

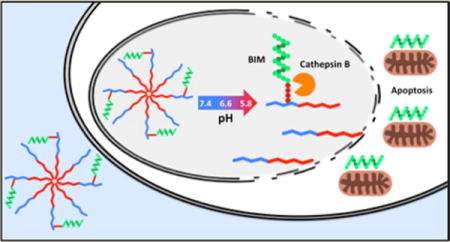
Abstract
Peptides derived from the third Bcl-2 homology domain (BH3) renormalize apoptotic signaling by antagonizing prosurvival Bcl-2 family members. These potential peptide drugs exhibit therapeutic activities but are limited by barriers including short circulation half-lives and poor penetration into cells. A diblock polymeric micelle carrier for the BIM BH3 peptide was recently described that demonstrated antitumor activity in a B-cell lymphoma xenograft model [Berguig et al., Mol. Ther. 2015, 23, 907–917]. However, the disulfide linkage used to conjugate the BIM peptide was shown to have nonoptimal blood stability. Here we describe a peptide macromonomer composed of BIM capped with a four amino acid cathepsin B substrate (FKFL) that possesses high blood stability and is cleaved to release the drug inside of target cells. Employing RAFT polymerization, the peptide macromonomer was directly integrated into a multifunctional diblock copolymer tailored for peptide delivery. The first polymer block was made as a macro-chain transfer agent (CTA) and composed of a pH-responsive endosomolytic formulation of N,N-diethylaminoethyl methacrylate (DEAEMA) and butyl methacrylate (BMA). The second polymer block was a copolymer of the peptide and polyethylene glycol methacrylate (PEGMA). PEGMA monomers of two sizes were investigated (300 Da and 950 Da). Protein gel analysis, high performance liquid chromatography, and coupled mass spectrometry (MS) showed that incubation with cathepsin B specifically cleaved the FKFL linker and released active BIM peptide with PEGMA300 but not with PEGMA950. MALDI-TOF MS showed that incubation of the peptide monomers alone in human serum resulted in partial cleavage at the FKFL linker after 12 h. However, formulation of the peptides into polymers protected against serum-mediated peptide degradation. Dynamic light scattering (DLS) demonstrated pH-dependent micelle disassembly (25 nm polymer micelles at pH 7.4 versus 6 nm unimers at pH 6.6), and a red blood cell lysis assay showed a corresponding increase in membrane destabilizing activity (<1% lysis at pH 7.4 versus 95% lysis at pH 6.6). The full carrier–drug system successfully induced apoptosis in SKOV3 ovarian cancer cells in a dose-dependent manner, in comparison to a control polymer containing a scrambled BIM peptide sequence. Mechanistic analysis verified target-dependent activation of caspase 3/7 activity (8.1-fold increase), and positive annexin V staining (72% increase). The increased blood stability of this enzyme-cleavable peptide polymer design, together with the direct polymerization approach that eliminated postsynthetic conjugation steps, suggests that this new carrier design could provide important benefits for intracellular peptide drug delivery.
Keywords: therapeutic peptide, apoptosis, cathepsin B, intracellular drug delivery, pH-responsive polymer, RAFT polymerization
Graphical abstract
INTRODUCTION
The overexpression of prosurvival B-cell lymphoma 2 (Bcl-2) proteins is a widespread mechanism of impaired apoptosis, tumorigenesis, and resistance to chemotherapy.1–4 Bcl-2 and its prosurvival relatives (Bcl-XL, Bcl-w, Mcl-1, Bcl-B, and Bfl1) inhibit apoptosis by binding and sequestering the proapoptotic proteins BAK and BAX, which normally initiate the apoptotic cascade by forming pores in the mitochondrial membrane and releasing cytochrome C.5,6 In recent years, advanced understanding of apoptotic signaling has enabled the development of inhibitors of the prosurvival Bcl-2 proteins that mimic the third Bcl-2 homology domain (BH3) of their physiological antagonists.7–13 Most notably, the selective Bcl-2 inhibitor ABT-199 showed encouraging results in early lymphoma clinical trials and was recently granted breakthrough therapy designation for the treatment of 17p deletion relapsed–refractory chronic lymphocytic leukemia.14–16 To date, other small molecule inhibitors with broader specificities (GX15-070, ABT-101, ABT-263, ABT-737) have demonstrated only modest clinical benefits with significant dose-limiting toxicities.
These findings demonstrate the promise of Bcl targets and corresponding proapoptotic peptides derived from the BH3 binding domain, which are advantageous in their simple design, high potency, and predictable target specificity.17–19 In particular, the peptide BIM is known to bind to all six prosurvival Bcl-2 proteins with high affinity,20,21 and TAT-BIM and stapled-BIM peptides have been found to induce apoptosis in cancer cell lines and murine xenograft models with therapeutic benefits.22,23 Unfortunately, a number of drug delivery barriers have limited the clinical application of peptide therapeutics such as BIM. Peptides have short circulation half-lives, are susceptible to degradation by extracellular proteases, and cross cell membranes with low efficiency, thus limiting access to intracellular targets such as Bcl-2.
Previously, RAFT polymerization was employed to synthesize a series of pH-responsive diblock copolymers that improve the pharmacokinetic properties of biologic drugs (peptides, proteins, siRNA, mRNA, etc.) and facilitate endosomal escape to the cell cytosol.24–33 One such carrier, conjugated to BIM through a reversible disulfide linkage, successfully enhanced its therapeutic activity in a murine xenograft model of human B-cell lymphoma.34 A major limitation of this design was found to be the reducible disulfide linkage that displayed poor blood stability and compromised the full potential of the polymer–BIM conjuguates.34 Disulfide linkers are designed to exploit the highly reducing cytosolic environment for intracellular drug release,35 but have often demonstrated poor stability in the bloodstream.36–39
Design of a polymer–peptide linker that is (a) highly stable in systemic circulation and (b) efficiently cleaved selectively in intracellular compartments could further enhance the quantity of BIM and other peptide/protein drugs reaching intracellular tumor targets. A peptide linker that is a selective substrate for a protease enriched in intracellular compartments is an alternative approach that meets both of the above criteria. Cathepsin B is a cysteine protease that is preferentially localized in endo/lysosomal compartments.40–43 Furthermore, cathepsin B labile linkers have been utilized previously for other drug delivery applications including antibody–drug conjugates (ADCs) and degradable nucleic acid delivery vehicles.44–47
A second drawback of the disulfide linkage is the added manufacturing complexity associated with postsynthetic peptide conjugation. Conjugation is especially challenging in the context of polyethylene glycol (PEG) grafts, which provide steric shielding properties, but also block access to pyridyl disulfide moieties during conjugation.
