

Abstract
The mechanistic target of rapamycin complex 1 (mTORC1) protein kinase is a master growth regulator that becomes activated at the lysosome in response to nutrient cues. Here we identify cholesterol, an essential building block for cellular growth, as a nutrient input that drives mTORC1 recruitment and activation at the lysosomal surface. The lysosomal transmembrane protein, SLC38A9, is required for mTORC1 activation by cholesterol through conserved cholesterol-responsive motifs. Moreover, SLC38A9 enables mTORC1 activation by cholesterol independently from its arginine sensing function. Conversely, the Niemann-Pick C1 (NPC1) protein, which regulates cholesterol export from the lysosome, binds to SLC38A9 and inhibits mTORC1 signaling through its sterol transport function. Thus, lysosomal cholesterol drives mTORC1 activation and growth signaling through the SLC38A9-NPC1 complex.
Cholesterol is an essential building block for membrane biogenesis, and rapidly proliferating cells rely on enhanced cholesterol synthesis and uptake to sustain their growth(1). Eukaryotic cells have evolved specialized protein machinery that senses cholesterol and adjusts the rate of cholesterol synthesis and uptake to match the cell’s specific metabolic needs (2, 3). Whether direct cholesterol sensing extends beyond the control of its own synthesis and, specifically, whether it signals to pathways that regulate the control of cellular growth and proliferation is currently uncertain (4, 5).
The lysosome is a major sorting station for dietary cholesterol. Low-density lipoproteins (LDL) carrying dietary cholesterol and fatty acids enter the cell by receptor-mediated endocytosis and are disassembled in the lysosomal lumen(3). A sterol transport system composed of the Niemann-Pick C1 (NPC1) and NPC2 proteins localize specifically at the late endosome or lysosome, where it binds free cholesterol and mediates its export to diverse cellular compartments including the plasma membrane and the endoplasmic reticulum (ER), through mechanisms that remain poorly understood, (6). The sterol trafficking function of NPC1 is essential for the correct execution of numerous cellular activities, and its inactivation results in a lipid storage disorder, Niemann-Pick type C, characterized by spleen and liver dysfunction and neurodegeneration (7, 8).
The lysosome has also emerged as the cellular site where the master growth regulator, mTORC1 kinase, is activated. Nutrient signals carried by free amino acids and glucose regulate mTORC1 activation through the Rag guanosine triphosphatases (GTPases). The Rag GTPases are obligate heterodimers of Rag A or B in complex with Rag C or D. GTP loading of Rag A or B and concomitant GTP hydrolysis by Rag C or D are thought to enable the Rag heterodimer to physically bind to mTORC1 and anchor it to the lysosomal surface(9–11). At the lysosome, another GTPase known as Rheb triggers the kinase activity of mTORC1 and enables substrate phosphorylation (11–13).
mTORC1 drives the synthesis of fatty acids and sterols by increasing the expression and proteolytic processing of the master lipogenic transcription factors, SREBP1c and SREBP2(14, 15), thereby promoting cell proliferation (16). Proliferating cells also actively take up exogenous lipids in the form of LDL(1, 3). Given the central role of mTORC1 in promoting lipid biosynthesis, it is reasonable that dietary lipids should feed back on mTORC1 activation. Accordingly, a high fat diet (HFD) stimulates mTORC1 activity in mice (16, 17). However, whether and how mTORC1 senses dietary lipids in a cell-autonomous manner remains unclear (4). Given the dual role of the lysosome as a nutrient gateway and a signaling platform for mTORC1 activation, we tested whether the lysosomal membrane contains dedicated machinery that senses and communicates cholesterol availability to mTORC1.
In cultured human embryonic kidney (HEK-293T) cells, depletion of cellular cholesterol with methyl-beta cyclodextrin (MCD) suppressed mTORC1 signaling, as measured by loss of phosphorylation of mTORC1 substrates S6-kinase 1 (S6K1, Thr 389) and 4E-binding protein 1 (4E-BP1, Ser 65), whereas exposure of cells to exogenous LDL reactivated mTORC1 in a dose-dependent manner (Fig. 1A). LDL-mediated mTORC1 activation occurred in a time-dependent manner and peaked at 1h (Fig. 1B top). This lag correlated with the time required for fluorescently labeled LDL particles to accumulate in the lysosome following endocytosis (Fig. 1B bottom). Pre-complexed MCD and cholesterol at a 1:1 molar ratio is a commonly used method to deliver cholesterol to membranes (18). Treatment of sterol-depleted cells with MCD:cholesterol for 1h activated mTORC1 signaling with a potency comparable to LDL in HEK-293T cells, Chinese hamster ovary (CHO) cells and mouse embryonic fibroblasts (MEFs) (Fig. 1C and Fig. S1A). As an additional readout of mTORC1 activity, we visualized the subcellular localization of green-fluorescent protein (GFP)-tagged Transcription Factor EB (TFEB), which translocates to the nucleus upon mTORC1 inactivation (12). In HEK-293T and CHO cells stably expressing TFEB-GFP that were sterol-depleted, TFEB was found primarily in the nucleus, whereas in cells refed with LDL or MCD:cholesterol, TFEB redistributed in the cytoplasm consistent with mTORC1 reactivation (Fig 1D, 1E, and Fig. S1B).
Fig. 1. Lysosomal cholesterol stimulates mTORC1 recruitment and signaling.
