nanoscale Cx43-Pkp2 subdomains by SMLM indicates an interactive hub (a ‘connexome’) for molecules that traditionally are classified as gap junction or desmosome to come together to control cardiac function.
Desmosomes
Desmosomes are the primary junction responsible for maintaining cell adhesion in epithelial tissues by resisting mechanical stress.95-97 Desmosomes consist of transmembrane cadherins and intracellular plaque proteins that bind to the intermediate filament cytoskeleton.98 By EM desmosomes appear as a pair of electron dense plaques along the plasma membrane, averaging ∼500 nm in length.5,99,100 The internal molecular architecture of desmosomes cannot be resolved by widefield or confocal microscopy, because they appear as diffraction limited spots.101
The resolution provided by SIM allowed for significant advancements in understanding desmosome dynamics and the pathogenesis of the autoimmune disease Pemphigus Vulgaris (PV). SIM identified co-localization of desmosomal cadherin desmoglein 3 (Dsg3) with lipid raft markers in primary human keratinocytes.102 This, combined with biochemistry, helped to identify a role for lipid rafts in desmosome assembly and disassembly dynamics. In PV patient tissue desmosome size was reduced and Dsg3 was found to be aberrantly clustered and localized in “linear arrays”.103 The mirror symmetry created by desmosomal plaques was visible in the SIM images, which allowed identification of desmosomes “split” along the adhesive interface at blister sites. Desmosome splitting was recapitulated in vitro by exposing cultured keratinocytes to PV IgG and to mechanical stress, demonstrating that mechanical stress can lead to desmosome splitting. SIM contributed insights into how protein organization, trafficking, and function are altered in PV patient tissue.
The increased resolution of dSTORM made it possible not only to resolve the plaque mirror symmetry, but also to quantify the organization of proteins within desmosomes.101 Plaque localization and length were measured for several core desmosomal proteins in primary keratinocytes, resulting in a molecular map. This revealed that the desmosomal proteins are not arranged completely perpendicular to the plasma membrane but instead splay outward into the cytosol, a level of organization not previously detected. In hyper-adhesive desmosomes, the molecular map revealed changes in desmoplakin orientation, with the C-terminal domain moving closer to the plasma membrane. These changes correlated with those measured in suprabasal versus basal cells in human skin sections. Together this indicates that desmosome molecular architecture and the organization of plaque proteins is critical for desmosome function.
Super-resolution studies of the intercalated disc in murine cardiomyocytes are also of interest when considering desmosome structure. AC associated defects in desmosomal proteins, including Pkp2, can result in disruption of the intercalated disc. dSTORM revealed a nanoscale retraction of the microtubule plus end from N-cadherin in Pkp2 deficient mice, suggesting a mechanism for AC that relies on Pkp2 integrity.104 Related work found that, like disruption of Pkp2, truncation of Cx43 dissociated the microtubule plus end from the junction site and resulted in mislocalization of NaV1.5, the sodium channel protein integral to cardiac function.105 The use of dSTORM in these studies linked electrical coupling, cell adhesion, and excitability in the heart through a common mechanism of microtubule attachment.
Super-resolution imaging of desmosomes has thus far explored the ultrastructure of the complex and demonstrated a correlation between structure and function. These approaches will allow quantification of desmosomal structure as it relates to disease, wound healing, and embryogenesis.
Hemidesmosomes
In stratified epithelia, hemidesmosomes anchor basal cells to the underlying basement membrane. Hemidesmosomes are distinct from desmosomes in their protein composition, but derive their name from their association with the intermediate filament cytoskeleton and a similar “spot-weld” like appearance as seen by EM.
Multi-color GSDIM revealed that the hemidesmosome integrin β4 was distributed alongside and not directly under the keratin fibers in cultured primary keratinocytes.106 Association with the linker protein plectin regulated the distance between β4 and keratin. Quantification showed that integrin BP180 and β4 codistributed in characteristic structures along keratin filaments. Interestingly, a zone at the cell periphery contained β4 but not BP180, indicating recruitment of plaque protein BP180 only to mature hemidesmosomes. BP180 and BP230 were found to be organized in distinct concentric structures. Quantification of the architecture in cultured keratinocytes and human skin sections were consistent. These results suggest a complex interdependence of hemidesmosome structure and protein function. Prior to this work, very little was known about the architecture of hemidesmosomes or how they associate with the keratin intermediate filament cytoskeleton. The resolution of GSDIM was critical for the determination of the molecular organization of the hemidesmosome, which could provide the building blocks for future work studying different functional states and possible disease mechanisms.