To address both the stability and conjugation limitations of the disulfide linkage, a synthetic strategy has been employed that creates a selectively cleavable BIM peptide macromonomer (Figure 1). The macromonomer contains the BIM peptide sequence capped with the well-characterized cathepsin B substrate Phe-Lys-Phe-Leu (FKFL).47–52 This substrate is flanked on either side by a six carbon spacer (6-aminohexanoic acid (Ahx)) to facilitate steric access of the cathepsin enzyme in the context of the surrounding PEG grafts. The peptide is functionalized on its N-terminus with methacrylamide and directly and stably polymerized into a multifunctional diblock copolymer. The first polymer block is composed of a pH-responsive formulation that drives micelle formation at physiological pH and destabilizes membranes at the acidic pH values found within endosomes.28,29 The second polymer block contains the peptide macromonomer and polyethylene glycol methacrylate (PEGMA) monomer for solubility and stability.34,53–55 Two different PEGMA monomers varying in length (300 Da and 950 Da) were investigated, but only PEGMA300 permitted cathepsin B access to the linker. In ovarian cancer cell cultures, the polymer was shown to facilitate intracellular BIM delivery and induce apoptosis in comparison to a scrambled BIM peptide sequence.
Figure 1.
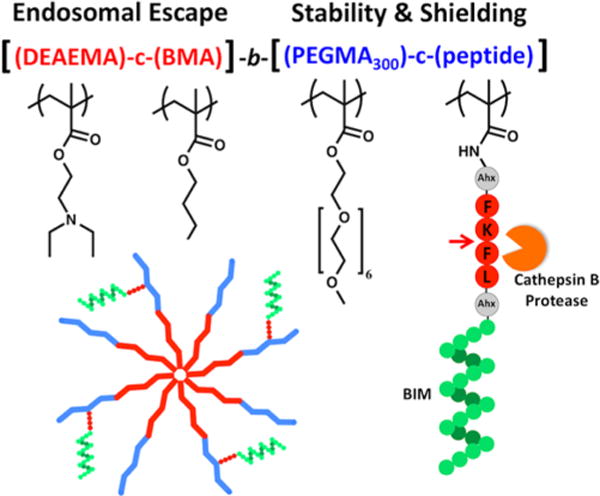
Design of a cathepsin B cleavable diblock copolymer for the intracellular delivery and release of the proapoptotic peptide BIM. The first block drives micelle formation at physiological pH (7.4) and destabilizes membranes and facilitates endosomal escape at acidic pH values (5.8–6.6). The second block contains PEG units for biocompatibility and stability and a methacrylamido-peptide macromonomer consisting of BIM capped with a four amino acid (FKFL) cathepsin B substrate flanked on either side by a six carbon spacer (aminohexanoic acid (Ahx)). Cleavage at the FKFL linker by cathepsin B specifically releases BIM inside the endo/lysosomes of target cells. The full chemical structure of the diblock copolymer is depicted in Figure S3.
EXPERIMENTAL SECTION
Synthesis and Characterization of Peptide Macro-monomers
Using an automated PS3 peptide synthesizer (Protein Technologies), the following peptides were synthesized on a solid support (rink amide MBHA resin (100–200 mesh), EMD Millipore) from FMOC protected (L) amino acids (EMD Millipore) and an FMOC protected 6-aminohexanoic acid (Ahx) spacer (AnaSpec): AhxFKFLAhxMR-PEIWIAQELRRIGDEFNAY (CathBIM), FLAhxMRPEI-WIAQELRRIGvDEFNAY (FLAhxBIM), and AhxFKFLAhx-LRMREIIDAYERQFGEPNIWA (CathScrBIM). For the synthesis of corresponding methacrylamido-peptide monomers, the amino termini of the peptides were deprotected and functionalized with N-succinimidyl methacrylate (TCI America), while still linked to the resin. Two peptide monomers were synthesized: MaCathBIM and MaCathScrBIM. The peptides/monomers were deprotected/cleaved from the resin by treatment with trifluoroacetic acid/triisopropylsilane/H2O (9.5:2.5:2.5, v/v/v) for 4 h and precipitated in cold ether. Crude peptides/monomers were purified by reverse phase high performance liquid chromatography (RP-HPLC) on a Jupiter 5 μm C18 300 Å column (Phenomenx) with an Agilent 1260 HPLC. Ion trap mass spectrometry with electrospray (Bruker Esquire) was used to confirm the molecular weights of the purified products.