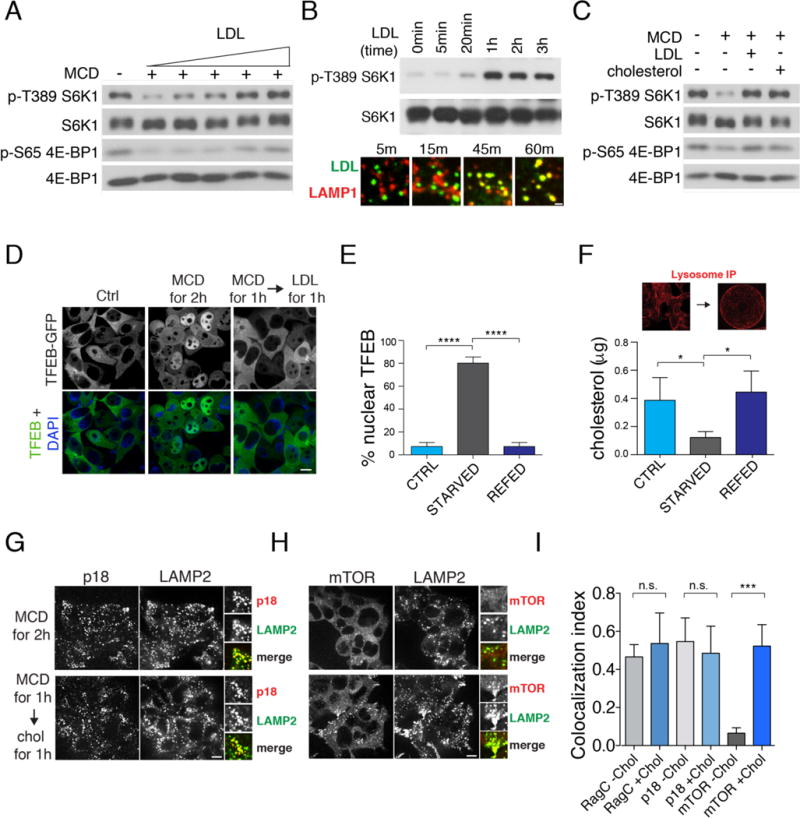
(A) Dose-dependent activation of mTORC1 by LDL. HEK-293T cells were depleted of sterol with methyl-beta cyclodextrin (MCD, 0.5% w/v) for 2 hours and stimulated for 2 hours with various concentrations (0-100 μg/ml) of LDL. Cell lysates were analyzed for phosphorylation status of S6K1 (T389) and 4E-BP1 (S65) and for total protein abundance. (B) (top) Time-dependent activation of mTORC1 by LDL. HEK-293T cells were depleted of sterol for 2 hours and re-stimulated with LDL (50 μg/ml) for the indicated times. Cell lysates were analyzed for phosphorylation status of S6K (T389). (bottom) Time-course of LDL delivery to the lysosome. Cells stably expressing LAMP1-mRFP-FLAGX2 (LRF) were treated with BODIPY-LDL for the indicated times. Scale bar, 0.5μm (C) Activation of mTORC1 signaling by cholesterol. HEK-293T cells were depleted of sterol for 2 hours and, where indicated, re-stimulated for 2 hours with 50μM MCD:cholesterol or with 50μg/ml LDL where indicated. Phosphorylation of S6K1 and 4E-BP1 are shown. (D) Regulation of TFEB nuclear localization by cholesterol. HEK-293T cells stably expressing TFEB-GFP were depleted of sterol and, where indicated, re-stimulated with LDL (50μg/ml). Scale bar, 10μm. (E) Quantification of TFEB-GFP localization from (D). Shown are mean + SD. N=800 cells. ANOVA: p < 0.0001 followed by Tukey’s t-test: ****p < 0.0001 (F) (top) Lysosomes from LRF-expressing HEK-293T cells were immuno-captured onto FLAG affinity beads. (bottom) Mass spectrometry measurement of unesterified cholesterol in immuno-captured lysosomes from LRF-expressing HEK-293T cells subjected to the indicated treatments. Shown are mean + SD. N=4 samples/condition. ANOVA: p < 0.05 followed by Tukey’s t-test: *p <0.05 (G) Cholesterol status does not affect Ragulator localization to LAMP2-positive lysosomes. HEK-293T cells were subjected to the indicated treatments, followed by immunofluorescence for endogenous p18 and LAMP2. Scale bar, 10μm. (H) Cholesterol regulates mTORC1 recruitment to LAMP2-positive lysosomes. HEK-293T cells were subjected to the indicated treatments, followed by immunofluorescence for endogenous mTOR and LAMP2. Scale bar, 10μm. (I) Quantification of RagC-LAMP2, p18-LAMP2 and mTOR-LAMP2 co-localization under cholesterol-depleted and cholesterol-stimulated conditions. Shown are mean + SD. N=15 cells/condition. ANOVA: p < 0.0001 followed by Tukey’s t-test: ***p < 0.001.
The stimulatory effect of cholesterol toward mTORC1 appeared to be highly specific, as it could not be recapitulated by treating sterol-depleted cells with high concentrations of various oxysterols, sterol-related compounds or with oleic acid, a fatty acid found in LDL(19) (Fig S1A, S1C and S1D). Moreover, inhibiting endogenous cholesterol synthesis with mevastatin also suppressed mTORC1 signaling, which fully recovered following washout of the drug for 1h (Fig S1E).