Conclusion
In the past decade, our ability to observe protein organization at the nanoscale has vastly improved. A variety of approaches have broken the diffraction barrier and enabled us to visualize structures in their cellular context as was previously accessible only by EM. Additional developments in optics, reconstruction algorithms, labeling methods, and fluorescent probes, will continue to advance super-resolution microscopy. Combining super-resolution microscopy with other advanced imaging modalities can also provide access to novel information. Examples of this combinatorial approach include correlative super-resolution and electron microscopy (CLEM),107,108 confocal,109 fluorescence polarization,110 fluorescence correlation spectroscopy (FCS),111,112 or atomic force microscopy (AFM).113
Super-resolution microscopy has allowed a more detailed visualization and quantification of the 3D molecular architecture of cell junctions in living or fixed cells and tissues. These techniques are highly suitable for studying macromolecular complexes and enable the investigation of novel questions about junction architecture and how complex structure relates to function.
Funding Statement
This work was supported by funding to ALM from NIH/NIAMS under grant R21AR066920 and the NSF under CAREER award 1553344.
Disclosure of potential conflict of interest
No potential conflicts of interest were disclosed.
References
- 1.Rübsam M, Broussard JA, Wickström SA, Nekrasova O, Green KJ, Niessen CM. Adherens junctions and desmosomes coordinate mechanics and signaling to orchestrate tissue morphogenesis and function: An evolutionary perspective. Cold Spring Harb Perspect Biol. 2017. doi: 10.1101/cshperspect.a029207. PMID:28893859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xia S, Kanchanawong P. Nanoscale mechanobiology of cell adhesions. Semin Cell Dev Biol. 2017;71:53–67. doi: 10.1016/j.semcdb.2017.07.029. PMID:28754443. [DOI] [PubMed] [Google Scholar]
- 3.Yano T, Kanoh H, Tamura A, Tsukita S. Apical cytoskeletons and junctional complexes as a combined system in epithelial cell sheets. Ann N Y Acad Sci. 2017;1405(1):32–43. doi: 10.1111/nyas.13432. PMID:28763830. [DOI] [PubMed] [Google Scholar]
- 4.Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T. Structure of the connexin 26 gap junction channel at 3.5 Å resolution. Nature. 2009;458:597–602. doi: 10.1038/nature07869. PMID:19340074. [DOI] [PubMed] [Google Scholar]
- 5.Tamarin A, Sreebny LM. An analysis of desmosome shape, size, and orientation by the use of histometric and densitometric methods with electron microscopy. J Cell Biol. 1963;18:125–34. doi: 10.1083/jcb.18.1.125. PMID:19866631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abbe E. The Relation of Aperture and Power in the Microscope*. J Royal Microscopical Soc. 1882;2:300–9. doi: 10.1111/j.1365-2818.1882.tb00190.x. [DOI] [Google Scholar]
- 7.Rayleigh L. LVI. Investigations in optics, with special reference to the spectroscope. Philos Mag. 1879;8:477–86. doi: 10.1080/14786447908639715. [DOI] [Google Scholar]
- 8.North AJ, Bardsley WG, Hyam J, Bornslaeger EA, Cordingley HC, Trinnaman B, Hatzfeld M, Green KJ, Magee AI, Garrod DR. Molecular map of the desmosomal plaque. J Cell Sci. 1999;112 (Pt 23):4325–36. PMID:10564650. [DOI] [PubMed] [Google Scholar]
- 9.Al-Amoudi A, Castaño-Diez D, Devos DP, Russell RB, Johnson GT, Frangakis AS. The three-dimensional molecular structure of the desmosomal plaque. Proc Natl Acad Sci U S A. 2011;108:6480–5. doi: 10.1073/pnas.1019469108. PMID:21464301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al-Amoudi A, Chang J-J, Leforestier A, McDowall A, Salamin LM, Norlén LPO, Richter K, Blanc NS, Studer D, Dubochet J. Cryo-electron microscopy of vitreous sections. EMBO J. 2004;23:3583–8. doi: 10.1038/sj.emboj.7600366. PMID:15318169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods. 2006;3:793–5. doi: 10.1038/nmeth929. PMID:16896339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–5. doi: 10.1126/science.1127344. PMID:16902090. [DOI] [PubMed] [Google Scholar]
- 13.Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett. 1994;19:780–2. doi: 10.1364/OL.19.000780. PMID:19844443. [DOI] [PubMed] [Google Scholar]