RAFT Synthesis of Diblock Copolymers
For synthesis of the diblock copolymers, a macroCTA consisting of N,N-diethylaminoethyl methacrylate (DEAEMA) and butyl methacrylate (BMA) was first polymerized under nitrogen in dioxane (50 wt % monomer) at 70°C for 6 h using 4-cyanopentanoic acid dithiobenzoate (CTP) as the chain transfer agent (CTA) and azobis(4-cyanopentanoic acid) (ABCVA) as the radical initiator. The molar composition of the reaction feed was 60% DEAEMA and 40% BMA and the initial monomer ([M]0) to CTA ([CTA]0) to initiator ([I]0) ratio was 200:1:0.1. The resulting macroCTA, poly-[(DEAEMA)-co-(BMA)], was purified by dialysis in acetone for 48 h, followed by dialysis in water for 24 h, and dried by lyophilization. The macroCTA was then employed for block copolymerization of PEGMA (MW = 300 Da, PEGMA300) and peptide macromonomer. Two different polymers were synthesized by varying the identity of the peptide monomer: poly[(DEAEMA)-co-(BMA)]-b-[(PEGMA300)-c-(MaCath-BIM)] (PolCathBIM) and poly[(DEAEMA)-co-(BMA)]-b-[(PEGMA300)-c-(MaCathScrBIM)] (PolCathScrBIM). The block copolymerizations were conducted for 18 h at 70°C under nitrogen in an equal by volume mixture of dimethyl sulfoxide (DMSO) and dioxane (20 wt % monomer and macroCTA). The molar composition of the monomer feed was 96% PEGMA300 and 4% peptide monomer, and the [M]0: [mCTA]0:[I]0 ratio was 45:1:0.1. The resulting diblock copolymers were precipitated 4× in a mixture of petroleum ether and diethyl ether (9:1, v/v) to remove unreacted PEGMA300 and solvents. To remove unreacted peptide, the polymers were redissolved in acetone, the peptide monomer was removed by centrifugation, and polymers were reprecipitated in petroleum ether. This two-step precipitation scheme was repeated 3×, and the purified diblock copolymers were lyophilized. For synthesis of PEGMA-peptide copolymers, the cathepsin B cleavable BIM peptide macromonomer (MaCathBIM) was copolymerized with PEGMA300 (Pol300) or PEGMA950 (Pol950) under nitrogen in DMSO (20 wt % monomer) for 24 h at 70°C using CTP and ABCVA. The molar composition of the reaction feed was 4% peptide monomer and 96% PEGMA, and the [M]0:[CTA]0:[I]0 ratios were 25:1:0.2 and 50:1:0.2 for Pol950 and Pol300, respectively. The resulting polymers were precipitated in diethyl ether, and the unreacted peptide monomer was subsequently precipitated in acetone. This two-step precipitation scheme was repeated 6×, and isolated polymers were lyophilized.
Polymer Characterization by Gel Permeation Chromatography (GPC), 1H NMR, and RP-HPLC
To measure the number-average molecular weights (Mn) and molar mass dispersities (Đ) of the polymers, GPC was conducted using Tosoh SEC TSK GEL α-3000 and α-4000 columns (Tosoh Bioscience), a 1200 series liquid chromatography system (Agilent), and a miniDAWN TREOS three-angle light scattering instrument with an Optilab TrEX refractive index detector (Wyatt Technology). The mobile phase was 0.1 wt % lithium bromide in HPLC-grade N,N-dimethylformamide at 60°C with a flow rate of 1 mL/min. The compositions of the macroCTA and diblock copolymers were determined by 1H NMR spectroscopy (Bruker Avance DRX 499) in CDCl3 and C2D6OS, respectively. For quantification of peptide content, reaction aliquots were collected at T0 and Tx, and monomer depletion was measured by RP-HPLC (abs 280 nm).
Formulation of Polymer Micelles
Aqueous polymer solutions were prepared in DMSO at 100 mg/mL and diluted into phosphate buffer saline (PBS) at 10 mg/mL. Serial dilutions were made, and absorbance at 282 nm was measured to determine extinction coefficients. DMSO was removed by centrifugal dialysis in PBS (Amicon Ultra, 5 mL, 3K MWCO, Millipore), and final polymer concentrations were determined by UV spectrometry.
Cathepsin B Cleavage Assay
Specific cleavage at the FKFL linker by cathepsin B was determined by adapting a method from Dubowchik et al.44 and Chu et al.47 Human liver cathepsin B (Enzo Life Sciences) was activated for 15 min in a solution of 0.158 mg/mL cathepsin B, 20 mM DTT, and 10 mM EDTA at 37°C. Peptide or polymer was then solubilized in reaction buffer (10 mM phosphate, 1 mM EDTA, pH 6.6, 37°C) and added to the enzyme solution for a final concentration of 1.28 μg/mL cathepsin B and 65 μM peptide/polymer. At various time points, reaction aliquots were removed, enzymatic activity was halted by addition of a thioprotease inhibitor (E-64 (Thermo Scientific), 26 μg/mL), and RP-HPLC and mass spectrometry were used to quantify cathepsin B cleavage of the FKFL linker. Cleavage of the polymers was also visualized by SDS–PAGE on 8–16% Tris-glycine gels (Bio-Rad). For protein gel analyses, reactions were conducted at 3× concentration and 160 μg of polymer was loaded per well.
Serum Stability of Peptide Macromonomers and Polymers
The stability of the peptide linker in the presence of serum proteases was evaluated by incubating the peptide monomers and polymers in human serum and analyzing the degradation products over time. A solution of peptide (40 mM, DMSO) or polymer (4 mM, PBS) was added to human serum to a final peptide/polymer concentration of 400 μM and incubated at 37°C. At various time points, 40 μL aliquots of the mixture were withdrawn and 40 μL of acetonitrile was added to precipitate serum proteins and halt protease degradation. Precipitated solutions were centrifuged at 13,000 rpm for 10 min, and supernatants were analyzed by MALDI-TOF MS using a Bruker Autoflex II.
Sizing of Micelles by Dynamic Light Scattering
The hydrodynamic diameters of the polymer micelles were determined by dynamic light scattering (DLS) using a Nanoseries Zetasizer (Malvern). Measurements were taken of 0.25 mg/mL polymer solutions in 100 mM phosphate buffer (supplemented with 150 mM NaCl) at pH values ranging from 5.8 to 7.4. Mean particle diameter is reported as the number-average ± the half width of three independently prepared formulations.
pH-Responsive Hemolysis Assay
The ability of the polymers to induce pH-dependent membrane destabilization was assessed via a red blood cell hemolysis assay as described previously.32,33 Briefly, polymers (60 μg/mL) were incubated for 1 h at 37°C with human red blood cells in a 100 mM phosphate buffer (supplemented with 150 mM NaCl) at pH values ranging from 5.8 to 7.4. Percent red blood cell lysis (hemolysis) was quantified by measuring hemoglobin release (abs 541 nm) relative to a 100% lysis control (0.1% Triton X-100).
Cell Culture
SKOV3 human ovarian cancer cells (ATCC) were maintained in RPMI 1640 medium with L-glutamine and HEPES supplemented with 10% FBS (GIBCO) and 1% penicillin/streptomycin (GIBCO). Cells were maintained in log-phase growth at 37°C and 5% CO2.
