

Abstract
Biosensors that transduce target chemical and biochemical inputs into genetic outputs are essential for bioengineering and synthetic biology. Current biosensor design strategies often suffer from a lack of signal-to-noise, requirements for extensive optimization for each new input, and poor performance in mammalian cells. Here we report the development of a proximity-dependent split RNA polymerase (RNAP) as a general platform for biosensor engineering. After discovering that interactions between fused proteins modulate the assembly of a split T7 RNAP, we optimized the split RNAP components for protein-protein interaction detection by phage-assisted continuous evolution (PACE). We then applied the resulting “activity-responsive RNAP” (AR) system to create light and small molecule activated biosensors, demonstrating the “plug-and-play” nature of the platform. Finally, we validated that ARs can interrogate multidimensional protein-protein interactions and trigger RNA nanostructure production, protein synthesis, and gene knockdown in mammalian systems, illustrating the versatility of ARs in synthetic biology applications.
Diverse areas of chemical biology and biotechnology, including directed evolution, synthetic biology, and bioengineering, require methods to link chemical and biochemical processes to defined genetic outputs1–4. RNA is a particularly useful output due to the extensive technologies available to drive cellular responses based on computed nucleic acid signals5–7. Nature utilizes transcription factors to detect target activities and drive genetic responses, and many naturally occurring transcription factors, such as the Lac and Tet repressors, have been repurposed as gene expression control elements4,8. However, because the scope of detection of such natural systems is limited, several engineered alternative technologies have been developed.
N-hybrid systems, such as those for the detection of protein-DNA (1-hybrid), protein-protein (2-hybrid), protein-RNA (3-hybrid), and protein-small molecule (3-hybrid) interactions, are among the most commonly deployed approaches for genetic sensor design9–12. The development of programmable DNA binding domains such as TALEs (Transcription activator-like effectors) and dCas9 have revolutionized 1- and 2-hybrid approaches8,13–15. However, a challenge with n-hybrids is that each system needs to be carefully tuned and optimized for each new interaction, and more complex multicomponent systems, such as 3-hybrids, often lack in sensitivity and signal-to-noise16,17. Although 1-hybrids, and in special cases, 2-hybrids, can be utilized for synthetic biology purposes, in general, these methods are not suitable for applications where a high level of control and dynamic range is required. A primary alternative to n-hybrid approaches is riboswitches, RNA-based elements that drive translational outputs based on an aptamer interacting with a target ligand18,19. Riboswitches have become an important tool for synthetic biologists for designing systems that respond to target inputs, which are generally small molecules, and in rare cases proteins20. While riboswitches can work well for targets that aptamers can be created for, protein-based biochemical activities are generally out of the scope of detection of such RNA-based parts, and translating these tools in mammalian systems often results in diminished performance17. Outside of these somewhat general strategies, a host of other synthetic biology parts have been developed for highly specialized activities2,21. Therefore, there is a need for a general method to transduce endogenous chemical and biochemical information into DNA or RNA for subsequent storage or integration with engineered regulatory systems.
Engineered polymerases have been proposed as a new strategy to respond to and measure endogenous biochemical processes22,23. Inspired by this concept, we recently unveiled protease-responsive RNAPs (PRs) as a new strategy to respond to protease activities by production of defined RNA outputs24. We showed that PRs can encode multidimensional protease activities in defined sequences of RNA in both prokaryotic and mammalian cells. RNAPs in principle provide a new platform for biosensor creation, but engineering such complex enzymes is challenging, and the inhibitor-based design strategy utilized for the PRs is fundamentally limited to protease activities.
We envisioned a new biosensor system based on recently reported “split” T7 RNAPs25,26, in which N- and C-terminal components of the T7 RNAP were shown to spontaneously assemble to form a functional RNAP enzyme. Rather than assembling spontaneously, however, we aimed to engineer ARs, whose assembly is dependent on fused interaction partners, similar to other protein fragment complementation technologies (PFC)27. PFC involves tethering two potential binding partners under investigation to two halves of a split protein marker, such as a fluorescent protein28. Generally, the protein binding partners must be expressed at significantly higher levels than endogenous conditions to achieve adequate signal, and critically, only two or fewer signals can be monitored at once29. More importantly, the fluorescent output is only for analysis and cannot be integrated into downstream synthetic control systems. If a split RNAP could be developed, then genetic biosensors could be easily created in a “plug-and-play” approach by simply swapping in target interacting domains, resulting in a GFP-like platform for biosensor engineering (Fig. 1a).
Figure 1. Design and biophysical feasibility of activity-responsive RNA polymerases (ARs) based on proximity-dependent split RNAPs.
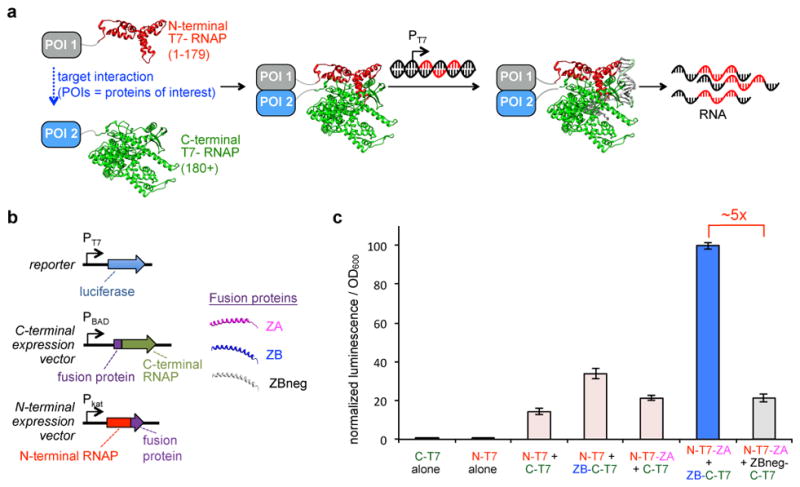
(a) Schematic depiction of AR design. Split T7 RNAP engineered such that it assembles into a functional RNAP when proteins of interest (POIs) fused to each half interact with one another, resulting in transcription of a user-defined sequence of RNA from a supplied DNA substrate. (b) Vectors designed to test split RNAPs in vivo, including a luciferase reporter vector and expression vectors for each of the two halves of the split RNAP. N-terminal split RNAP (red) and C-terminal split RNAP (green) were fused to anti-parallel leucine zipper peptide fusions, ZA (pink), ZB (blue), or ZBneg (gray). ZA and ZB form a tight interaction with one another; ZBneg has three point mutations compared to ZB that dramatically weaken the interaction. (c) Transcriptional output of split RNAPs with fusion proteins assayed in E. coli using the vectors shown in (b). Cells induced for 2 h with arabinose and then analyzed for luminescence (error bars std. error, n = 4). Fusion of the peptides does not interfere with RNAP assembly. Transcription is enhanced if fused peptides interact.