Changes in cholesterol levels can alter the activity of signaling pathways originating at the plasma membrane such as phosphoinositide-3 kinase (PI3K) and the protein kinase Akt, which acts upstream of mTORC1 (4, 12). However, in mouse embryonic fibroblasts (MEFs) in which PI3K-Akt input to mTORC1 is constitutively active due to lack of the Tuberous Sclerosis Complex 2 (TSC2) gene (15, 20), sterol depletion still inhibited mTORC1 in a dose-dependent manner (Fig. S2A). Thus, cholesterol concentrations affect mTORC1 activity through a mechanism that is, at least in part, distinct from growth factor signaling.
Because the lysosome is the site of mTORC1 activation, we tested whether global sterol depletion via MCD led to changes in the lysosomal cholesterol pool. Indeed, mass spectrometry analysis of intact lysosomes immuno-purified from HEK-293T cells stably expressing an affinity tag composed of the lysosomal transmembrane protein LAMP1 fused to mRFP and FLAGX2 (LRF) (Fig. 1F top) (21) showed that MCD treatment led to a 60% decrease in lysosomal cholesterol compared to unstarved cells. Importantly, steady-state levels of cholesterol in the lysosome were restored upon re-feeding with MCD:cholesterol for 1h (Fig. 1F bottom). Thus, the lysosomal cholesterol pool is in rapid equilibrium with the plasma membrane pool and reflects global changes in cellular sterol levels (18).
Based on these results, we examined whether cholesterol affects mTORC1 localization to the lysosomal surface, a step regulated by the nucleotide state of the Rag GTPases and by their scaffold, the Ragulator complex (10, 12). Manipulating cholesterol levels did not disrupt the integrity of the Ragulator-Rag GTPase complex, because RagC and the Ragulator subunit p18 localized to LAMP2-containing lysosomes irrespective of cholesterol status (Fig 1G, 1I and S2B). In contrast, cholesterol depletion caused the dissociation of mTOR from LAMP2-positive lysosomes, whereas restimulation with either MCD:cholesterol or LDL restored co-localization of mTOR and LAMP2 (Fig 1H, 1I and Fig. S2C). Although this pattern was reminiscent of that induced by amino acid starvation and refeeding(10, 11, 21), manipulation of cholesterol levels did not significantly alter intracellular amino acid levels (Fig. S3A). Collectively, these results indicate that lysosomal cholesterol functions upstream of the mTORC1 scaffolding complex independent of amino acids.
To determine whether cholesterol regulates the Rag GTPases, we used a co-immunoprecipitation assay that reads out the activation state of Rag GTPases as a function of their affinity for Ragulator. The Ragulator-Rag GTPase interaction is weakened by amino acids. This effect is thought to reflect GTP loading of RagA or RagB by Ragulator, enabling RagA/B to bind to mTORC1 (21, 22). In HEK-293T cells that were cholesterol-depleted and then restimulated with MCD:cholesterol, FLAG-tagged Ragulator subunit p14 bound to endogenous RagA and RagC more weakly than in cholesterol-depleted cells (Fig. 2A). Thus, cholesterol has the same activating effect as amino acids on the mTORC1 scaffolding complex.
Fig. 2. Cholesterol regulates the Ragulator-Rag GTPase complex in cells and in vitro.
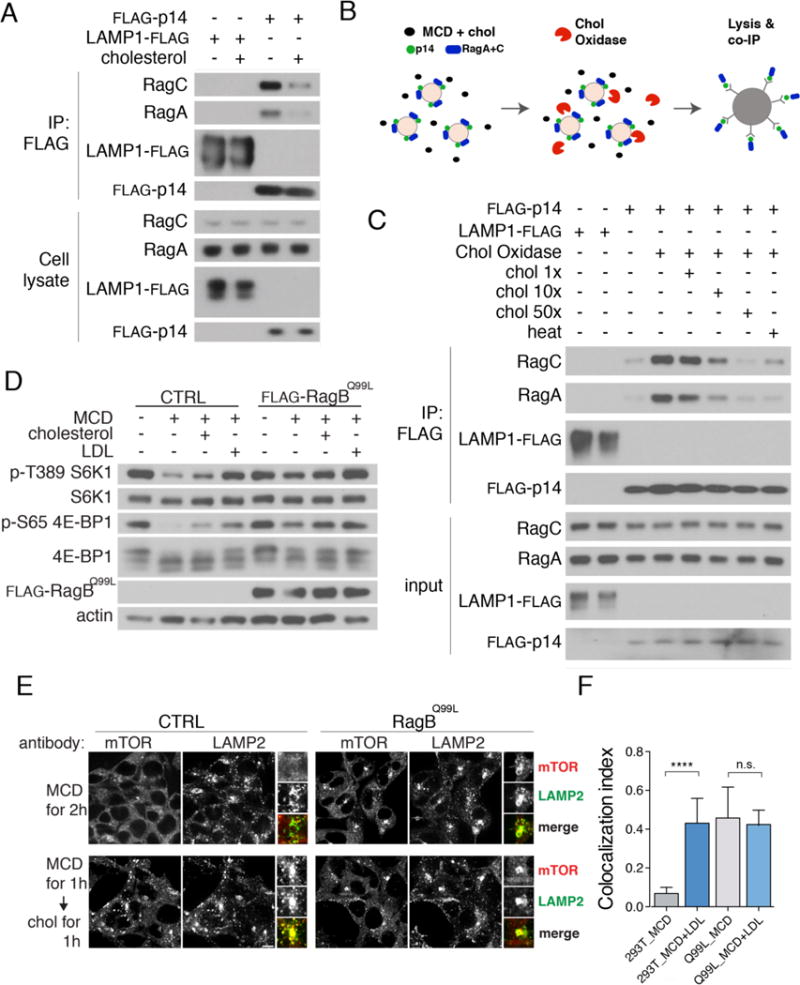
(A) Cholesterol regulates the interaction between Ragulator and Rag GTPases in cells. HEK-293T cells stably expressing FLAG-p14 or LRF were sterol-depleted for 2h and, where indicated, restimulated with MCD:cholesterol for 2h. After lysis, samples were subjected to FLAG immunoprecipitation and immunoblotting for the indicated proteins. (B) Organelle-based in vitro assay. A light organelle preparation from cells stably expressing FLAG-tagged mTORC1 pathway components is treated with cholesterol oxidase in the absence or presence of MCD-cholesterol complex. Interaction with endogenous binding partners is determined by FLAG immunoprecipitation and western blotting following detergent solubilization. (C) Cholesterol regulates the interaction between Ragulator and Rag GTPases in vitro. Light organelle fractions stably expressing FLAG-p14 were treated with