Cell Viability Assay
The cell killing activity of the polymers was initially evaluated using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay (MTS) (Promega). SKOV3 cells were plated in a 96-well plate at a density of 7,000 cells per well and allowed to adhere for 24 h. Cells were then incubated with 100 μL of polymer solution at concentrations ranging from 0 to 10 μM for 96 h. At 96 h, cell viability was quantified by adding 20 μL of [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) reagent to each well, incubating for 30 min, and measuring the absorbance at 490 nm using a plate reader. All experimental groups were run in triplicate, and the results from three independent experiments were averaged.
Caspase-3/7 Activity Assay
Caspase 3/7 activity was measured using a SensoLyte Homogenous AMC Caspase-3/7 Assay Kit (AnaSpec). SKOV3 cells were plated in a black 96-well plate with a clear bottom at a density of 7,000 per well and then treated with polymer (0–10 μM) for 72 h. Assay reagents were mixed with cell culture medium as per the manufacturer’s instructions and incubated for 24 h, and fluorescence (ex/em = 380 nm/500 nm) was measured using a plate reader. Percent caspase 3/7 activity was calculated relative to untreated cell cultures.
Annexin V Apoptosis Assay
Induction of apoptosis was measured with an FITC Annexin V/Dead Cell Apoptosis Kit (Invitrogen) as per the manufacturer’s instructions. SKOV3s cells were plated in 6-well plates at a density of 120,000 cells per well and treated with 10 μM polymer solutions for 72 h. After 72 h, cells were trypsinized, rinsed in PBS, centrifuged, stained with FITC–annexin V, and analyzed on a BD LSRII flow cytometer.
RESULTS
Synthesis and Characterization of a Cathepsin B Cleavable Diblock Copolymer for Intracellular BIM Delivery
A cathepsin B cleavable BIM peptide macromonomer (MaCathBIM) was synthesized containing BIM capped with the cathepsin B substrate FKFL flanked on either side by a six-carbon spacer (Ahx). The peptide was functionalized on its N-terminus with methacrylamide for polymerization. A cathepsin B cleavable peptide macromonomer containing a scrambled BIM sequence (MaCathScrBIM) was synthesized as a control. The molecular weights of the peptide macromonomers (3437 Da) were confirmed by mass spectrometry (Figure S1).
Using RAFT polymerization, the peptide macromonomers were directly integrated into polymers designed to enhance peptide delivery. Targeted and experimentally determined polymer characteristics are summarized in Table 1. GPC and 1H NMR characterization data are included in Figures S2 and S3
Table 1.
Characterization of the Cathepsin B Cleavable Diblock Copolymers (PolCathBIM and PolCathScrBIM) and Peptide-PEGMA copolymers (Pol950 and Pol300)a
| pH-responsive block
|
hydrophilic block
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| % feed/comp
|
% feed/comp
|
|||||||||
| polymer | DP | DEAEMA | BMA | Mn | PDI | DP | PEGMA | peptide | Mn | Đ |
| PolCathBIM | 200 | 60/55 | 40/45 | 16,600 | 1.07 | 45 | 96/97 | 4/3 | 28,700 | 1.12 |
| PolCathScrBIM | 200 | 60/55 | 40/45 | 16,600 | 1.07 | 45 | 96/98 | 4/2 | 29,200 | 1.07 |
| Pol950 | 25 | 96/95 | 4/5 | 19,000 | 1.07 | |||||
| Pol300 | 50 | 96/96 | 4/4 | 18,800 | 1.08 | |||||
For the synthesis of diblock copolymers, a poly[(DEAEMA)-co-(BMA)] macroCTA was synthesized and employed for block copolymerization of PEGMA300 and MaCathBIM (PolCathBIM) or MaCathScrBIM (PolCathScrBIM). To investigate the effect of PEGMA chain length on cathepsin B cleavage, MaCathBIM peptide monomer was copolymerized with either PEGMA950 (Pol950) or PEGMA300 (Pol300). Polymer molecular weights (Mns) and molar mass dispersities (Đ) were determined by GPC. Molar compositions were determined by 1H NMR and RP-HPLC analysis of reaction aliquots taken at t0 and tx.
For the synthesis of pH-responsive diblock copolymers, a poly[(DEAEMA)-co(BMA)] macroCTA was synthesized with a molecular weight (Mn) of 16600 g/mol and a narrow molar mass dispersity (Đ) of 1.07. A polymer formulation of 60% DEAEMA and 40% BMA was targeted, as this composition has been shown to possess optimal pH-responsive membrane destabilizing activity and trigger the endosomal release of biologic drugs.28,29 For the second polymer block, PEGMA300 (96 mol %) and peptide macromonomer (4 mol %) were copolymerized. PEGMA300 was chosen as the hydrophilic stabilizing segment because the resultant copolymer, which contains the peptide macromonomer, is less sterically shielded from cathepsin B than longer PEGMA segments. Additionally, micelles with PEGMA300 hydrophilic stabilizers show enhanced pH-dependent endosomolytic activity relative to longer PEGMA950-based materials.34 Diblock copolymers containing either MaCathBIM (PolCathBIM) or MaCathScrBIM peptide (PolCathScrBIM) were synthesized. The molecular weights and Đs of PolCathBIM and PolCathScrBIM were determined to be 28700 Da and 29300 Da, and 1.12 and 1.07, respectively. RP-HPLC analysis of aliquots collected at reaction start and end times was used to determine the peptide content of the polymers. PolCathBIM and PolCathScrBIM were found to contain 0.9 and 0.8 peptide units per polymer chain, respectively.
To evaluate the effect of PEGMA monomer length on cathepsin B access to the FKFL linker, two copolymers were also synthesized containing the cathepsin B cleavable BIM peptide monomer (MaCathBIM) and either PEGMA950 (Pol950) or PEGMA300 (Pol300). PEGMA950 and PEGMA300 contain 19 and 6 ethylene oxide units, respectively. The molar compositions of the copolymer feeds were 4% peptide monomer and 96% PEGMA. In order to produce polymers of roughly equal size, the degrees of polymerization (DP) were 25 and 50 for Pol950 and Pol300, respectively. The molecular weights of the copolymers were approximately 19000 Da, and molar mass dispersities were less than or equal to 1.08.