In this study, we engineered the first proximity dependent split T7 RNAP sensor using continuous molecular evolution. We then demonstrated the versatility and ease of use of the platform by creating robust light and small molecule-responsive genetic sensors. To illustrate the power of polymerase-based biosensors, we showed that multidimensional protein-protein interactions (PPIs) can be monitored using ARs. Finally, we validated that ARs can trigger RNA nanostructure formation, protein synthesis, and gene knockdown in mammalian cells using a small molecule-triggered AR biosensor. The AR platform dramatically simplifies and expands genetic circuit creation, and opens up new opportunities in protein engineering, synthetic biology, and bioengineering.
RESULTS
Biophysical feasibility of proximity-dependent split RNAP
Two key biophysical criteria for our proposed AR strategy are that fused protein domains do not sterically interfere with the split RNAP and that interactions of fused domains can influence the RNAP assembly process. We chose to deploy T7 RNAP split at position 179 because, 1) the N-terminal half is small, 2) structural data indicates this position is solvent exposed and removed from the DNA-binding face of the protein, and 3) mutations that influence DNA promoter specificity are C-terminal to this position26,30,31. First, we validated that the two RNAP halves spontaneously assemble using an Escherichia coli (E. coli) luciferase reporter system (Fig. 1b, Supplementary Results, Supplementary Fig. 1, Supplementary Table 1). Next, we utilized leucine zipper peptides that form a tight interaction with one another (ZA and ZB)32,33 as PPI partners. Fusion of ZA or ZB to the N-terminal or C-terminal RNAP, respectively, did not dramatically affect spontaneous RNAP assembly, indicating that the split RNAP can tolerate fusions. However, fusion of both RNAP halves to the interaction partners resulted in a ~5-fold enhancement in transcription, indicating additional pendant interactions enhance split RNAP assembly (Fig. 1c). A triple mutant of ZB that weakens the interaction (ZBneg) confirms the enhancement in transcription is due to the fused PPI. We note that T7 RNAP undergoes large conformational changes during the course of its enzymatic activity,34 including dramatic structural changes in the N-terminal RNAP half. It is therefore non-obvious that the RNAP could tolerate fusions, or that the interactions between fusions could modulate the assembly process while maintaining enzymatic viability. However, given that these preliminary data confirm the split enzyme is amenable to controlled assembly, we turned our attention to engineering the split RNAP to be more dependent on the fused interaction partners.
Development of an evolution to optimize split RNAPs
In order to develop split RNAPs as a platform for biosensor design, the RNAP assembly process needs to be more dependent on fused interaction partners. This involves tuning the assembly of the two RNAP halves while maintaining all other aspects of RNAP enzymatic function, including DNA binding, nucleotide binding, and RNA synthesis. Such a protein engineering problem presents substantial challenges, which can in principle be overcome by molecular evolution. Therefore, we chose to deploy PACE, a rapid evolution system35, which has been used to evolve RNAP promoter specificity, protein-DNA interactions, protease activities, and protein-protein interactions3,30,35–40. Briefly, PACE involves providing an evolving gene of interest to M13 bacteriophage, linking the life cycle of the phage to a desired activity of interest to be evolved in the target gene, and then propagating the virus until the activity evolves. Expression of gIII, a required phage gene, is the basis of the life cycle link.
We envisioned a new PACE system for the evolution of selective assembly for split RNAPs using the leucine zipper peptides as a model PPI. In this system, phage would carry an evolving N-terminal RNAP variant fused to ZA, and the host E. coli cells would express two different C-terminal variants, each with orthogonal DNA promoter specificity, and each fused to either ZB or ZBneg. Assembly of the evolving phage-carried ZA-fused N-terminal RNAP with the ZB-fused C-terminal RNAP in the host cells would result in enhanced phage propagation, while assembly with the ZBneg-fused C-terminal RNAP would decrease phage propagation. We postulated this simultaneous dual positive/negative selection would result in the most robust evolutionary outcomes. Critically, we would use relatively long (6–8 amino acids), unstructured linkers to tether the fusion proteins to the RNAP halves, enforcing the evolution of a mechanism for proximity-dependent RNAP assembly that is less dependent on geometry and linker composition, which we hypothesized would result in a more versatile biosensor platform.
As shown in Fig. 2a, to develop this new PACE system, we engineered M13 phage by replacing gIII with N-terminal T7 RNAP fused to ZA, which is the target of the evolution. E. coli cells were engineered with a “Positive Accessory Plasmid” (posAP) that expresses a ZB-fused C-terminal T7 RNAP variant (C-term CGG RNAP), which contains 7 point mutations that allow it to act selectively on the “CGG” promoter over the T7 promoter25,31, and CGG promoter-driven gIII (Supplementary Fig. 1i, 1j). A second “Negative Accessory Plasmid” (negAP) was also engineered for the E. coli cells, which expresses the wildtype C-terminal T7 RNAP fused to ZBneg and a dominant negative form of gIII under control of the T7 promoter37 (Supplementary Fig. 1k). Therefore, if an evolving N-terminal RNAP variant assembles efficiently with both of the T7 and CGG C-terminal RNAP halves regardless of the fusion protein, phage production will be blocked. However, phage encoding N-terminal variants that selectively assemble with the ZB fused CGG C-terminal half will replicate more efficiently, and continue to mutate until the interaction is optimized (Fig. 2b). The key is that the only difference between the positive and negative selection is whether or not the fused peptides interact based on 3 point mutations in ZBneg compared to ZB, and the 7 mutations that alter DNA binding of the CGG C-terminal RNAP, which are not at the protein-protein interface and are not expected to alter RNAP assembly.
Figure 2. Evolution of a proximity-dependent split RNAP for protein-protein interaction detection.

(a) Vectors for PACE system for proximity dependent RNAPs. (b) Schematic of mechanism of PACE system for proximity dependent RNAPs. Phage carry an evolving N-terminal RNAP fused to ZA, which is given a constant choice of assembling with either a C-terminal RNAP variant fused to the ZB binding partner that produces gIII and allows phage replication, or a C-terminal RNAP variant that is fused to the non-interacting ZBneg partner, which poisons phage production by producing a dominant negative form of gIII (gIIIneg). (c) Schematic of the evolution parameters used during PACE. The RBS strengths50 controlling the expression of the C-terminal RNAPs, the RBSs controlling the output gIII or gIIIneg, and the copy number of the posAP were carefully tuned during the course of the 29 days of evolution. (d) Mapping the mutations of N-29-1 onto T7 RNAP crystal structures (top panel: initiation complex, PDB 1QLN; bottom panel: elongation complex, PDB 1h38). (e) Transcriptional reporter assay of the two primary N-terminal split RNAP genotypes fused to ZA or the evolved ZA double mutant, interacting with either the C-terminal RNAP alone (pink), the C-terminal RNAP fused to ZB (blue), or the C-terminal RNAP fused to ZBneg (grey) (error bars std. error, n = 4). Note: data plotted on a logarithmic scale to show background.