cholesterol oxidase (2U/ml) along with increasing concentrations of MCD:cholesterol as indicated. Samples were subjected to lysis and FLAG immunoprecipitation, followed by immunoblotting for the indicated proteins. (D) HEK-293T cells stably expressing the constitutively active RagBQ99L mutant, along with control HEK-293T, were sterol-depleted for 2h, or depleted and restimulated for 2h. Cell lysates were immunoblotted for the indicated proteins and phospho-proteins. (E) Control and RagBQ99L -expressing HEK-293T cells were sterol-depleted for 2h, or depleted and, where indicated, restimulated for 1h, followed by immunofluorescence staining for endogenous mTOR and LAMP2. Scale bar, 10μm. (F) Quantification of mTOR-LAMP2 co-localization in control HEK-293T and in HEK-293T expressing RagBQ99L under sterol-depleted and sterol-stimulated conditions. Shown are mean + SD. N=15 cells/condition. ANOVA: p < 0.0001 followed by Tukey’s t-test: ****p < 0.0001.
We reconstituted cholesterol signaling in vitro by modifying a cell-free assay that monitors amino acid-dependent activation of the Rag GTPases (21) (Fig. 2B and Fig. S4A). Light organelle fractions from HEK-293T cells stably expressing FLAG-p14 were treated with recombinant cholesterol oxidase, a bacterial enzyme that converts cholesterol into cholest-4-en-3-one, effectively depleting cholesterol from the lysosomal limiting membrane (23) (Fig. 2B). Notably, cholest-4-en-3-one failed to activate mTORC1 signaling when delivered to HEK-293T cells (Fig. S1D). Conversion of cholesterol to cholest-4-en-3-one in vitro enhanced the binding of FLAG-p14 to RagA and RagC, consistent with Rag GTPase inactivation (Fig. 2C). The increased association of p14 with RagA and RagC was blocked by free cholesterol in a dose-dependent manner, or by heat-inactivation of the cholesterol oxidase enzyme (Fig. 2C). Similarly, depleting cholesterol in the cell-free assay with MCD enhanced interaction of Ragulator with Rag GTPases over that observed in the replete state. (Fig. S4A and S4B). Thus, both in cells and using an in vitro reconstituted organelle system, lysosomal cholesterol modulates protein-protein interactions within the mTORC1 scaffolding complex in a manner consistent with an activating effect toward the Rag GTPases.
To further test the hypothesis that cholesterol functions upstream of the Rag GTPases, we used HEK-293T and CHO lines stably expressing ‘active’ Rag GTPase mutants that constitutively target mTORC1 to the lysosomal surface independent of amino acid status (9, 11). In HEK-293T and CHO cells expressing active Rag GTPase mutants, mTORC1 signaling was stronger than in control cells and largely resistant to cholesterol depletion, either alone or in combination with arginine depletion (Fig. 2D, S4C and S4D). Moreover, in these cells mTOR remained clustered on LAMP2-positive lysosomes upon sterol starvation, whereas the same treatment led to dissociation of mTOR from the lysosomes of control cells (Fig. 2E and 2F). Thus, like amino acids, cholesterol affects the function of the mTORC1 scaffolding complex by modulating the activation state of the Rag GTPases.
A search for cholesterol-regulated motifs within the mTORC1 scaffolding complex identified a putative Cholesterol Recognition Amino acid Consensus (CRAC) motif within transmembrane domain 8 of the lysosomal Na+-coupled amino acid transporter, SLC38A9 (Fig. 3A). SLC38A9 is implicated in the regulation of mTORC1 by amino acids, particularly by arginine (24, 25). The consensus CRAC sequence: (L/V)-X1–5-Y- X1–5-(K/R) of SLC38A9 is conserved across vertebrates (23, 26) (Fig. 3A). SLC38A9 is the only member of the SLC38 Na+ coupled amino acid transporter family that contains a CRAC motif (Fig. S5A). Moreover, the CRAC motif is immediately preceded by a similar but inverted cholesterol-recognition motif known as CARC, such that the tandem motifs together span the lipid bilayer (Fig. 3A). CRAC and CARC motifs have been described in cholesterol-regulated proteins such as caveolins and the Ca2+ channel Orai1 (23).
Fig. 3. SLC38A9 mediates mTORC1 activation by lysosomal cholesterol.