Cathepsin B Mediated Release of the Proapoptotic Peptide BIM
Cathepsin B is known to cleave the FKFL substrate between the lysine and C-terminal phenylalanine residues.47 Consequently, cathepsin B cleavage of the peptide macromonomers (MW 3437 Da) at the FKFL linker should yield a 2980 Da product consisting of BIM or scrambled BIM modified on the N-terminus with FLAhx. To ensure that the FLAhx modification did not impact BIM’s proapoptotic activity, the ability of FLAhxBIM to induce cytochrome C release from the mitochondria of granta-519 tumor cells was measured and compared to unmodified BIM. At a concentration of 10 μM, both FLAhxBIM and BIM induced >90% cytochrome C release (Figure S4) in comparison to a positive 1% Triton X-100 control. A negative control protein did not induce any measurable release of cytochrome C.
To confirm cathepsin B mediated cleavage of the FKFL linker, peptide macromonomers and diblock copolymers were incubated with human liver cathepsin B enzyme and degradation products were analyzed over time by RP-HPLC and MS (Figure 2A). Cathepsin B rapidly and specifically cleaved MaCathBIM and MaCathScrBIM at the FKFL linker, with 100% cleavage observed by 20 min. Likewise, cathepsin B cleaved PolCathBIM and PolCathScrBIM at the FKFL linker to release the desired (2980 Da) peptide product, although the kinetics of polymer cleavage were significantly slower than for the peptide monomers alone. In combination, these findings suggest that cathepsin B will release active BIM peptide from its polymeric carrier within the endosomes of cancer cells.
Figure 2.
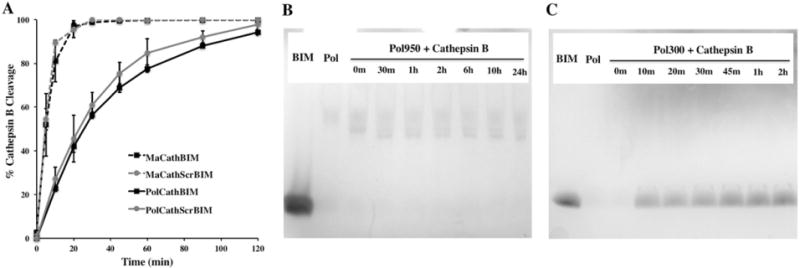
Cathepsin B cleavage of the FKFL peptide linker. (A) Peptide monomers (MaCathBIM and MaCathScrBIM) and diblock copolymers made with PEGMA300 (PolCathBIM and PolCathScrBIM) were incubated with human liver cathepsin B (1.28 ug/mL) in 10 mM phosphate buffer (pH 6.6) at a concentration of 65 μM. At various time points the reactions were stopped by addition of E-64 thioprotease inhibitor (26 ug/mL) and reaction products were analyzed by RP-HPLC and MS. Cathepsin B rapidly and specifically cleaved the FKFL linker to release BIM. (B, C) Protein gel analysis of peptide-PEGMA copolymers (Pol300 and Pol950) following incubation with cathepsin B showed that (B) PEGMA950 hindered cathepsin B access to the FKFL linker, while (C) PEGMA300 permitted cleavage and peptide release from the polymer backbone. For protein gel analyses, enzymatic reactions were run at 3× concentrations and 3.6 nmol of polymer was loaded per well.
To evaluate the effect of PEGMA chain length on cathepsin B access to the FKFL linker, cathepsin B cleavage of Pol300 and Pol950 was analyzed by protein gel electrophoresis. Cathepsin B failed to cleave Pol950 at the FKFL linker with the BIM peptide band absent at time points ranging from 0 min to 24 h (Figure 2B). In contrast, cathepsin B cleaved Pol300 similarly to the diblock copolymers (Figure 2C). It appears the longer-chain PEGMA950 sterically blocks cathepsin B access to copolymerized MaCathBIM peptide.
Stability of the FKFL Linker in Human Serum
To investigate the susceptibility of the FKFL peptide linker to extracellular protease degradation, the peptide macromonomers (MaCathBIM and MaCathScrbIM) and their corresponding diblock copolymers (PolCathBIM and PolCathScrBIM) were incubated in human serum at 37°C. At time points ranging from 0 to 12 h, peptide products were extracted into acetonitrile and analyzed by MALDI-TOF MS. For both MaCathBIM and MaCathScrBIM, intact peptide monomer (3437 Da) was detectable after 12 h. However, low levels of cleavage at the FKFL linker did occur, with the slow appearance of a 2980 Da peak observed over time. The polymers were found to protect against degradation at the FKFL linker, as the 2980 Da peak was absent at all time points. Representative spectra after 12 h are shown in Figure 3.
Figure 3.

Stability of the FKFL linker in human serum. Peptide monomers/polymers (400 μM) were incubated in human serum, and peptide products were extracted and analyzed by MALDI-TOF MS. Spectra at 12 h are shown. (A, C) Peptide monomers (3437 Da) underwent low levels of cleavage at the FKFL linker to release FLAhxBIM, as indicated by the appearance of a 2980 Da peak. (B, D) In contrast, formulation into polymers prevented undesired cleavage of the FKFL linker by serum proteases.
Micelle Sizing and pH-Responsive Membrane Destabilizing Activity
To confirm pH-dependent micelle formation, dynamic light scattering (DLS) was employed to measure particle sizes in phosphate buffers of varying pH (5.8–7.4) (Figure 4A). At pH 7.4, PolCathBIM and PolCathScrBIM assembled into micelles with hydrodynamic diameters of 25 and 26 nm, respectively. As the pH was decreased stepwise to 5.8, the micelles disassembled into unimers with diameters of 6 nm. Previously, the micellar morphology of closely related diblock copolymers was extensively characterized as a function of pH and buffer composition using surface tension and cryo-TEM measurements.56
Figure 4.