Evolution of a proximity dependent split RNAP using PACE
After cloning and validating the system components, we initiated PACE. The positive and negative selection pressures were modified by carefully tuning the system components (Fig. 2c, Supplementary Table 2) and the progress of the evolution was monitored by activity-dependent plaque assays and genetic analysis of the evolving phage (Supplementary Table 3). Specifically, we altered the concentrations of the on-target and off-target interactions, and the strength of selection of the RNA output, by tuning the ribosome binding sites (RBSs) controlling each system component. After each 3–4 days of PACE on a given target, we would use activity-dependent plaque assays to decide the subsequent evolutionary targets. This process continued for 29 days, after which time the N-terminal RNAP converged on two main genotypes, a 6-mutation (N-29-1) variant and a 7-mutation (N-29-8) variant, with several of the mutations near the interface between the two halves of the split RNAP (Fig. 2d). Several mutations that are highly enriched exist in either solvent-exposed regions of the structure, or even less predictably, at the protein-DNA interface34,41,42. This suggests epistatic interactions between mutations that tune the protein-protein interface with key mutations elsewhere in the protein, further illustrating why our unbiased directed evolution strategy is the optimal approach for tuning a complex molecular machine like an RNAP.
Assaying the two primary variants that emerged from PACE in the luciferase transcription reporter system revealed that the background level of transcription with the ZBneg control was dramatically decreased (Fig. 2e). Further genetic analysis revealed that the ZA leucine zipper protein, which we assumed was already fully optimized for interaction with ZB, also evolved during PACE, converging on two leucine to isoleucine mutations. Inclusion of the two ZA mutations into N-29-1 and N-29-8 resulted in a dramatic enhancement in assembly of the split RNAP with the ZA-ZB partners, but maintained low levels of background with the ZBneg control, with variant N-29-1 showing a 44-fold increase in RNA synthesis based on the interaction (Fig. 2e). This observation demonstrates that this new split RNAP PACE system can be deployed to optimize biomolecular interactions, similar to recent 2-hybrid PACE systems36.
We hypothesized that the actual background of the evolved split RNAP was lower than what was being measured due to the two isoleucine mutations in ZA effecting the interaction between ZA and ZBneg. To test this, we assayed the transcriptional output of N-29-1 and N-29-8 fused to ZA with the C-terminal RNAP without any fusion. Indeed, we found a dramatically lower background, indicating that there is a measurable affinity for ZA (L13I, L20I) and ZBneg. Therefore, the actual dynamic range of N-29-1 is >350-fold based on the target PPI. These results indicate that the assembly of the evolved RNAP is not only dependent on fused interaction partners, but is dynamically sensitive to the affinity of the interaction. Most importantly, PACE yielded the N-terminal RNAP variant N-29-1 for the AR strategy.
ARs as light and small molecule biosensors
With the optimized and validated AR system for PPIs in hand, we next sought to explore the generality of the approach by developing an inducible PPI systems, and therefore targeted light and small molecule-activated AR sensors. To create a light-activated RNAP, we appended the light-oxygen-voltage 2 (LOV2)-SsrA fusion variant and SspB from the recently developed improved light induced dimer (iLID-nano) system43 to the N-29-1 and C-terminal RNAP, respectively (Fig. 3a), without any additional optimization of linkers, geometry, or concentrations, and assayed the fusions in E. coli. Illumination with blue LED light, which induces dimerization of the iLID-nano system, resulted in a 26-fold enhancement in transcriptional output of the light-activated AR, while control fusions do not show a light response (Fig. 3b). This dynamic range of this non-optimized system is impressive because the reported light induced difference in affinity of the iLID-nano system proteins is only 36-fold43.
Figure 3. Small molecule and light-responsive ARs.
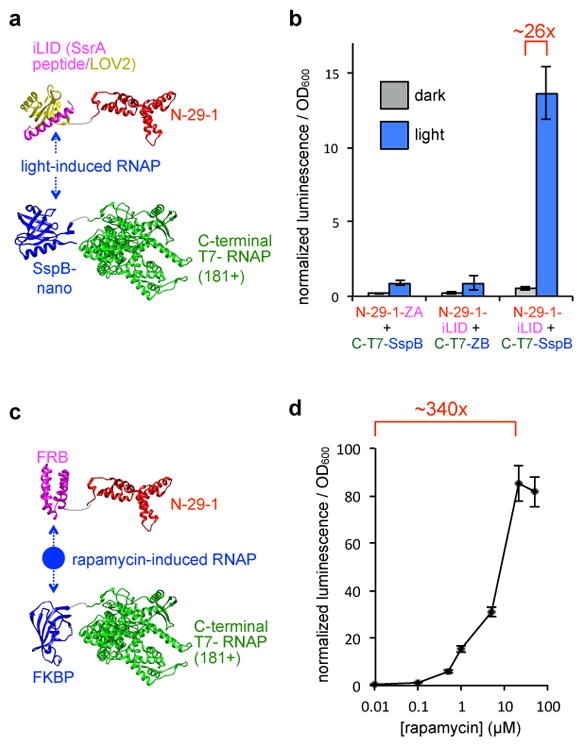
(a) Schematic of light activated RNAP design using the iLID-nano system fused to the evolved split RNAP. (b) Transcription response of the light-activated RNAP system in E. coli. Cells were transformed with expression vectors for the two halves of the light-inducible RNAP and a reporter vector, and then either kept in the dark or illuminated with blue LED light for 3 h prior to transcriptional analysis (error bars std. error, n = 4). (c) Schematic of small molecule responsive RNAP design using FRB and FKBP fused to the evolved split RNAP. (d) Transcription response of the rapamycin-inducible RNAP system in E. coli. Cells were transformed with expression vectors for the two halves of the small molecule-inducible RNAP and a reporter vector, and then induced with varying concentrations of rapamycin for 3 h prior to transcription analysis (error bars std. error, n = 4).
Next, to further explore the versatility of ARs to detect a 3-hybrid-like interaction, we sought to engineer a small molecule-activated RNAP. The rapamycin-induced dimerization of FRB and FKBP has served as a workhorse small molecule-induced dimerization system for a multitude of applications44, and served as the next target for our split RNAP system. While there are existing methods to control RNA synthesis with small molecules in prokaryotic systems, there are far fewer that function well in mammalian systems45,46, and many have issues with signal-to-noise and background. We replaced the peptide fusions on the E. coli expression vectors for the C-terminal RNAP and N-29-1 with the FKBP and FRB, respectively, again without any optimization of linkers, concentration, geometry, or any other system component (Fig. 3c). As seen in Fig. 3d, a dramatic, dose-responsive increase in RNA synthesis is observed in the in vivo luciferase transcription reporter assay upon treatment with rapamycin, with a 340-fold enhancement in RNA synthesis and essentially undetectable background.