- 14.Neil MAA, Juškaitis R, Wilson T. Method of obtaining optical sectioning by using structured light in a conventional microscope. Opt Lett, OL. 1997;22:1905–7. doi: 10.1364/OL.22.001905. [DOI] [PubMed] [Google Scholar]
- 15.Godin AG, Lounis B, Cognet L. Super-resolution microscopy approaches for live cell imaging. Biophys J. 2014;107:1777–84. doi: 10.1016/j.bpj.2014.08.028. PMID:25418158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durisic N, Cuervo LL, Lakadamyali M. Quantitative super-resolution microscopy: pitfalls and strategies for image analysis. Curr Opin Chem Biol. 2014;20:22–8. doi: 10.1016/j.cbpa.2014.04.005. PMID:24793374. [DOI] [PubMed] [Google Scholar]
- 17.Schermelleh L, Heintzmann R, Leonhardt H. A guide to super-resolution fluorescence microscopy. J Cell Biol. 2010;190:165–75. doi: 10.1083/jcb.201002018. PMID:20643879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hell SW. Microscopy and its focal switch. Nat Methods. 2009;6:24–32. doi: 10.1038/nmeth.1291. PMID:19116611. [DOI] [PubMed] [Google Scholar]
- 19.Wegel E, Göhler A, Christoffer Lagerholm B, Wainman A, Uphoff S, Kaufmann R, Dobbie IM. Imaging cellular structures in super-resolution with SIM, STED and Localisation Microscopy: A practical comparison. Sci Rep. 2016;6:27290. doi: 10.1038/srep27290. PMID:27264341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert TJ, Waters JC. Navigating challenges in the application of superresolution microscopy. J Cell Biol. 2017;216:53–63. doi: 10.1083/jcb.201610011. PMID:27920217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sydor AM, Czymmek KJ, Puchner EM, Mennella V. Super-resolution microscopy: From single molecules to supramolecular assemblies. Trends Cell Biol. 2015;25:730–48. doi: 10.1016/j.tcb.2015.10.004. PMID:26546293. [DOI] [PubMed] [Google Scholar]
- 22.Dempsey GT. A user's guide to localization-based super-resolution fluorescence imaging. Methods Cell Biol. 2013;114:561–92. doi: 10.1016/B978-0-12-407761-4.00024-5. PMID:23931523. [DOI] [PubMed] [Google Scholar]
- 23.Gustafsson MG. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc. 2000;198:82–7. doi: 10.1046/j.1365-2818.2000.00710.x. PMID:10810003. [DOI] [PubMed] [Google Scholar]
- 24.Heintzmann R, Jovin TM, Cremer C. Saturated patterned excitation microscopy–a concept for optical resolution improvement. J Opt Soc Am A Opt Image Sci Vis. 2002;19:1599–609. doi: 10.1364/JOSAA.19.001599. PMID:12152701. [DOI] [PubMed] [Google Scholar]
- 25.Heintzmann R. Saturated patterned excitation microscopy with two-dimensional excitation patterns. Micron. 2003;34:283–91. doi: 10.1016/S0968-4328(03)00053-2. PMID:12932771. [DOI] [PubMed] [Google Scholar]
- 26.Li D, Shao L, Chen B-C, Zhang X, Zhang M, Moses B, Milkie DE, Beach JR, Hammer JA 3rd, Pasham M, et al.. Advanced imaging. extended-resolution structured illumination imaging of endocytic and cytoskeletal dynamics. Science. 2015;349:aab3500. doi: 10.1126/science.aab3500. PMID:26315442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gustafsson MGL. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci U S A. 2005;102:13081–6. doi: 10.1073/pnas.0406877102. PMID:16141335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Demmerle J, Innocent C, North AJ, Ball G, Müller M, Miron E, Matsuda A, Dobbie IM, Markaki Y, Schermelleh L. Strategic and practical guidelines for successful structured illumination microscopy. Nat Protoc. 2017;12:988–1010. doi: 10.1038/nprot.2017.019. PMID:28406496. [DOI] [PubMed] [Google Scholar]
- 29.Lee CW, Vitriol EA, Shim S, Wise AL, Velayutham RP, Zheng JQ. Dynamic localization of G-actin during membrane protrusion in neuronal motility. Curr Biol. 2013;23:1046–56. doi: 10.1016/j.cub.2013.04.057. PMID:23746641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirano Y, Matsuda A, Hiraoka Y. Recent advancements in structured-illumination microscopy toward live-cell imaging. Microscopy. 2015;64:237–49. doi: 10.1093/jmicro/dfv034. PMID:26133185. [DOI] [PubMed] [Google Scholar]