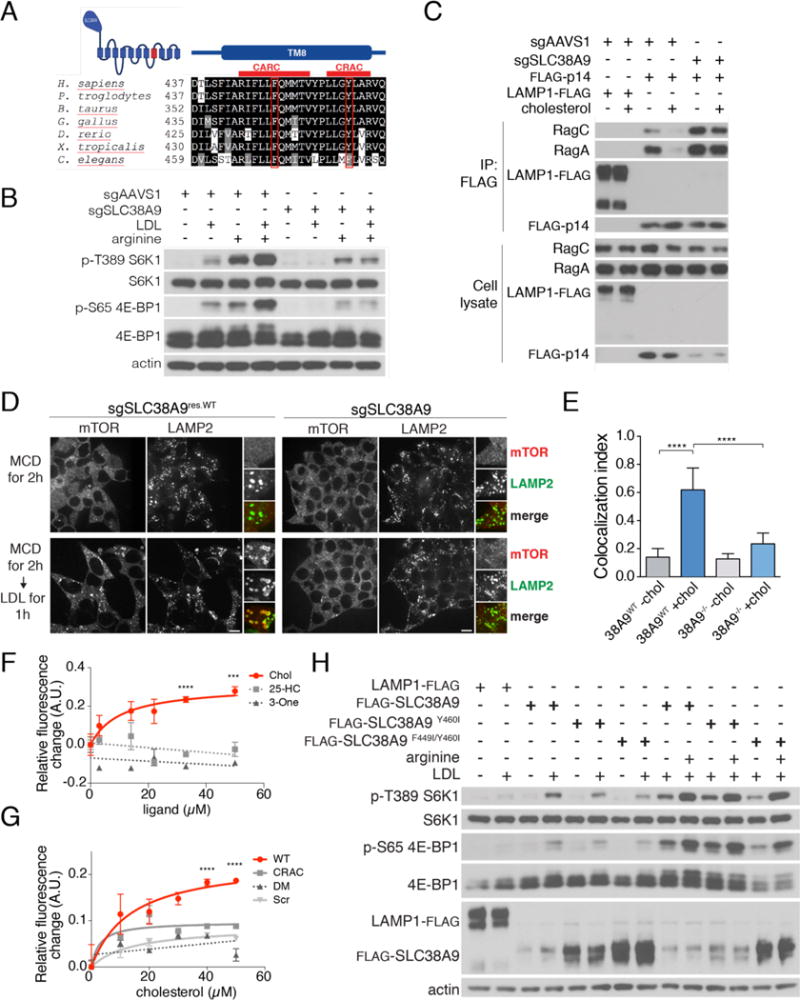
(A) Conservation analysis of transmembrane helix 8 (TM8) of SLC38A9, with the CARC and CRAC motifs indicated by red lines. The essential Phe and Tyr within the CARC and CRAC motif, respectively, are boxed in red. (B) Control or SLC38A9-deleted HEK-293T cells were double-starved for cholesterol and arginine for 2h and, where indicated, restimulated with LDL, arginine or both for 2h. Cell lysates were immunoblotted for the indicated proteins and phospho-proteins. (C) SLC38A9 is required for cholesterol modulation of the Ragulator-Rag GTPase interaction. Control or SLC38A9-deleted HEK-293T stably expressing FLAG-p14 or LRF were subjected to cholesterol starvation for 2h and, where indicated, restimulated with MCD:cholesterol for 2h. Cells were lysed and subjected to FLAG immunoprecipitation and immunoblotting for the indicated proteins. (D) SLC38A9-deleted or FLAG-SLC38A9 rescued HEK-293T cells were subjected to the indicated treatments, followed by immunofluorescence for endogenous mTOR and LAMP2. Scale bar, 10μm. (E) Quantification of mTOR-LAMP2 co-localization from the experiment in (D). Shown are mean + SD. N=15 cells/condition. ANOVA: p < 0.0001 followed by Tukey’s t-test: ****p < 0.0001. (F) Titration curves displaying changes in the intrinsic fluorescence of the SLC38A9 CARC-CRAC peptide (2.5μM) in the presence of increasing concentrations of cholesterol, 25-hydroxycholesterol (25-HC) or cholest-4-en-3-one (3-one). Shown are mean ± SEM. N=3 samples/condition. ANOVA: ****p < 0.0001, ***p < 0.001 (G) Titration curves displaying changes in the intrinsic fluorescence of the wild-type, CRAC-mutated and CARC-CRAC mutated (DM) peptides, or a control ‘scrambled’ peptide (Scr, all peptides at 2.5μM), in the presence of increasing concentrations of cholesterol. Shown are mean ± SEM. N=3 samples/condition. ANOVA: ****p < 0.0001. (H) SLC38A9-deleted HEK-293T cells stably expressing the indicated wild-type and mutant FLAG-SLC38A9 constructs were starved for the indicated nutrients for 2 hours, or starved and re-stimulated with LDL or arginine for 2h or 30 min, respectively. Cell lysates were immunoblotted for the indicated proteins and phospho-proteins.
To test whether SLC38A9 could interact with cholesterol, we synthesized an ultraviolet light (UV)-photoactivatable cholesterol analogue, 7-Azi-27-yne (herein called LKM-38), bearing a diazirine group in position 7 and an alkyne handle on the alkyl side chain (Fig. S5B). Human fibroblasts stably expressing FLAG-SLC38A9 were incubated with LKM-38 and then exposed to UV light to induce covalent crosslinking. SLC38A9 was immunoprecipitated through the FLAG tag, and associated LKM-38 was detected by copper(I)-catalyzed click chemistry, resulting in conjugation of a rhodamine-X azide (ROX) dye (Fig. S5B). A fluorescent signal was detectable at the expected molecular size for immunoprecipitated SLC38A9 (Fig. S5C) and was competed in a dose-dependent manner when unmodified cholesterol was included in the incubation step, (Fig. S5C and S5D). Notably, unlike several sterol-related compounds we tested (Fig. S1D and S1E), LKM-38 activated mTORC1 signaling with similar potency to cholesterol (Fig. S5E).