Compared with traditional n-hybrid strategies that require extensive linker and geometric optimization36, the ARs appeared to be much less dependent on linkers. We experimentally interrogated the effect of linker length on the split RNAP assembly process using the rapamycin-inducible AR as a prototype, by varying the linker lengths of the fusions proteins from two to fourteen amino acids. Transcriptional assays with the panel of fusion proteins revealed minimal linker dependency, and in fact, the longer linkers performed slightly better (Supplementary Fig. 2). This might be due to the large conformational changes that occur at the split site (Fig. 2d), which may be better accommodated by longer linkers. Regardless of the mechanism, these data demonstrate the ability to simply swap in new binding domains to the AR system with minimal optimization to create biosensors. Finally, we cloned two split GFP sensors33,47 into the rapamycin AR vector system for comparison, which, under the same experimental conditions, revealed no detectible signal, further illustrating the advantages of ARs (Supplementary Fig. 3).
ARs can monitor multidimensional PPI networks
One core advantage of an RNAP-based biosensor for analyzing endogenous molecular interactions is that the signals are encoded in an output RNA. Fluorescent-based split biosensors have challenges with spectral overlap, differential binding affinities, and problems with linker dependencies, limiting their ability to perform multidimensional analysis48. RNA, however, potentially permits highly multidimensional analysis. To explore this possibility, we next sought to test whether ARs with orthogonal RNA outputs could be used to monitor dynamic, multidimensional PPIs. We engineered a well-controlled, synthetic trimolecular PPI network with an inducible change in interactions to validate the concept. As shown in Fig. 4a, we engineered N-29-1 fused to both ZA and FRB (“FZ-N”), which we could then deploy with C-terminal “CGG” RNAP variant fused to ZB or ZBneg (“Z-CG” or “Zneg-CG”) and C-terminal T7 RNAP fused to FKBP (“F-C7”). Therefore, interaction between FZ-N and Z-CG should produce an RNA signal from the CGG promoter, while interaction between FZ-N and F-C7 should produce an RNA signal from the T7 promoter (Fig. 4b).
Figure 4. Multidimensional PPI detection by ARs.

(a) Design of a synthetic trimolecular protein interaction network, using N-29-1 fused to ZA and FRB (“FZ-N”), and two different binding partners, C-terminal CGG RNAP fused to ZB or ZBneg (“Z-CG” pr “Zneg-CG), and C-terminal T7 RNAP fused to FKBP (“F-C7”). In the absence of rapamycin, FZ-N/Z-CG should be the dominant PPI, driving a CGG-promoter output. In the presence of rapamycin, the FZ-N/F-C7 PPI should also be present. (b) Vectors used to monitor the two PPIs simultaneously as shown in (a). Vectors designed that simultaneously express FZ-N, Z-CG, and F-C7, along with T7 promoter-driven luciferase and CGG-promoter-driven DsRed outputs. (c) Simultaneous monitoring both PPIs in the same cells. E. coli transformed with the expression vectors as shown, induced with either DMSO or 10 μM rapamycin for 5 h, then analyzed for luminescence and DsRed fluorescence (error bars std. dev., n = 4).
We first validated the engineered trimolecular model using our luciferase reporter system with only a single interaction at a time, revealing that each interaction can be monitored and selectively acts only on the prescribed promoter (Supplementary Fig. 4). Next, to simultaneously monitor both interactions in the same cells, we redesigned the vector system to express both F-C7 and Z-CG/Zneg-CG along with FZ-N, and produce DsRed from the CGG promoter and luciferase from the T7 promoter, allowing both transcriptional outputs to be simultaneously monitored. As seen in Fig. 4c, the FZ-N/Z-CG interaction is dependent on the zipper peptides and is unresponsive to rapamycin, while the FZ-N/F-C7 interaction is induced upon rapamycin addition. These data confirm that orthogonal C-terminal variants can be used along with N-29-1 to monitor multidimensional PPI networks in live cells.
ARs can control RNA synthesis in mammalian cells
Finally, we assayed the ability of the ARs to function in mammalian cells, using the rapamycin-inducible system as an exemplar. We generated the “rapa-T7” vector, which expresses both the rapamycin-inducible AR along with a T7 promoter-driven gene of interest (goi) output (Fig. 5a). To validate that RNA was being generated and to measure the kinetics of AR activation, we first deployed a fluorescent aptamer (F30-2xdBroccoli)49 as the goi output of the rapa-T7 vector, allowing the visualization of RNA synthesis using fluorescence microscopy. Treatment of HEK293T cells transfected with the rapa-T7-F30-2xdBroccoli vector with 100 nM rapamycin for 30 min results in a robust enhancement in intracellular fluorescence (Fig. 5b, Supplementary Fig. 5), demonstrating the fast kinetics of a T7 RNAP-based biosensor. Next, to assay whether we could trigger protein production and to assay processivity of the evolved split RNAP, we set IRES-driven mRNA for green fluorescent protein (GFP) as the goi on the rapa-T7 vector. Again, the background fluorescence level of rapa-T7-mRNA(GFP) transfected cells was low in the absence of rapamycin, but addition of 10 nM rapamycin resulted in a dramatic enhancement in GFP fluorescence (Fig. 5c, Supplementary Fig. 6). Finally, to test dose-responsiveness and whether ARs could trigger genetic changes to the cell, we tested whether RNAi is a viable output. For this, we set shRNA targeting GFP as the goi in the rapa-T7 vector, cotransfected cells with both the GFP expression vector and the rapa-T7-shRNA(GFP) vector, and analyzed GFP production by flow cytometry. Induction with rapamycin resulted in a dose-dependent knockdown of GFP signal (Fig. 5d). Collectively, these results demonstrate that ARs function in mammalian cells and can trigger a variety of different outputs via the RNA signal.
Figure 5. ARs can trigger a variety of outputs in mammalian cells.

(a) Design of the “rapa-T7” vector for rapamycin induced transgene expression in mammalian systems. (b) Validation of the rapa-T7 vector with a fluorescent RNA aptamer as the output. HEK293T cells were transfected with the rapa-T7-F30-2xdBroccoli vector (pJin141) and induced with 0 or 100 nM rapamycin for 30 min in the presence of 20 μM DHFBI-1T then analyzed by fluorescence microscopy. 100 μm scale bar shown. (c) Validation of the rapa-T7 vector with mRNA as the output. HEK293T cells transfected with the rapa-T7-mRNA(GFP) vector (p6-8) were induced with 0 or 10 nM rapamycin overnight and then analyzed by fluorescence microscopy (100 μm scale bar shown). (d) Validation of the rapa-T7 vector with shRNA as the output. HEK293T cells transfected with a GFP expression vector and a rapa-T7-shRNA(GFP) vector (pJin140) and induced with varying concentrations of rapamycin for 44 h. GFP fluorescence analyzed by flow cytometry (error bars std. error, n = 3).
DISCUSSION
Here we present the design, optimization, and deployment of a new split RNAP as a versatile biosensor platform. After discovering that fused PPIs can modulate the assembly of T7 RNAP split at position 179 to a modest degree, we developed a new PACE system to evolve an optimal proximity-dependent split RNAP. 29 days of PACE yielded a variant with a 350-fold dynamic range based on fused leucine zipper peptide PPIs. We then explored the versatility of the split RNAP system by generating light and small molecule activated RNAPs by simply swapping in different combinations of fusion proteins. Without any optimization, the resulting sensors had between 26 and >300-fold dynamic range. Moreover, we demonstrated that multidimensional PPI networks can be monitored using orthogonal RNAP sensors with different RNA outputs using a model trimolecular PPI system. Finally, we demonstrated the versatility of RNAP-based sensors, by showing that a small molecule-driven RNA output can be used to synthesize fluorescent RNA nanostructures, generate proteins, or knockdown genes in mammalian cells.