- 31.York AG, Parekh SH, Nogare DD, Fischer RS, Temprine K, Mione M, Chitnis AB, Combs CA, Shroff H. Resolution doubling in live, multicellular organisms via multifocal structured illumination microscopy. Nat Methods. 2012;9:749–54. doi: 10.1038/nmeth.2025. PMID:22581372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olshausen PV, Soufo HJD, Wicker K, Heintzmann R, Graumann PL, Rohrbach A. Superresolution imaging of dynamic MreB Filaments in B. subtilis—a multiple-motor-driven transport? Biophys J. 2013;105:1171–81. doi: 10.1016/j.bpj.2013.07.038. PMID:24010660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schermelleh L, Carlton PM, Haase S, Shao L, Winoto L, Kner P, Burke B, Cardoso MC, Agard DA, Gustafsson MGL, et al.. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science. 2008;320:1332–6. doi: 10.1126/science.1156947. PMID:18535242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gustafsson MGL, Shao L, Carlton PM, Wang CJR, Golubovskaya IN, Cande WZ, Agard DA, Sedat JW. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys J. 2008;94:4957–70. doi: 10.1529/biophysj.107.120345. PMID:18326650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shao L, Kner P, Rego EH, Gustafsson MGL. Super-resolution 3D microscopy of live whole cells using structured illumination. Nat Methods. 2011;8:1044–6. doi: 10.1038/nmeth.1734. PMID:22002026. [DOI] [PubMed] [Google Scholar]
- 36.Heilemann M, van de Linde S, Schüttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, Sauer M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Ed Engl. 2008;47:6172–6. doi: 10.1002/anie.200802376. PMID:18646237. [DOI] [PubMed] [Google Scholar]
- 37.Fölling J, Bossi M, Bock H, Medda R, Wurm CA, Hein B, Jakobs S, Eggeling C, Hell SW. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat Methods. 2008;5:943–5. doi: 10.1038/nmeth.1257. PMID:18794861. [DOI] [PubMed] [Google Scholar]
- 38.Sharonov A, Hochstrasser RM. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc Natl Acad Sci U S A. 2006;103:18911–6. doi: 10.1073/pnas.0609643104. PMID:17142314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jungmann R, Steinhauer C, Scheible M, Kuzyk A, Tinnefeld P, Simmel FC. Single-molecule kinetics and super-resolution microscopy by fluorescence imaging of transient binding on DNA origami. Nano Lett. 2010;10:4756–61. doi: 10.1021/nl103427w. PMID:20957983. [DOI] [PubMed] [Google Scholar]
- 40.Lemmer P, Gunkel M, Baddeley D, Kaufmann R, Urich A, Weiland Y, Reymann J, Müller P, Hausmann M, Cremer C. SPDM: light microscopy with single-molecule resolution at the nanoscale. Appl Phys B. 2008;93:1–12. doi: 10.1007/s00340-008-3152-x. [DOI] [Google Scholar]
- 41.Dertinger T, Colyer R, Iyer G, Weiss S, Enderlein J. Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI). Proc Natl Acad Sci U S A 2009;106:22287–92. doi: 10.1073/pnas.0907866106. PMID:20018714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moerner WE, Orrit M, Wild UP, Basché T. Frontmatter, in Single-Molecule Optical Detection, Imaging and Spectroscopy, VCH Verlagsgesellschaft mbH, Weinheim, Germany. doi: 10.1002/9783527614714.fmatter 1996. [DOI] [Google Scholar]
- 43.Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods. 2011;8:1027–36. doi: 10.1038/nmeth.1768. PMID:22056676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Durisic N, Laparra-Cuervo L, Sandoval-Álvarez A, Borbely JS, Lakadamyali M. Single-molecule evaluation of fluorescent protein photoactivation efficiency using an in vivo nanotemplate. Nat Methods. 2014;11:156–62. doi: 10.1038/nmeth.2784. PMID:24390439. [DOI] [PubMed] [Google Scholar]
- 45.Deschout H, Lukes T, Sharipov A, Szlag D, Feletti L, Vandenberg W, Dedecker P, Hofkens J, Leutenegger M, Lasser T, et al.. Complementarity of PALM and SOFI for super-resolution live-cell imaging of focal adhesions. Nat Commun. 2016;7:13693. doi: 10.1038/ncomms13693. PMID:27991512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones SA, Shim S-H, He J, Zhuang X. Fast, three-dimensional super-resolution imaging of live cells. Nat Methods. 2011;8:499–508. doi: 10.1038/nmeth.1605. PMID:21552254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shroff H, Galbraith CG, Galbraith JA, Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods. 2008;5:417–23. doi: 10.1038/nmeth.1202. PMID:18408726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Henriques R, Griffiths C, Hesper Rego E, Mhlanga MM. PALM and STORM: unlocking live-cell super-resolution. Biopolymers. 2011;95:322–31. doi: 10.1002/bip.21586. PMID:21254001. [DOI] [PubMed] [Google Scholar]