Depleting SLC38A9 by siRNA or its deletion by CRISPR-Cas9 genome editing (25) largely abolished mTORC1 activation by LDL, as well as by arginine as previously reported (Fig. 3B and S5F). Mechanistically, SLC38A9 loss induced a tighter interaction of FLAG-p14 with RagC and RagA than in control cells, indicative of complex inactivation, and this interaction could not be weakened by addition of cholesterol (Fig. 3C). Moreover, cells lacking SLC38A9 failed to induce recruitment of mTORC1 to LAMP2-containing lysosomes in response to LDL (Fig. 3D and 3E), further suggesting that the mTORC1 scaffolding complex senses cholesterol, at least in part, via SLC38A9. Overexpressing full-length SLC38A9 or its N-terminal Ragulator-binding domain alone strongly boosted mTORC1 signaling upon stimulation with LDL/cholesterol, an effect that was not seen with SLC38A9.2, a shorter isoform that lacks the N-terminal domain (Fig. S6A). Thus, SLC38A9 translates increases in the lysosomal concentration of cholesterol into activation of mTORC1 via the Rag GTPases.
To dissect the specific role of the CARC-CRAC domain of SLC38A9, we carried out in vitro binding assays using a peptide that encompasses the two motifs (amino acids 444-464). As described for other CRAC-containing proteins(23), titrating the peptide with increasing concentrations of cholesterol yielded a dose-dependent, saturable increase of the 350nm fluorescence emission of a Trp residue within the peptide (Fig. 3F), suggesting a conformational change induced by cholesterol. In contrast, 25-hydroxycholesterol and cholest-4-en-3-one, which do not activate mTORC1 in cells (Fig. S1D), failed to increase the fluorescence emission of the CARC-CRAC motif (Fig. 3F). Mutating the obligate Tyr460 of the CRAC motif to Ile reduced the cholesterol-induced fluorescence change, whereas a double mutant of the CARC Phe449 and CRAC Tyr460 abolished it altogether (Fig. 3G).
Next, we tested the ability of wild-type, Y460I (CRAC) mutant or F449I-Y460I (CARC-CRAC) double mutant SLC38A9 to rescue mTORC1 activity in SLC38A9-depleted cells. Wild-type SLC38A9 restored mTORC1 activity (Fig. 3H) and lysosomal localization (Fig. S5G and S5H) in response to LDL, whereas the single CRAC and the double CARC-CRAC mutants did not (Fig. 3H, S5G and S5H). Notably, neither mutant disrupted arginine-mediated activation of mTORC1, indicating a specific role for the CARC-CRAC motif in cholesterol-dependent signaling (Fig. 3H). While both the CRAC and the CARC-CRAC mutant proteins localized normally to LAMP2-containing lysosomes (Fig. S6, B-E), they showed increased interactions with both Rag GTPase and Ragulator subunits, a pattern consistent with their failure to activate the mTORC1 scaffolding complex (24, 25) (Fig. S6F). Taken together, these results indicate that SLC38A9 is necessary for mTORC1 activation by lysosomal cholesterol and that the tandem CARC and CRAC motifs are important in this process.
The activating effect of LDL-derived cholesterol on mTORC1 implies that lysosomal proteins that function in cholesterol transport may participate in mTORC1 regulation. FLAG-tagged NPC1, a bone fide cholesterol transporter of the lysosomal limiting membrane, immuno-precipitated multiple components of the mTORC1 scaffolding complex, including Ragulator, Rag GTPases, SLC38A9 and the vacuolar H+-ATPase (v-ATPase) (Fig. 4A, S7A and S7B). Conversely, FLAG-SLC38A9 associated with endogenous NPC1 along with Rag GTPase and Ragulator subunits (Fig. 4B). Binding of NPC1-FLAG to endogenous v-ATPase, RagC, RagA and p18 was reduced in SLC38A9-null cells, indicating that NPC1 may associate with the scaffolding complex via SLC38A9 (Fig. S7C).
Fig. 4. NPC1 interacts with SLC38A9 and mediates mTORC1 inhibition upon cholesterol depletion.