A challenge with traditional n-hybrid approaches is that linker lengths, compositions, and geometries of all of the parts need to be carefully tuned for each new interaction to be interrogated, measured, selected, or evolved. In practice, this often means months of cloning, screening, and careful optimization, and for some targets, steric or geometric concerns preclude detection. With this in mind, we sought to create the ARs in a manner that is less dependent on these variables, which we postulated would streamline the process of engineering new sensors. From a first approximation, tuning the assembly of a split RNAP to be more dependent on fusion proteins would involve weakening the affinity of the two RNAP halves. However, such a thermodynamic design would likely result in a high dependency on linkers and composition of the fusion proteins, which would make up the loss of binding energy. Therefore, we deliberately made the evolution of the ARs more challenging by including long (6–8 amino acids), flexible linkers tethering the fusion zipper peptides to each half of the split RNAP. In this way, the RNAP assembly process would likely evolve altered thermodynamics and kinetics of assembly, because the flexible linkers would not allow sufficient thermodynamic gain in binding energy between the fusion proteins. Further studies delineating the mechanism of assembly of the evolved proximity-dependent RNAPs will test this hypothesis and improve engineering efforts, and are currently ongoing. However, the fact that the AR system worked so well with diverse new binding partners suggests that whatever the evolved mechanism of RNAP assembly is, it resulted in a system that functions as we anticipated. Moreover, the fact that linker length had little effect on RNAP activity suggests thermodynamics are not the primary driving force.
Although PACE is an exceptionally powerful directed evolution platform, a fundamental limitation of the technology, and all in vivo evolution systems, is the challenge of designing genetic circuits that link target activities to fitness3. ARs provide a new, robust mechanism to link target activities to gene expression and fitness for applications in directed evolution. Now, with N-terminal RNAP variants that assemble in a manner dependent on fused molecular interactions in hand, we are exploring whether other target protein interaction domains can replace the leucine zipper peptides for engineering by continuous evolution. Even more broadly, our system can also be adopted to other 3-hybrid like approaches, where different types of “baits” are displayed on the C-terminal RNAP half, such as small molecules or RNA. Finally, because this new PACE system utilizes continuous and simultaneous dual positive and negative selection pressures, the approach should have advantages in terms of evolving selectivity in target proteins, as off target interactions can be displayed on the orthogonal C-terminal RNAP and drive negative selection.
The AR system provides a new approach to monitor and respond to molecular interactions, molecules, and external cues in live cells. Combined with the ability to generate ARs with orthogonal DNA promoter specificity, these tools open the possibility of performing highly multidimensional interaction network analysis, presaging a new approach to cell analysis using high-throughput sequencing23,24. ARs should have advantages in terms of sensitivity due to the signal amplification of the RNA output made possible by PCR. Unlike fluorescent-based technologies, which are solely for analysis, the RNA outputs of the ARs can be engineered to store information, or can drive cell fate changes based on measured events. Engineering AR-based gene circuits that simultaneously measure and compute both pathologically relevant endogenous events and external cues provides a new strategy to develop “smart” genetic therapies. Although we demonstrated PPI, small molecule, and light activated systems here, in principle, all of the existing fluorescent protein-based probes could be immediately transported into the AR system for integration with synthetic biology applications. We anticipate that ARs will provided a simplified and more robust strategy for engineering gene circuits for applications in screening, directed evolution, and synthetic biology.
ONLINE METHODS
Cloning
All plasmids were constructed by Gibson Assembly51 from PCR products generated using Q5 Hot Start DNA Polymerase (NEB) or Phusion Polymerase. Phage were cloned by Gibson Assembly of the split N-terminal RNAP-ZA fusion into a previously optimized SP phage backbone38 and transfromation into 1059 cells37, which supply gIII in an activity-independnent manner. After overnight growth in media, the supernatant was isolated by centrifugation and plaque assays were performed on 1059 cells. Single plaques were selected for overnight growth and sequencing to identify clonal phage samples with the correct insert. All plasmids and phage were sequenced at the University of Chicago Comprehensive Cancer Center DNA Sequencing and Genotyping Facility. All new vectors are described in Supplementary Table 1 and maps for each plasmid are shown in Supplementary Fig. 1.
Sequence of split RNAP fusions
Shown below are the structures and sequences of the leucine zipper peptide fusions, iLID-nano light-induced dimerization fusions, and rapamycin-induced dimerization constructs used in this study. Linkers are colored red and the fusion proteins are colored green. The three point mutations between ZB and ZBneg are underlined and were obtained from previous studies33, and the two mutations in ZA that evolved during PACE are also underlined.
| N-ZA: | RNAP(1-179)–GGSGSGSS–ALKKELQANKKELAQLKWELQALKKELAQ |
| C-ZB: | MASEQLEKKLQALEKKLAQLEWKNQALEKKLAQ–TSGGSG–RNAP(180+) |
| C-ZBneg: | MASEQLEKELQALEKELAQLKWKNQALEKKLAQ–TSGGSG–RNAP(180+) |
| N-ZA-(L13I,L20I): | N-28-1(1-179)–GGSGSGSS–ALKKELQANKKEIAQLKWEIQALKKELAQ |
| N-term-iLID: | N-28-1(1-179)–GGSGSGSS–iLID |
| SspB-C-term: | SspB nano–TSGGSG–RNAP(180+) |
| N-term-FRB: | N-28-1(1-179)–GGSGSGSS–FRB |
| FKBP-C-term: | FKBP–TSGGSG–RNAP(180+) |
| N-ZA-FRB: | RNAP(1-179)–GGSGSGSS–ZA-GGSAGGSG-FRB |
In vivo transcription assays of split RNAPs
N- and C-terminal halves of the RNAP were cloned into expression vectors, with the N-terminal RNAP under a constitutive promoter and the C-terminal RNAP under the arabinose-inducible promoter. S1030 cells37 were transformed by electroporation with three plasmids: (i) an N-terminal RNAP expression plasmid, (ii) a C-terminal expression plasmid, and (iii) a reporter plasmid that encodes luciferase under control of the T7 promoter. The transformed cells were then plated onto agar plates (15 g/L in LB) with 50 μg/mL carbenicillin, 50 μg/mL spectinomycin, 33 μg/mL chloramphenicol, and 10 mM glucose. Single colonies were then grown to saturation overnight at 37 °C, and then each well of a 96-well deep well plate containing 0.54 mL of LB with antibiotics and 10 mM arabinose was innoculated with 60 μL of the overnight culture. After growth with shaking at 37 °C for 2 h, 150 μL of each culture was transferred to a 96-well black wall, clear bottom plate (Costar), and luminescence and OD600 was measured on a Synergy H4 Hybrid Reader (BioTek). The data were analyzed by dividing the luminscence values by the background-corrected OD600 value, then subtracting out the background from the reporter vector alone. All values were then normalized to the wild-type split RNAP fused to ZA and ZB (data shown in Fig. 1c), which was assigned an arbitrary value of 100, allowing the values from each luminescence plot to be compared to one another. For the light-activated system, the experiment was performed identically except upon outgrowth for 3 h, cells in the light condition were illuminated with a homemade blue LED lightbox and cultured at 25 °C, and cells in the dark condition were cultured at 37°C; this was done to maintain similar termperatures in both conditions due to heat output from the light source. For the rapamycin inducible system, the experiment was performed identically, except upon outgrowth, rapamycin was added for 3 h, and then luminescence analyzed. Sample size (n = 4 biological replicates for each condition) was determined by previous work using a similar in vivo luciferase reporter system, and provided excellent reproducibility both between biological replicates on a given day and between days of experimental replicates.