- 49.Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–3. doi: 10.1126/science.1153529. PMID:18174397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Willig KI, Rizzoli SO, Westphal V, Jahn R, Hell SW. STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature. 2006;440:935–9. doi: 10.1038/nature04592. PMID:16612384. [DOI] [PubMed] [Google Scholar]
- 51.Klar TA, Jakobs S, Dyba M, Egner A, Hell SW. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc Natl Acad Sci U S A. 2000;97:8206–10. doi: 10.1073/pnas.97.15.8206. PMID:10899992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harke B, Bianchini P, Vicidomini G, Galiani S, Diaspro A. Stimulated Emission Depletion (STED) Microscopy. Encyclopedia Biophys. 2013;1:2470–5. doi: 10.1007/978-3-642-16712-6_835. [DOI] [Google Scholar]
- 53.Harke B, Keller J, Ullal CK, Westphal V, Schönle A, Hell SW. Resolution scaling in STED microscopy. Opt Express. 2008;16:4154–62. doi: 10.1364/OE.16.004154. PMID:18542512. [DOI] [PubMed] [Google Scholar]
- 54.Rittweger E, Han KY, Irvine SE, Eggeling C, Hell SW. STED microscopy reveals crystal colour centres with nanometric resolution. Nat Photonics. 2009;3:144–7. doi: 10.1038/nphoton.2009.2. [DOI] [Google Scholar]
- 55.Hein B, Willig KI, Wurm CA, Westphal V, Jakobs S, Hell SW. Stimulated emission depletion nanoscopy of living cells using SNAP-tag fusion proteins. Biophys J. 2010;98:158–63. doi: 10.1016/j.bpj.2009.09.053. PMID:20074516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Curry N, Ghézali G, Kaminski Schierle GS, Rouach N, Kaminski CF. Correlative STED and Atomic Force Microscopy on Live Astrocytes Reveals Plasticity of Cytoskeletal Structure and Membrane Physical Properties during Polarized Migration. Front Cell Neurosci. 2017;11:104. doi: 10.3389/fncel.2017.00104. PMID:28469559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bianchini P, Cardarelli F, Di Luca M, Diaspro A, Bizzarri R. Nanoscale protein diffusion by STED-based pair correlation analysis. PLoS One. 2014;9:e99619. doi: 10.1371/journal.pone.0099619. PMID:24967681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Galdeen SA, North AJ. Live Cell Fluorescence Microscopy Techniques. Methods Mol Biol. 2011;769:205–22. doi: 10.1007/978-1-61779-207-6_14. PMID:21748678. [DOI] [PubMed] [Google Scholar]
- 59.Brown CM. Fluorescence microscopy – avoiding the pitfalls. J Cell Sci. 2007;120:1703–5. doi: 10.1242/jcs.03433. PMID:17502480. [DOI] [PubMed] [Google Scholar]
- 60.Weber K, Rathke PC, Osborn M. Cytoplasmic microtubular images in glutaraldehyde-fixed tissue culture cells by electron microscopy and by immunofluorescence microscopy. Proc Natl Acad Sci U S A. 1978;75:1820–4. doi: 10.1073/pnas.75.4.1820. PMID:417343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–53. doi: 10.1126/science.1146598. PMID:17702910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pástor MVD. Direct immunofluorescent labeling of cells. Methods Mol Biol. 2009;588:135–42. [DOI] [PubMed] [Google Scholar]
- 63.Brown JK, Pemberton AD, Wright SH, Miller HRP. Primary antibody–fab fragment complexes: A flexible alternative to traditional direct and indirect immunolabeling techniques. J Histochem Cytochem. 2004;52:1219–30. doi: 10.1369/jhc.3A6200.2004. PMID:15314089. [DOI] [PubMed] [Google Scholar]
- 64.Beghein E, Gettemans J. Nanobody technology: A versatile toolkit for microscopic imaging, protein–protein interaction analysis, and protein function exploration. Front Immunol. 2017;8:771. doi: 10.3389/fimmu.2017.00771. PMID:28725224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rothbauer U, Zolghadr K, Tillib S, Nowak D, Schermelleh L, Gahl A, Backmann N, Conrath K, Muyldermans S, Cardoso MC, et al.. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat Methods. 2006;3:887–9. doi: 10.1038/nmeth953. PMID:17060912. [DOI] [PubMed] [Google Scholar]