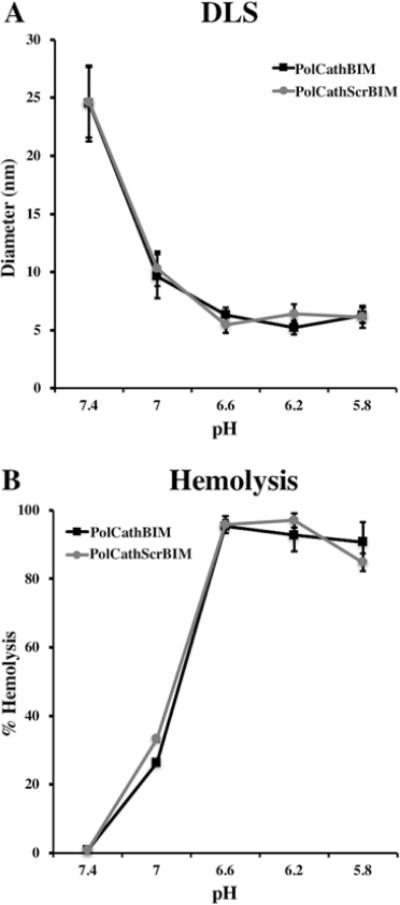
The diblock copolymers demonstrate pH-responsive membrane destabilizing activity. (A) Dynamic light scattering (DLS) was employed to measure particle diameters in phosphate buffers of varying pH. At pH 7.4 the polymers formed micelles, which disassembled into unimers of smaller diameter at more acidic pH values. (B) A red blood cell lysis assay demonstrated a corresponding increase in membrane destabilizing activity with a decrease in pH. Red blood cells were incubated with polymers (60 μg/mL), and % heme release (abs 541 nm) was quantified relative to a 0.1% Triton X-100 positive control.
The polymers’ corresponding pH-responsive membrane destabilizing activity was evaluated using a red blood cell hemolysis assay (Figure 4B). The polymers (60 μg/mL) were incubated with red blood cells at varying pH values, and the release of heme groups (hemolysis) was measured spectrophotometrically (abs 541 nm). At pH 7.4, no significant red blood cell lysis was observed. However, as the pH was lowered to the values found in early (6.6) and late endosomes (5.8), a sharp increase in hemolysis was observed, with >95% lysis observed at pH 6.6. These findings demonstrate the potential of these polymers as pH-responsive intracellular delivery vehicles.
Cancer Cell Viability
To assess the biological activity of PolCathBIM, a cytotoxicity study was conducted in SKOV3 ovarian cancer cells (Figure 5A) and 3T3 fibroblasts (Figure 5B) using an MTS cell viability assay. PolCathBIM potently induced SKOV3 cell death in a dose dependent fashion. After 96 h of treatment with 10 μM PolCathBIM, only 25% of cells remained viable. In contrast, PolCathScrBIM exhibited minimal toxicity with greater than 90% cell viability observed at all polymer concentrations. In 3T3 fibroblasts, no BIM-mediated cell death was observed. These findings suggest that BIM delivery specifically kills cancer cells with dysregulated Bcl-2 signaling.
Figure 5.
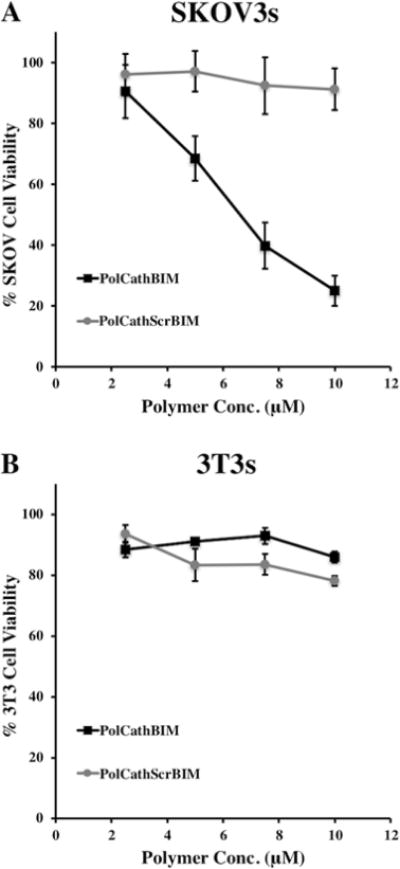
PolCathBIM induces cell death in cultures of SKOV3 ovarian cancer cells. (A) In a dose-responsive manner, PolCathBIM reduced the viability of SKOV3 cells at 96 h as measured by MTS assay. (B) In contrast, no activity was observed in noncancerous 3T3 cell cultures after 120 h of treatment.
Induction of Apoptotic Markers
Caspase 3/7 Activity and Annexin V Staining. Activation of caspase-3 and caspase-7 is a critical step in the execution of apoptosis, which was measured using a profluorescent enzyme substrate. SKOV3 cells treated with polymer containing the active BIM sequence (PolCathBIM) exhibited a dose-dependent increase in caspase 3/7 activity (Figure 6A), with 10 μM PolCathBIM resulting in more than 8-fold greater activity. In contrast, cells treated with PolCathScrBIM showed no change relative to untreated cultures.
Figure 6.
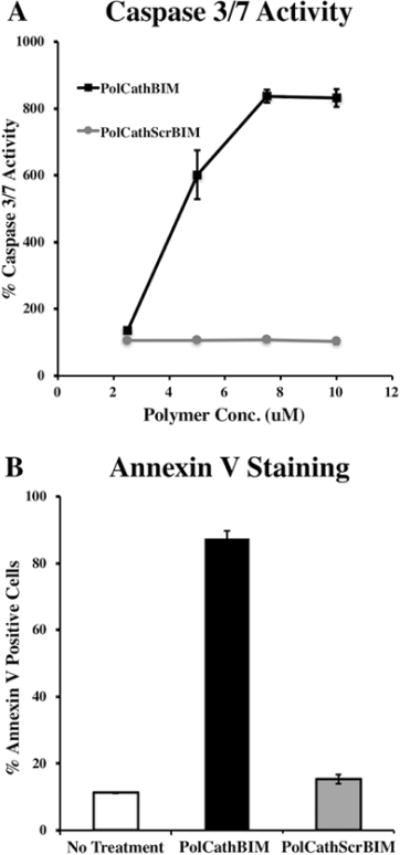
PolCathBIM induces expression of apoptotic markers in SKOV3 ovarian cancer cells. (A) After 72 h of polymer treatment, activation of caspase signaling was measured by addition of a profluorescent caspase 3/7 substrate to cell cultures. Percent caspase 3/7 activity is reported relative to untreated cells. (B) After 72 h of treatment with polymer (10 μM), FITC–annexin V dye was added to SKOV3 cultures and flow cytometry was used to measure percent of cells stained positive.