Phage-assisted continuous evolution (PACE)
PACE was carried out to evolve the split T7 RNAP variants using a modified version of previously described methods24. E. coli strain S1030 were transformed by electroporation with combinations of the Positive Accessory Plasmid (posAP), Negative Accessory Plasmid (negAP), and Mutagenesis Plasmid (MP)38. 5 mL starter cultures were grown overnight in LB supplemented with antibiotics and 10 mM glucose. Chemostats (100 mL sterile bottles) containing 80 mL of Davis rich media37 were inoculated with 2 mL of starter culture and grown at 37 °C with magnetic stir-bar agitation. At approximately OD600 1.0, fresh Davis rich media was pumped in at 60–80 mL h−1, with a waste needle set at 80 mL. 10 μL of phage were used to seed a fresh lagoon (25 mL flask with a rubber septum). To intiate the evolution, a monoclonal phage population was utilized. Waste needles were set to maintain the lagoon volume at 15–20 mL, and host cell cultures were flowed in at 15–17 mL h−1. Arabinose (10% w/v in water) was added directly to lagoons via syringe pump at 1.0 mL h−1 to induce mutagenesis. A lagoon sample was taken from the waste withdrawal line every 24 h, centrifuged, and the supernatant was stored at 4 °C. A schematic of the complete evolutionary protocol is shown in Supplementary Table 2. After the completion of each leg of the evolution, activity-dependent plaque assays were used to select the next evolutionary target, and the PACE experiment was again intitiated as described, using 10 μL of phage from the previous endpoint of the evolution. The strength of the positive and negative selection pressures were varied by altering the ribosome binding sites (RBSs)50 controlling the expression of each of the C-terminal target fusions, gIII, and gIIIneg. Mixed selection pressures, indicated by listing multiple posAP/negAP sets for a given timepoint, were utilized as appropriate to enhance the likelihood of successful evolution.24,30,40
Sequence and acitivity analysis of variants from PACE
Phage samples were boiled for 10 min to lyse the phage and release the genomes. PCR was then used to amplifiy the DNA library containing the N-terminal RNAP variants, which was then subcloned into vector p3-7. Single colonies were picked from the transformation and subjected to analysis by Sanger sequencing. The results of the sequence analysis during the course of the evolution are shown in Supplementary Table 3. Variants N-29-1 and N-29-8 cloned into vector p3-7 were subjected to analysis by the luciferases assays as shown in Fig. 2e. In order to sequence any potential mutations that occurred in the peptide fusion in the phage, single plaques from an activity-independent plaque assay were picked, grown overnight, boiled, and then analyzed by Sanger sequencing.
In vivo split GFP assay
S1030 cells were transfomred by electroporation with two plasmids: (i) a constituitive N-terminal GFP-FRB fusion, and (ii) an arabionse inducible FKBP-C-terminal GFP fusion. The transformed cells were then plated onto agar plates (15 g/L in LB) with 50 μg/mL spectinomycin, 33 μg/mL chloramphenicol, and 10 mM glucose. Single colonies were then grown to saturation overnight at 37 °C, and then each well of a 96-well deep well plate containing 0.54 mL of LB with antibiotics, 10 mM glucose or 10 mM arabinose, and varying concentrations of rapamycin (0 nM, 1 nM, 10 nM, 0.1 μM, 1 μM, 20 μM, and 100 μM) was innoculated with 60 μL of the overnight culture. After growth with shaking at 37 °C for 3 h, 6 h, and 30 h, 150 μL of each culture was transferred to a 96-well deep well plate. The cultures were centrifuged (10 min, 25 ° C, 2000 rcf) and resuspended in 1 mL of PBS three times before suspending them in 150 μL of PBS and transferring them to a 96-well black wall, clear bottom plate (Costar), and GFP fluorescence (ex. 485/20 nm, em. 516/20 nm) and OD600 was measured on a Synergy Neo2 Microplate Reader (BioTek). The data were analyzed by dividing the background-corrected GFP fluorescence values by the background-corrected OD600 value. All values were then normalized to the 0 nM rapamycin (sample size n = 5 biological replicates for each condition).
Dual reporter PPI in vivo detection assays
FRB and ZA peptide were fused to the N-terminal RNAP(N-29-1) in an E. coli expression vector to form pJin200. FKBP was fused to the C-terminal of T7 RNAP and ZB or ZBneg was fused to C-terminal of CGG RNAP in one vector to construct the dual C-terminal vectors pJin207 and pJin208. Then the CGG promoter driven DsRed-Express2 circuit was added to p2-22 which contained T7 promoter driven luciferase gene to generate pJin216 as the dual reporter vector. To test the multidimensional system, pJin200, pJin216, and either pJin207 or pJin208 were transformed into s1030 cells. The transformed cells were then plated onto agar plates (15 g/L in LB) with 50 μg/mL carbenicillin, 50 μg/mL spectinomycin, 33 μg/mL chloramphenicol, and 10 mM glucose. Single colonies were cultured overnight in LB liquid media with the same antibiotics and 10 mM glucose. The next day, 50 μl of overnight cultures were transferred to a deep 96 well plate contained the LB media with same antibiotics, 10 mM Arabinose with 0 or 20 μM rapamycin. After shaking at 37 °C for 5 h, 150 μL of each culture was transferred to a 96-well deep well plate. The samples were centrifuged, washed and prepared in the same way as described in split GFP assay. Then the OD600, luminescence and DsRed-Express2 fluorescence (ex. 555/15 nm, em. 590/15 nm) were measured on a Synergy Neo2 Microplate Reader (BioTek). The data were analyzed by dividing the background-corrected luminescence values and DsRed-Express2 fluorescence background-corrected values by the background-corrected OD600 value. All luminescence values were normalized to the 0 μM rapamycin of pJin200/pJin216/pJin207 while DsRed-Express2 fluorescence values were normalized to the 0 μM rapamycin of pJin200/pJin216/pJin208 (sample size n = 5 biological replicates for each condition).