- 66.Andrea Hartsock WJN. Adherens and tight junctions: Structure, function and connections to the actin Cytoskeleton. Biochim Biophys Acta. 2008;1778:660. doi: 10.1016/j.bbamem.2007.07.012. PMID:17854762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zihni C, Balda MS, Matter K. Signalling at tight junctions during epithelial differentiation and microbial pathogenesis. J Cell Sci. 2014;127:3401–13. doi: 10.1242/jcs.145029. PMID:25125573. [DOI] [PubMed] [Google Scholar]
- 68.Günzel D, Fromm M. Claudins and other tight junction proteins. Comprehensive Physiol. 2012;2(3):1819–52. [DOI] [PubMed] [Google Scholar]
- 69.Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol. 2007;127:2525–32. doi: 10.1038/sj.jid.5700865. PMID:17934504. [DOI] [PubMed] [Google Scholar]
- 70.Sluysmans S, Vasileva E, Spadaro D, Shah J, Rouaud F, Citi S. The role of apical cell-cell junctions and associated cytoskeleton in mechanotransduction. Biol Cell. 2017;109:139–61. doi: 10.1111/boc.201600075. PMID:28220498. [DOI] [PubMed] [Google Scholar]
- 71.Kaufmann R, Piontek J, Grüll F, Kirchgessner M, Rossa J, Wolburg H, Blasig IE, Cremer C. Visualization and quantitative analysis of reconstituted tight junctions using localization microscopy. PLoS One. 2012;7:e31128. doi: 10.1371/journal.pone.0031128. PMID:22319608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schlingmann B, Overgaard CE, Molina SA, Lynn KS, Mitchell LA, Dorsainvil White S, Mattheyses AL, Guidot DM, Capaldo CT, Koval M. Regulation of claudin/zonula occludens-1 complexes by hetero-claudin interactions. Nat Commun. 2016;7:12276. doi: 10.1038/ncomms12276. PMID:27452368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koval M, Molina SA, Schlingmann B. Use of Super-resolution immunofluorescence microscopy to analyze tight junction protein interactions in situ. Microsc Microanal. 2016;22:1014–5. doi: 10.1017/S1431927616005912. [DOI] [Google Scholar]
- 74.Siljamäki E, Raiko L, Toriseva M, Nissinen L, Näreoja T, Peltonen J, Kähäri V-M, Peltonen S. p38δ mitogen-activated protein kinase regulates the expression of tight junction protein ZO-1 in differentiating human epidermal keratinocytes. Arch Dermatol Res. 2014;306:131–41. doi: 10.1007/s00403-013-1391-0. PMID:23856837. [DOI] [PubMed] [Google Scholar]
- 75.Gehne N, Lamik A, Lehmann M, Haseloff RF, Andjelkovic AV, Blasig IE. Cross-over endocytosis of claudins is mediated by interactions via their extracellular loops. PLoS One. 2017;12:e0182106. doi: 10.1371/journal.pone.0182106. PMID:28813441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Van Itallie CM, Tietgens AJ, Anderson JM. Visualizing the dynamic coupling of claudin strands to the actin cytoskeleton through ZO-1. Mol Biol Cell. 2017;28:524–34. doi: 10.1091/mbc.E16-10-0698. PMID:27974639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perez-Moreno M, Jamora C, Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell. 2003;112:535–48. doi: 10.1016/S0092-8674(03)00108-9. PMID:12600316. [DOI] [PubMed] [Google Scholar]
- 78.Ivanov AI, Naydenov NG. Dynamics and regulation of epithelial adherens junctions: recent discoveries and controversies. Int Rev Cell Mol Biol. 2013;303:27–99. doi: 10.1016/B978-0-12-407697-6.00002-7. PMID:23445808. [DOI] [PubMed] [Google Scholar]
- 79.Yap AS, Gomez GA, Parton RG. Adherens Junctions Revisualized: Organizing Cadherins as Nanoassemblies. Dev Cell. 2015;35:12–20. doi: 10.1016/j.devcel.2015.09.012. PMID:26460944. [DOI] [PubMed] [Google Scholar]
- 80.Indra I, Hong S, Troyanovsky R, Kormos B, Troyanovsky S. The adherens junction: a mosaic of cadherin and nectin clusters bundled by actin filaments. J Invest Dermatol. 2013;133:2546–54. doi: 10.1038/jid.2013.200. PMID:23639974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Truong Quang B-A, Mani M, Markova O, Lecuit T, Lenne P-F. Principles of E-cadherin supramolecular organization in vivo. Curr Biol. 2013;23:2197–207. doi: 10.1016/j.cub.2013.09.015. PMID:24184100. [DOI] [PubMed] [Google Scholar]
- 82.Wu Y, Kanchanawong P, Zaidel-Bar R. Actin-delimited adhesion-independent clustering of E-cadherin forms the nanoscale building blocks of adherens junctions. Dev Cell. 2015;32:139–54. doi: 10.1016/j.devcel.2014.12.003. PMID:25600236. [DOI] [PubMed] [Google Scholar]