Early in apoptosis, phosphatidylserine (PS) is translocated from the inner to outer plasma membrane, becoming exposed extracellularly. Consequently, apoptotic cells can be identified by staining with the human anticoagulant annexin V, which binds to PS with high affinity. Treatment with PolCathBIM (10 μM) significantly increased annexin V staining of SKOV3s, with 87% of cells staining positive after 72 h (Figure 6B). In contrast, only 15% of cells treated with PolCathScrBIM stained positive. In combination, these findings indicate that PolCathBIM kills cancer cells by inducing apoptosis.
DISCUSSION
Peptides targeting prosurvival Bcl-2 proteins have been investigated as potential therapeutics for inducing apoptosis and/or restoring chemosensitivity in cancer. Before their activities can be tested clinically, a number of key limitations must be addressed including stability and half-life, tumor targeting, and efficient intracellular delivery. Due to their large size and polarity, peptides are unable to diffuse across cell membranes and reach intracellular drug targets on their own. In previous work, RAFT polymerization was employed to synthesize diblock copolymers tailored for peptide delivery.33,34
The reported polymers contained a hydrophilic block for biocompatibility and peptide shielding in circulation, a pH-responsive block for escape from endosomes, and pyridyl disulfide groups for reversible disulfide conjugation to cysteine-containing drugs. These polymers successfully enhanced the intracellular delivery and activity of proapoptotic peptides in cancer cell cultures and a murine xenograft model of B-cell lymphoma.33,34 It was the objective of this work to increase the stability of the polymer–peptide linkage through incorporation of a more stable peptide linker that is specifically cleaved within the endo/lysosomes of cancer cells. This objective was achieved through the synthesis and RAFT polymerization of a peptide macromonomer containing the proapoptotic peptide BIM capped with a cathepsin B labile peptide linker (Figure 1).
Reducible disulfide linkers have been widely investigated for drug delivery. They aim to harness the high redox potential difference between the oxidizing extracellular environment and reducing intracellular environment for specific release inside cells. Unfortunately, disulfide linkers have often proven to be insufficiently stable in systemic circulation. In a pharmacokinetic study conducted in mice using dual-radiolabeled polymer–BIM conjugates, the peptide to polymer ratio in the blood dropped rapidly over the course of just hours and leveled off at ∼45% of initial conjugation.34 This drop occurred as the disulfide linker was reduced and BIM was released and cleared from circulation. These findings are consistent with reports using disulfide linkers in antibody–drug conjugates (ADCs), which estimate a linker half-life on the order of 24–48 h.36–39
Enzyme labile peptide linkers, specifically those cleaved by endo/lysosomal proteases, offer an alternative. These linkers are chemically stable in the bloodstream yet are rapidly and specifically cleaved upon internalization into target cells. Cathepsin B, a ubiquitous cysteine protease, is a demonstrated candidate for this application. Cathepsin B is primarily localized in endo/lysosomes,57 its cleavage substrates have been well-characterized,47–52 and it is upregulated in a wide variety of cancers.57 While cathepsin B can be secreted in a variety of cancer cells,57 it has previously been employed for the selective intracellular degradation of a polymer conjugate of the angiogenesis inhibitor TNP-47057,58 and a nucleic acid delivery vehicle.47 In addition, selective intracellular cleavage by cathepsin B is likely aided by the lysosomotropic nature of polymer drug conjugates.57,59
As a linker, this work employed a four amino acid cathepsin B substrate, FKFL, that is rapidly cleaved between the lysine and C-terminal phenylalanine residues.47 The substrate was flanked on each side by a six carbon spacer, 6-aminohexanoic acid (Ahx), to spatially separate the site of enzymatic cleavage from the peptide drug and polymer backbone. The N-terminus of the BIM peptide was capped with this linker, and the linker–peptide sequence was subsequently functionalized with its N-terminus with methacrylamide for direct polymerization. Consequently, cathepsin B cleavage at the FKFL substrate would release a BIM peptide product modified on its N-terminus with FLAhx. To ensure that this modification did not abrogate the proapoptotic activity of the BIM peptide, the ability of BIM and FLAhxBIM to induce the release of cytochrome C from the mitochondria of tumor cells was compared. Reassuringly, both BIM and FLAhxBIM exhibited potent cytochrome C releasing activity in comparison to a negative control protein (Figure S4). Initially, there was also concern that the BIM peptide itself might undergo degradation by cathepsin B in the endo/lysosomal environment. Consequently, quantitative cathepsin B degradation of both BIM and the peptide monomer MaAhxFKFLAhxBIM was studied at pHs ranging from 5.0 to 7.2 using LC–MS. At pH 5.0, cathepsin B was found to cleave BIM between arginine (R) 64 and isoleucine (I) 65 (Figure S5A). However, cleavage of BIM occurred at a relatively slow rate, with only 50% of peptide cleaved after 30 min and 98% cleaved after 2 h. In addition, cathepsin B cleavage of BIM did not occur at higher pH values (6.6 and 7.4). In contrast, incubation of MaAhxFKFLAhxBIM with cathepsin B resulted in rapid cleavage of the FKFL linker (approximately 99% cleavage after 20 min) that was insensitive to changes in pH (Figure S5B). These results suggest that a window of opportunity exists for BIM to escape from endo/lysosomes between linker cleavage and endopeptide degradation.
The MaAhxFKFLAhxBIM macromonomer was directly polymerized into a pH-responsive diblock copolymer (Pol-CathBIM) designed to enhance BIM’s pharmacokinetic and tumor delivery properties. The pH-responsive hydrophobic block consisted of DEAEMA and BMA, and the hydrophilic block contained peptide and PEGMA300. This design bypasses the need for conjugation of peptide drugs in postsynthetic steps. It also abrogates the difficulties of conjugating peptides to polymers displaying sterically stabilizing PEG grafts that make conjugation more difficult. However, the PEG grafts may also sterically impact the access of enzymes to the linker, and thus the length of the PEG chains relative to the position of the linker in the peptide macromonomer construct must also be optimized. The rate of enzymatic cleavage was impacted in the polymer compared to the macromonomer, but greater than 94% of polymer cleavage was still complete by 2 h with PEGMA300 (Figure 2A). Protein gel analysis showed that PEGMA950 effectively blocked cathepsin B access to the FKFL linker, while PEGMA300 did not (Figure 2B,C). Previous in vivo studies with the disulfide linked BIM peptide indicated that in those carriers the PEGMA950 constructs displayed better pharmacokinetics and tumor biodistribution. Thus, these steric aspects require some competing optimization pressures. In the future, placing a longer and/or more hydrophilic spacer between the polymer backbone and enzyme substrate may permit cathepsin B access in the context of PEGMA950. Alternatively, block polymerization or copolymerization of peptide, PEGMA300, and PEGMA950 might combine the strengths of the individual monomers.