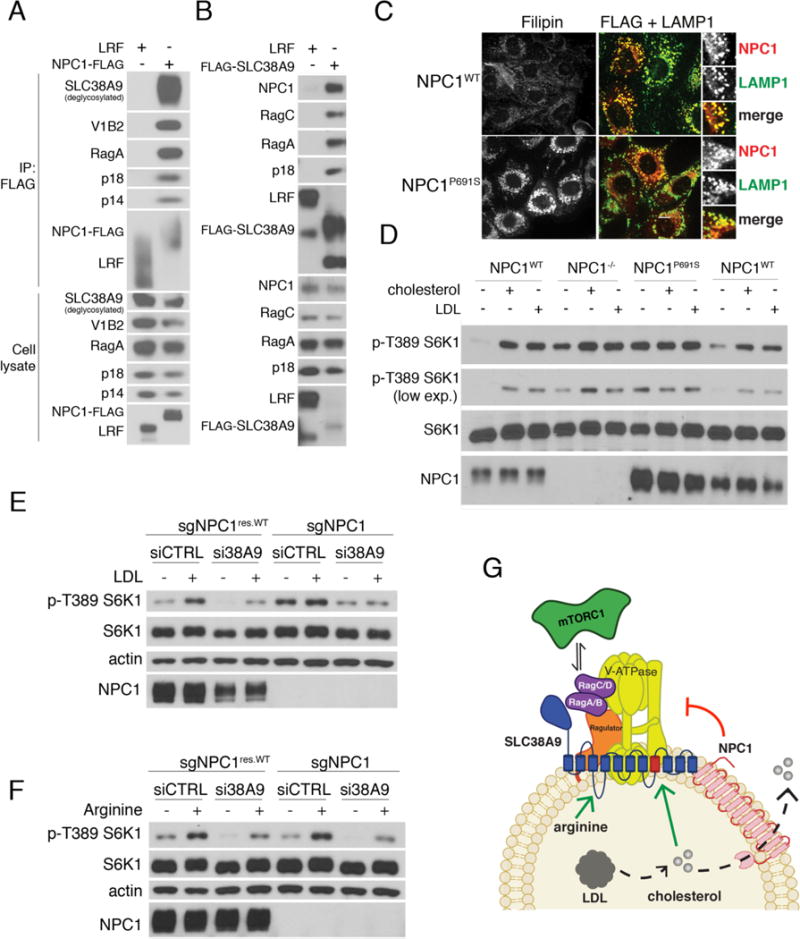
(A) Binding of NPC1 to the mTORC1 scaffolding complex. HEK-293T cells stably expressing LAMP1-FLAG or NPC1-FLAG were lysed and subjected to FLAG immunoprecipitation and immunoblotting for the indicated proteins. (B) HEK-293T cells stably expressing LAMP1-FLAG or FLAG-SLC38A9 were lysed and subjected to FLAG immunoprecipitation (IP) and immunoblotting for the indicated proteins. (C) NPC1−/− MEFs stably expressing NPC1WT-FLAG or NPC1P691S-FLAG were stained with the cholesterol-staining agent filipin or subjected to double immunofluorescence for FLAG and LAMP1, as indicated. Scale bar, 10μm (D) Requirement of the sterol-sensing domain (SSD) of NPC1 for cholesterol regulation of mTORC1. MEFs with the indicated genotypes were depleted of sterol for 2 hours or depleted and restimulated with MCD:cholesterol for 1 hour. Cell lysates were immunoblotted for the indicated proteins and phospho-proteins. (E) HEK-293T cells lacking NPC1 were treated with either scrambled or SLC38A9-targeting siRNA, depleted of sterol for 2 hours or depleted and re-stimulated with LDL for 2 hours. Cell lysates were immunoblotted for the indicated proteins and phospho-proteins. (F) HEK-293T cells lacking NPC1 were treated with either scrambled or SLC38A9-targeting siRNA, starved for arginine for 1 hour, or starved and re-stimulated with arginine for 30 min. Cell lysates were immunoblotted for the indicated proteins and phospho-proteins. (G) Model for mTORC1 regulation by LDL-derived cholesterol. SLC38A9 stimulates Rag GTPase activation in response to cholesterol. NPC1 binds to SLC38A9 and inhibits cholesterol-mediated mTORC1 activation via its sterol transport activity.
CRISPR-Cas9-mediated deletion of NPC1 in HEK-293T cells rendered mTORC1 signaling largely resistant to sterol depletion (Fig. S8A). Also, in CHO cells and MEFs bearing homozygous deletion of the NPC1 gene, mTORC1 signaling was constitutively active upon MCD-mediated cholesterol depletion, and could not be further stimulated by addition of cholesterol or LDL (Fig. S8B-D). Reintroduction of NPC1 in NPC1−/− cells was sufficient to restore cholesterol-mediated regulation of S6K1 and 4E-BP1 phosphorylation (Fig S8B). In wild-type cells, mTOR was dispersed from lysosomes upon sterol depletion but re-clustered if cholesterol was restored, whereas in NPC1-deleted cells mTOR remained associated with LAMP2-positive lysosomes under sterol-depleted conditions (Fig. S8E and S8F). The requirement for NPC1 appeared to be specific to mTORC1, as NPC1 status had no effect on the signaling response of mTORC2 (Akt, Ser 473) and extracellular signal-regulated kinase (ERK, Thr 202/204) 1/2 to sterol levels (Fig. S9A).
Transmembrane helices 3 to 7 of NPC1 contain a domain of unknown function that is similar in sequence to the sterol-sensing domain (SSD) of HMG-CoA reductase (HMGCR) and SREBP cleavage activating protein (SCAP) (27). Mutating proline 691, which is invariant within the SSD of SCAP and HMGCR, to serine, interferes with the sterol transport function of NPC1, leading to accumulation of lysosomal cholesterol (28). FLAG-tagged NPC1P691S expressed in NPC1−/− MEFs correctly targeted to lysosomes, but it failed to inhibit S6K1 phosphorylation upon cholesterol depletion, implicating the SSD of NPC1 in mTORC1 regulation (Fig. 4C and 4D).
Knocking down SLC38A9 in NPC1-null cells largely abrogated the constitutive mTORC1 signaling caused by loss of NPC1. Thus, SLC38A9 appears to communicate sterol abundance to mTORC1 downstream of or in parallel with NPC1 (Fig. 4E). Moreover, mTORC1 response to arginine or total amino acids was indistinguishable in control cells and cells lacking NPC1 and was identically affected by SLC38A9 depletion, consistent with a specific role of NPC1 in cholesterol-dependent but not arginine-dependent regulation (Fig. 4F, S9B and S9C).