Cell culture
HEK293T cells (ATCC) were maintained in DMEM (high glucose, L-glutamine, phenol red, sodium pyruvate; obtained from Gibco or Hyclone) supplemented with 10% fetal bovine serum (FBS, Gibco/Life Technologies, Qualified US origin) and 1% penecillin/streptomycin (P/S, Gibco/Life Technologies). As HEK293T cells are listed in the database of commonly misidentified cell lines maintained by ICLAC (http://iclac.org/databases/cross-contaminations/), we obtained fresh cells from ATCC, which were frozen down at an early passage (passage 5) in individual aliquots. The cells were then used for less than 25 passages for all experiments. Multiple biological replicates were performed with cells from different passages and freshly thawed aliquots. There was no testing for mycoplasma infection or further authentification because early passage cells were used for all experiments.
Imaging mammalian AR activation by fluorescence microscopy
HEK293T cells cultured in DMEM (high glucose, glutamine, phenol red, pyruvate; Gibco/Life Technolgies) supplemented with 10% fetal bovine serum (FBS, Gibco/Life Technolgies, Qualified US origin) were plated on an 8-well coverglass slide (Labtek) and transfected with 600 ng of a rapa-T7 vector (pJin141 or p6-8) using 1.5 μL of Lipofectamine 2000 (ThermoFisher Scientific) using the standard protocol. For the rapa-T7-F30-2xdBroccoli (pJin141) experiments, 100 nM raprmycin or DMSO control was added along with 20 μM DHFB1-1T for 30 min prior to imaging. For the rapa-T7-mRNA(GFP) (p6-8) experiments, 10 nM rapamycin or DMSO control was added to the sample 20 h after transfection, and then incubated for an additional 24 h. The cells were imaged on an Olympus BX53 microscope using a GFP filter set and a 10x objective. Each image for a given condition was processed using identical conditions to adjust brightness and contrast to a level where background fluorescence was observed for control samples in ImageJ (Wayne Rasband, NIH).
Flow cytometry
HEK293T cells were cultured in DMEM (high glucose, glutamine, phenol red, pyruvate; Gibco/Life Technolgies) supplemented with 10% fetal bovine serum (FBS, Gibco/Life Technolgies). The day prior to transfection, cells were passaged and plated at ~50,000 cells per well in a 48 well plate (NEST Biotechnology). After 19 hours, 50 ng of the RFP plasmid (p3-62), 200 ng of the GFP plasmid (p1-53), and 400 ng of the rapa-T7-shRNA(GFP) vector (pJin140) were transfected into cells using 1.5 μL of Lipofectamine 2000 (ThermoFisher Scientific) using the standard protocol. 30 min after transfection, DMEM supplemented with FBS and either DMSO or rapamycin was added to the wells so the final rapamycin concentrations were 0 nM, 0.001 nM, 0.01 nM, 0.1 nM, 1 nM, and 10 nM. 29 h after transfection, the media was replaced with fresh media including the correct concentration of rapamycin. 44 h after transfection, the cells were trypsinized and suspended in DMEM supplemented with FBS and rapamycin, and then analyzed on a LSR-Fortessa 4–15 (BD digital instrument, 488 nM laser, 530/30 nM filter for GFP, and 610/20 nM filter for RFP). Mean GFP fluorescence intensity was calculated for HEK293T cells expressing RFP using FloJo software. Reported values are average mean GFP fluorescence intensity values taken from three separate replicate samples. Sample size (n = 3 biological replicates for each condition) was determined by initial trial experiments to find the spread in the data.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
Supplementary Material
Acknowledgments
This work was supported by the University of Chicago, the Cancer Research Foundation, the National Institute of General Medical Sciences of the National Institutes of Health (R35 GM119840), the University of Chicago Medicine Comprehensive Cancer Center (#P30 CA14599), and the National Center for Advancing Translational Sciences of the National Institutes of Health (UL1 TR000430). J.Z. was supported by a Chemical Biology Training Grant from the US National Institutes of Health (T32GM008720). We thank Ian Chronis, Younghoon Koh, and Daniel Ahn for technical assistance, and David Liu, Brian McNaughton, Alexander Dieter, Benjamin Glick, Michael Glotzer, Yamuna Krishnan, Joe Thornton, and Yossi Weizmann for supplying equipment and materials.
Footnotes
AUTHOR CONTRIBUTIONS
J.Z., J.P., and B.C.D. cloned and validated all materials and performed transcription assays. J.P. and B.C.D. performed PACE experiments. J.Z. and J.P performed cell culture experiments. J.Z., J.P., and B.C.D. designed experimental strategies and wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors have filed a provisional patent application on the AR system.
References
- 1.Ruder WC, Lu T, Collins JJ. Synthetic biology moving into the clinic. Science. 2011;333:1248–1252. doi: 10.1126/science.1206843. [DOI] [PubMed] [Google Scholar]
- 2.Lienert F, Lohmueller JJ, Garg A, Silver PA. Synthetic biology in mammalian cells: next generation research tools and therapeutics. Nat Rev Mol Cell Biol. 2014;15:95–107. doi: 10.1038/nrm3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Packer MS, Liu DR. Methods for the directed evolution of proteins. Nat Rev Genet. 2015;16:379–394. doi: 10.1038/nrg3927. [DOI] [PubMed] [Google Scholar]
- 4.Zhang J, Jensen MK, Keasling JD. Development of biosensors and their application in metabolic engineering. Current Opinion in Chemical Biology. 2015;28:1–8. doi: 10.1016/j.cbpa.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 5.Benenson Y. RNA-based computation in live cells. Curr Opin Biotechnol. 2009;20:471–478. doi: 10.1016/j.copbio.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Green AA, Silver PA, Collins JJ, Yin P. Toehold switches: de-novo-designed regulators of gene expression. Cell. 2014;159:925–939. doi: 10.1016/j.cell.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brophy JA, Voigt CA. Principles of genetic circuit design. Nat Methods. 2014;11:508–520. doi: 10.1038/nmeth.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Copeland MF, Politz MC, Johnson CB, Markley AL, Pfleger BF. A transcription activator-like effector (TALE) induction system mediated by proteolysis. Nat Chem Biol. 2016 doi: 10.1038/nchembio.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Putz U, Skehel P, Kuhl D. A tri-hybrid system for the analysis and detection of RNA--protein interactions. Nucleic Acids Res. 1996;24:4838–4840. doi: 10.1093/nar/24.23.4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 11.SenGupta DJ, et al. A three-hybrid system to detect RNA-protein interactions in vivo. Proc Natl Acad Sci U S A. 1996;93:8496–8501. doi: 10.1073/pnas.93.16.8496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker K, et al. Chemical complementation: a reaction-independent genetic assay for enzyme catalysis. Proc Natl Acad Sci U S A. 2002;99:16537–16542. doi: 10.1073/pnas.262420099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konermann S, et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500:472–476. doi: 10.1038/nature12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polstein LR, Gersbach CA. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat Chem Biol. 2015;11:198–200. doi: 10.1038/nchembio.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nunez JK, Harrington LB, Doudna JA. Chemical and Biophysical Modulation of Cas9 for Tunable Genome Engineering. ACS Chem Biol. 2016;11:681–688. doi: 10.1021/acschembio.5b01019. [DOI] [PubMed] [Google Scholar]