- 83.Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, Waterman CM. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468:580–4. doi: 10.1038/nature09621. PMID:21107430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shtengel G, Galbraith JA, Galbraith CG, Lippincott-Schwartz J, Gillette JM, Manley S, Sougrat R, Waterman CM, Kanchanawong P, Davidson MW, et al.. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc Natl Acad Sci. 2009;106:3125–30. doi: 10.1073/pnas.0813131106. PMID:19202073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bertocchi C, Wang Y, Ravasio A, Hara Y, Wu Y, Sailov T, Baird MA, Davidson MW, Zaidel-Bar R, Toyama Y, et al.. Nanoscale architecture of cadherin-based cell adhesions. Nat Cell Biol. 2017;19:28–37. doi: 10.1038/ncb3456. PMID:27992406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goodenough DA, Paul DL. Gap Junctions. Cold Spring Harb Perspect Biol. 2009;1:a002576. doi: 10.1101/cshperspect.a002576. PMID:20066080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kemperman MH, Hoefsloot LH, C W R. Hearing loss and connexin 26. JRSM Short Rep. 2002;95:171–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu W, Edin F, Blom H, Magnusson P, Schrott-Fischer A, Glueckert R, Santi PA, Li H, Laurell G, Rask-Andersen H. Super-resolution structured illumination fluorescence microscopy of the lateral wall of the cochlea: the Connexin26/30 proteins are separately expressed in man. Cell Tissue Res. 2016;365:13–27. doi: 10.1007/s00441-016-2359-0. PMID:26941236. [DOI] [PubMed] [Google Scholar]
- 89.Marziano NK, Casalotti SO, Portelli AE, Becker DL, Forge A. Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum Mol Genet. 2003;12:805–12. doi: 10.1093/hmg/ddg076. PMID:12668604. [DOI] [PubMed] [Google Scholar]
- 90.Ortolano S, Di Pasquale G, Crispino G, Anselmi F, Mammano F, Chiorini JA. Coordinated control of connexin 26 and connexin 30 at the regulatory and functional level in the inner ear. Proc Natl Acad Sci U S A. 2008;105:18776–81. doi: 10.1073/pnas.0800831105. PMID:19047647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu W, Li H, Edin F, Brännström J, Glueckert R, Schrott-Fischer A, Molnar M, Pacholsky D, Pfaller K, Rask-Andersen H. Molecular composition and distribution of gap junctions in the sensory epithelium of the human cochlea—a super-resolution structured illumination microscopy (SR-SIM) study. Ups J Med Sci. 2017;122:1–11. doi: 10.1080/03009734.2017.1322645. PMID:28256956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kant S, Holthöfer B, Magin TM, Krusche CA, Leube RE. Desmoglein 2-dependent Arrhythmogenic Cardiomyopathy is caused by a loss of adhesive function. Circ Cardiovasc Genet. 2015;8:553–63. doi: 10.1161/CIRCGENETICS.114.000974. PMID:26085008. [DOI] [PubMed] [Google Scholar]
- 93.Agullo-Pascual E, Cerrone M, Delmar M. Arrhythmogenic cardiomyopathy and Brugada syndrome: diseases of the connexome. FEBS Lett. 2014;588:1322–30. doi: 10.1016/j.febslet.2014.02.008. PMID:24548564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Agullo-Pascual E, Reid DA, Keegan S, Sidhu M, Fenyö D, Rothenberg E, Delmar M. Super-resolution fluorescence microscopy of the cardiac connexome reveals plakophilin-2 inside the connexin43 plaque. Cardiovasc Res. 2013;100:231–40. doi: 10.1093/cvr/cvt191. PMID:23929525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kowalczyk AP, Green KJ. Structure, function, and regulation of desmosomes. Prog Mol Biol Transl Sci. 2013;116:95–118. doi: 10.1016/B978-0-12-394311-8.00005-4. PMID:23481192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Johnson JL, Najor NA, Green KJ. Desmosomes: regulators of cellular signaling and adhesion in epidermal health and disease. Cold Spring Harb Perspect Med. 2014;4:a015297. doi: 10.1101/cshperspect.a015297. PMID:25368015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Berika M, Garrod D. Desmosomal adhesion in vivo. Cell Commun Adhes. 2014;21:65–75. doi: 10.3109/15419061.2013.876018. PMID:24460202. [DOI] [PubMed] [Google Scholar]
- 98.Garrod D, Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta. 2008;1778:572–87. doi: 10.1016/j.bbamem.2007.07.014. PMID:17854763. [DOI] [PubMed] [Google Scholar]