This work identifies dedicated machinery that couples cholesterol trafficking through the lysosome to regulation of cellular growth signaling. LDL-derived cholesterol affects mTORC1 through the combined action of a positive regulator, SLC38A9, and a negative regulator, NPC1 (Fig. 4G). SLC38A9 conveys increases in lysosomal cholesterol through its conserved CARC and CRAC motifs, leading to lysosomal recruitment and activation of mTORC1. In contrast, NPC1 associates with the mTORC1 scaffolding complex and translates cholesterol depletion into mTORC1 inhibition. By interacting with the mTORC1 scaffolding complex, and possibly by regulating the available cholesterol pool, NPC1 enables mTORC1 to sense changes in dietary lipid supply. Consequently, loss of NPC1 may effectively decouple mTORC1 signaling from variations in sterol levels in vivo, potentially impacting downstream metabolic processes. Consistent with this notion, NPC1 is a risk allele for early-onset obesity in humans, and NPC1 haploinsufficiency in mice has been associated with weight gain and insulin resistance, which are hallmarks of mTORC1 hyperactivation (29, 30). Thus, the cholesterol-mTORC1 axis described here may play a role in metabolic homeostasis both in normal and disease states.
Supplementary Material
Acknowledgments
We thank all members of the Zoncu Lab for helpful insights; David M. Sabatini, Michael Rape and Andreas Stahl for critical reading of the manuscript; David M. Sabatini, Greg Wyant and Shuyu Wang for providing SLC38A9 constructs and SLC38A9-deleted cell lines; Linton Traub for providing the polyclonal antibody against NPC1; Sovan Sarkar for providing the NPC1WT and NPC1−/− MEFs; the Innovative Genomics Initiative and the QB3 High Throughput Snotecreening Facility for assistance generating the Crispr/Cas9 NPC1-deleted HEK-293T cells. This work was supported by the NIH Director’s New Innovator Award (1DP2CA195761-01), the Pew-Stewart Scholarship for Cancer Research, the Damon Runyon-Rachleff Innovation Award and the Edward Mallinckrodt, Jr. Foundation Grant to R.Z., a National Institutes of Health R01 HL067773 to D.O. and D.G.; a National Science Foundation Graduate Research Fellowship (DGE 1106400) to A.M.T. B.M.C. is a Howard Hughes Medical Institute Gilliam Fellow.
REFERENCES AND NOTES
- 1.Nohturfft A, Zhang SC. Coordination of lipid metabolism in membrane biogenesis. Annual review of cell and developmental biology. 2009;25:539–566. doi: 10.1146/annurev.cellbio.24.110707.175344. [DOI] [PubMed] [Google Scholar]
- 2.Espenshade PJ, Hughes AL. Regulation of sterol synthesis in eukaryotes. Annual review of genetics. 2007;41:401–427. doi: 10.1146/annurev.genet.41.110306.130315. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015;161:161–172. doi: 10.1016/j.cell.2015.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nature cell biology. 2013;15:555–564. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalaany NY, Mangelsdorf DJ. LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annual review of physiology. 2006;68:159–191. doi: 10.1146/annurev.physiol.68.033104.152158. [DOI] [PubMed] [Google Scholar]
- 6.Kwon HJ, et al. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137:1213–1224. doi: 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi T, et al. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nature cell biology. 1999;1:113–118. doi: 10.1038/10084. [DOI] [PubMed] [Google Scholar]
- 8.Ory DS. The niemann-pick disease genes; regulators of cellular cholesterol homeostasis. Trends in cardiovascular medicine. 2004;14:66–72. doi: 10.1016/j.tcm.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nature cell biology. 2008;10:935–945. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sancak Y, et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sancak Y, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perera RM, Zoncu R. The Lysosome as a Regulatory Hub. Annual review of cell and developmental biology. 2016;32:223–253. doi: 10.1146/annurev-cellbio-111315-125125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menon S, et al. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell. 2014;156:771–785. doi: 10.1016/j.cell.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Owen JL, et al. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:16184–16189. doi: 10.1073/pnas.1213343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yecies JL, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell metabolism. 2011;14:21–32. doi: 10.1016/j.cmet.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duvel K, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Um SH, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 18.Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell metabolism. 2008;8:512–521. doi: 10.1016/j.cmet.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berneis KK, Krauss RM. Metabolic origins and clinical significance of LDL heterogeneity. Journal of lipid research. 2002;43:1363–1379. doi: 10.1194/jlr.r200004-jlr200. [DOI] [PubMed] [Google Scholar]
- 20.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Current biology: CB. 2003;13:1259–1268. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 21.Zoncu R, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012;150:1196–1208. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Derler I, et al. Cholesterol modulates Orai1 channel function. Science signaling. 2016;9:ra10. doi: 10.1126/scisignal.aad7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rebsamen M, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature. 2015;519:477–481. doi: 10.1038/nature14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang S, et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science. 2015;347:188–194. doi: 10.1126/science.1257132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fantini J, Barrantes FJ. How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Frontiers in physiology. 2013;4:31. doi: 10.3389/fphys.2013.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 28.Millard EE, et al. The sterol-sensing domain of the Niemann-Pick C1 (NPC1) protein regulates trafficking of low density lipoprotein cholesterol. The Journal of biological chemistry. 2005;280:28581–28590. doi: 10.1074/jbc.M414024200. [DOI] [PubMed] [Google Scholar]
- 29.Jelinek D, et al. Npc1 haploinsufficiency promotes weight gain and metabolic features associated with insulin resistance. Human molecular genetics. 2011;20:312–321. doi: 10.1093/hmg/ddq466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meyre D, et al. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nature genetics. 2009;41:157–159. doi: 10.1038/ng.301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.