- 16.Martin F. Fifteen years of the yeast three-hybrid system: RNA-protein interactions under investigation. Methods. 2012;58:367–375. doi: 10.1016/j.ymeth.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 17.Church GM, Elowitz MB, Smolke CD, Voigt CA, Weiss R. Realizing the potential of synthetic biology. Nat Rev Mol Cell Biol. 2014;15:289–294. doi: 10.1038/nrm3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yen L, et al. Exogenous control of mammalian gene expression through modulation of RNA self-cleavage. Nature. 2004;431:471–476. doi: 10.1038/nature02844. [DOI] [PubMed] [Google Scholar]
- 19.Winkler WC, Breaker RR. Regulation of bacterial gene expression by riboswitches. Annu Rev Microbiol. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- 20.Culler SJ, Hoff KG, Smolke CD. Reprogramming cellular behavior with RNA controllers responsive to endogenous proteins. Science. 2010;330:1251–1255. doi: 10.1126/science.1192128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng J, et al. A general strategy to construct small molecule biosensors in eukaryotes. eLife. 2015;4:e10606. doi: 10.7554/eLife.10606. DOI: http://dx.doi.org/10.7554/eLife.10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JH, et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 2014;343:1360–1363. doi: 10.1126/science.1250212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zamft BM, et al. Measuring cation dependent DNA polymerase fidelity landscapes by deep sequencing. PLoS One. 2012;7:e43876. doi: 10.1371/journal.pone.0043876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pu J, Chronis I, Ahn D, Dickinson BC. A Panel of Protease-Responsive RNA Polymerases Respond to Biochemical Signals by Production of Defined RNA Outputs in Live Cells. J Am Chem Soc. 2015;137:15996–15999. doi: 10.1021/jacs.5b10290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Segall-Shapiro TH, Meyer AJ, Ellington AD, Sontag ED, Voigt CA. A ‘resource allocator’ for transcription based on a highly fragmented T7 RNA polymerase. Mol Syst Biol. 2014;10:742. doi: 10.15252/msb.20145299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shis DL, Bennett MR. Library of synthetic transcriptional AND gates built with split T7 RNA polymerase mutants. Proc Natl Acad Sci U S A. 2013;110:5028–5033. doi: 10.1073/pnas.1220157110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shekhawat SS, Ghosh I. Split-protein systems: beyond binary protein-protein interactions. Curr Opin Chem Biol. 2011;15:789–797. doi: 10.1016/j.cbpa.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kerppola TK. Visualization of molecular interactions using bimolecular fluorescence complementation analysis: characteristics of protein fragment complementation. Chem Soc Rev. 2009;38:2876–2886. doi: 10.1039/b909638h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu CD, Kerppola TK. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat Biotechnol. 2003;21:539–545. doi: 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dickinson BC, Leconte AM, Allen B, Esvelt KM, Liu DR. Experimental interrogation of the path dependence and stochasticity of protein evolution using phage-assisted continuous evolution. Proc Natl Acad Sci U S A. 2013;110:9007–9012. doi: 10.1073/pnas.1220670110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ellefson JW, et al. Directed evolution of genetic parts and circuits by compartmentalized partnered replication. Nat Biotechnol. 2014;32:97–101. doi: 10.1038/nbt.2714. [DOI] [PubMed] [Google Scholar]
- 32.Ghosh I, Hamilton AD, Regan L. Antiparallel Leucine Zipper-Directed Protein Reassembly: Application to the Green Fluorescent Protein. J Am Chem Soc. 2000;122:5658–5659. [Google Scholar]
- 33.Magliery TJ, et al. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: scope and mechanism. J Am Chem Soc. 2005;127:146–157. doi: 10.1021/ja046699g. [DOI] [PubMed] [Google Scholar]
- 34.Steitz TA. The structural changes of T7 RNA polymerase from transcription initiation to elongation. Curr Opin Struct Biol. 2009;19:683–690. doi: 10.1016/j.sbi.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature. 2011;472:499–503. doi: 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Badran AH, et al. Continuous evolution of Bacillus thuringiensis toxins overcomes insect resistance. Nature. 2016 doi: 10.1038/nature17938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlson JC, Badran AH, Guggiana-Nilo DA, Liu DR. Negative selection and stringency modulation in phage-assisted continuous evolution. Nat Chem Biol. 2014;10:216–222. doi: 10.1038/nchembio.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dickinson BC, Packer MS, Badran AH, Liu DR. A system for the continuous directed evolution of proteases rapidly reveals drug-resistance mutations. Nat Commun. 2014;5:5352. doi: 10.1038/ncomms6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hubbard BP, et al. Continuous directed evolution of DNA-binding proteins to improve TALEN specificity. Nat Methods. 2015;12:939–942. doi: 10.1038/nmeth.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leconte AM, et al. A population-based experimental model for protein evolution: effects of mutation rate and selection stringency on evolutionary outcomes. Biochemistry. 2013;52:1490–1499. doi: 10.1021/bi3016185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheetham GM, Steitz TA. Structure of a transcribing T7 RNA polymerase initiation complex. Science. 1999;286:2305–2309. doi: 10.1126/science.286.5448.2305. [DOI] [PubMed] [Google Scholar]
- 42.Tahirov TH, et al. Structure of a T7 RNA polymerase elongation complex at 2.9 A resolution. Nature. 2002;420:43–50. doi: 10.1038/nature01129. [DOI] [PubMed] [Google Scholar]
- 43.Guntas G, et al. Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc Natl Acad Sci U S A. 2015;112:112–117. doi: 10.1073/pnas.1417910112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rivera VM, et al. A humanized system for pharmacologic control of gene expression. Nat Med. 1996;2:1028–1032. doi: 10.1038/nm0996-1028. [DOI] [PubMed] [Google Scholar]
- 45.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paulmurugan R, et al. A novel estrogen receptor intramolecular folding-based titratable transgene expression system. Mol Ther. 2009;17:1703–1711. doi: 10.1038/mt.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blakeley BD, Chapman AM, McNaughton BR. Split-superpositive GFP reassembly is a fast, efficient, and robust method for detecting protein-protein interactions in vivo. Mol Biosyst. 2012;8:2036–2040. doi: 10.1039/c2mb25130b. [DOI] [PubMed] [Google Scholar]
- 48.Kerppola TK. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys. 2008;37:465–487. doi: 10.1146/annurev.biophys.37.032807.125842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Filonov GS, Kam CW, Song W, Jaffrey SR. In-gel imaging of RNA processing using broccoli reveals optimal aptamer expression strategies. Chem Biol. 2015;22:649–660. doi: 10.1016/j.chembiol.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ringquist S, et al. Translation initiation in Escherichia coli: sequences within the ribosome-binding site. Mol Microbiol. 1992;6:1219–1229. doi: 10.1111/j.1365-2958.1992.tb01561.x. [DOI] [PubMed] [Google Scholar]
- 51.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are available from the corresponding author on reasonable request.