- 99.Odland GF. The fine structure of the interrelationship of cells in the human epidermis. J Biophys Biochem Cytol. 1958;4:529–38. doi: 10.1083/jcb.4.5.529. PMID:13587545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Selby CC. An electron microscope study of the epidermis of mammalian skin in thin sections: I. Dermo-Epidermal junction and basal cell layer. J Cell Biol. 1955;1:429–44. doi: 10.1083/jcb.1.5.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stahley SN, Bartle EI, Atkinson CE, Kowalczyk AP, Mattheyses AL. Molecular organization of the desmosome as revealed by direct stochastic optical reconstruction microscopy. J Cell Sci. 2016;129:2897–904. doi: 10.1242/jcs.185785. PMID:27505428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stahley SN, Saito M, Faundez V, Koval M, Mattheyses AL, Kowalczyk AP. Desmosome assembly and disassembly are membrane raft-dependent. PLoS One. 2014;9:e87809. doi: 10.1371/journal.pone.0087809. PMID:24498201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stahley SN, Warren MF, Feldman RJ, Mattheyses AL, Kowalczyk AP. Super resolution microscopy reveals altered desmosome organization, endocytosis and desmosome splitting in pemphigus vulgaris epidermis. J Dermatol Sci. 2016;84:e25. doi: 10.1016/j.jdermsci.2016.08.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cerrone M, Delmar M. Desmosomes and the sodium channel complex: implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends Cardiovasc Med. 2014;24:184–90. doi: 10.1016/j.tcm.2014.02.001. PMID:24656989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Agullo-Pascual E, Lin X, Leo-Macias A, Zhang M, Liang F-X, Li Z, Pfenniger A, Lübkemeier I, Keegan S, Fenyö D, et al.. Super-resolution imaging reveals that loss of the C-terminus of connexin43 limits microtubule plus-end capture and NaV1.5 localization at the intercalated disc. Cardiovasc Res. 2014;104:371–81. doi: 10.1093/cvr/cvu195. PMID:25139742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nahidiazar L, Kreft M, van den Broek B, Secades P, Manders EMM, Sonnenberg A, Jalink K. The molecular architecture of hemidesmosomes, as revealed with super-resolution microscopy. J Cell Sci. 2015;128:3714–9. doi: 10.1242/jcs.171892. PMID:26330528. [DOI] [PubMed] [Google Scholar]
- 107.Johnson E, Kaufmann R. Correlative in-resin super-resolution fluorescence and electron microscopy of cultured cells. Methods Mol Biol. 2017;1663:163–77. doi: 10.1007/978-1-4939-7265-4_14. PMID:28924667. [DOI] [PubMed] [Google Scholar]
- 108.Perkovic M, Kunz M, Endesfelder U, Bunse S, Wigge C, Yu Z, Hodirnau V-V, Scheffer MP, Seybert A, Malkusch S, et al.. Correlative light- and electron microscopy with chemical tags. J Struct Biol. 2014;186:205–13. doi: 10.1016/j.jsb.2014.03.018. PMID:24698954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Soeller C, Hou Y, Jayasinghe ID, Baddeley D, Crossman D. Correlative single-molecule localization microscopy and confocal microscopy. Methods Mol Biol. 2017;1663:205–17. doi: 10.1007/978-1-4939-7265-4_17. PMID:28924670. [DOI] [PubMed] [Google Scholar]
- 110.Valades Cruz CA, Shaban HA, Kress A, Bertaux N, Monneret S, Mavrakis M, Savatier J, Brasselet S. Quantitative nanoscale imaging of orientational order in biological filaments by polarized superresolution microscopy. Proc Natl Acad Sci U S A. 2016;113:E820–8. doi: 10.1073/pnas.1516811113. PMID:26831082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sarangi NKP II, Ayappa KG, Visweswariah SS, Basu JK. Super-resolution stimulated emission depletion-fluorescence correlation spectroscopy reveals nanoscale membrane reorganization induced by pore-forming proteins. Langmuir. 2016;32:9649–57. doi: 10.1021/acs.langmuir.6b01848. PMID:27564541. [DOI] [PubMed] [Google Scholar]
- 112.Vicidomini G, Ta H, Honigmann A, Mueller V, Clausen MP, Waithe D, Galiani S, Sezgin E, Diaspro A, Hell SW, et al.. STED-FLCS: An advanced tool to reveal spatiotemporal heterogeneity of molecular membrane dynamics. Nano Lett. 2015;15:5912–8. doi: 10.1021/acs.nanolett.5b02001. PMID:26235350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bondia P, Casado S, Flors C. Correlative super-resolution fluorescence imaging and atomic force microscopy for the characterization of biological samples. Methods Mol Biol. 2017;1663:105–13. doi: 10.1007/978-1-4939-7265-4_9. PMID:28924662. [DOI] [PubMed] [Google Scholar]