To assess the blood stability of the FKFL linker, the peptide monomers and PEGMA300 diblock copolymers (PolCathBIM and PolCathScrBIM) were incubated in human serum and the degradation products were analyzed over time by MALDI-TOF MS (Figure 3). The peptide monomers were found to cleave at the FKFL linker, but only at very low levels. Most importantly, extracellular degradation by serum proteases was abrogated by formulation into polymers, with cleavage products undetectable even at later time points. In combination, these findings suggest that the diblock copolymer design strikes an advantageous balance between protecting linker degradation in circulation and permitting cathepsin B access and peptide release upon internalization. The described macromonomer is proof-of-concept of this initial design, and investigation of alternative cathepsin B substrates may further enhance circulation stability. In particular, the dipeptide valine-citrulline is known to increase the circulation half-life of ADCs in comparison to phenylalanine-lysine.60
The cathepsin B cleavable BIM polymer potently induced cell death in SKOV3 ovarian cancer cells, in comparison to a control polymer containing an inactive peptide sequence (Figure 5A). A corresponding increase in caspase 3/7 activity and PS externalization (annexin V staining) was observed, indicating that the mechanism of cell death is BIM-mediated activation of the apoptotic cascade (Figure 6). In contrast, the BIM-containing polymer did not exhibit bioactivity in 3T3 fibroblast cultures (Figure 5B). This demonstrates the potential of BIM for killing cancer cells while sparing healthy tissues.
In this report, an enzyme-cleavable diblock copolymer was validated through initial in vitro characterization as an intracellular peptide carrier. The carrier offers two key advantages over previously employed disulfide linkages. First, the cathepsin-labile peptide linker is highly stable in human serum and still efficiently cleaved inside target cells. Second, peptide incorporation via RAFT polymerization eliminates postsynthetic conjugation and substantially simplifies material manufacturing. Consequently, this work elucidates initial design strategies that balance blood stability and enzyme accessibility in the polymeric carrier. As such it represents an initial step toward realizing the promise of a number of therapeutic peptides with intracellular drug targets.
Supplementary Material
Acknowledgments
The work was supported by the following NIH grants: F30 CA183263 (H.B.K.), T32 GM0072666 (H.B.K.), R01 CA158921-04 (D.H.), R21EB014572 (P.S.S.), R01CA109663 (O.W.P.), R01CA154897 (O.W.P.), R01CA076287 (O.W.P.), David and Patricia Giuliani Family Foundation (O.W.P.), and Giuliani/Press Endowed Chair in Cancer Research (O.W.P.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.6b01178.
Cytochrome C release assay methods and MS, GPC, 1H NMR, cytochrome C release, and cathepsin B cleavage results (PDF)
ORCID
Patrick S. Stayton: 0000-0001-6939-6371
Notes
The authors declare the following competing financial interest(s): Related polymer compositions to those reported in this manuscript have been part of issued patents and patent applications previously licensed to PhaseRx Inc. by the University of Washington. Dr. Stayton and Dr. Press are co-founders with others in PhaseRx and and have a significant financial interest in this company. However, PhaseRx was not involved in any way with the work presented in this manuscript.
References
- 1.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–2. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 2.Reed JC, Kitada S, Takayama S, Miyashita T. Regulation of chemoresistance by the bcl-2 oncoprotein in non-Hodgkin’s lymphoma and lymphocytic leukemia cell lines. Ann Oncol. 1994;5(Suppl. 1):S61–S65. doi: 10.1093/annonc/5.suppl_1.s61. [DOI] [PubMed] [Google Scholar]
- 3.Gascoyne RD, Adomat SA, Krajewski S, Krajewska M, Horsman DE, Tolcher AW, O’Reilly SE, Hoskins P, Coldman AJ, Reed JC, Connors JM. Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin’s lymphoma. Blood. 1997;90:244–51. [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30:3667–83. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lomonosova E, Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene. 2008;27:S2–19. doi: 10.1038/onc.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem. 2003;46:4259–64. doi: 10.1021/jm030190z. [DOI] [PubMed] [Google Scholar]
- 8.Becattini B, Kitada S, Leone M, Monosov E, Chandler S, Zhai D, Kipps TJ, Reed JC, Pellecchia M. Rational design and real time, in-cell detection of the proapoptotic activity of a novel compound targeting Bcl-X(L) Chem Biol. 2004;11:389–95. doi: 10.1016/j.chembiol.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 9.Verhaegen M, Bauer JA, Martín de la Vega C, Wang G, Wolter KG, Brenner JC, Nikolovska-Coleska Z, Bengtson A, Nair R, Elder JT, Van Brocklin M, Carey TE, Bradford CR, Wang S, Soengas MS. A novel BH3 mimetic reveals a mitogen-activated protein kinase-dependent mechanism of melanoma cell death controlled by p53 and reactive oxygen species. Cancer Res. 2006;66:11348–59. doi: 10.1158/0008-5472.CAN-06-1748. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, Goulet D, Viallet J, Bélec L, Billot X, Acoca S, Purisima E, Wiegmans A, Cluse L, Johnstone RW, Beauparlant P, Shore GC. Small molecule obatoclax (GX15–070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A. 2007;104:19512–7. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joudeh J, Claxton D. Obatoclax mesylate: pharmacology and potential for therapy of hematological neoplasms. Expert Opin Invest Drugs. 2012;21:363–73. doi: 10.1517/13543784.2012.652302. [DOI] [PubMed] [Google Scholar]
- 12.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 13.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, Yang X, Zhang H, Fesik S, Rosenberg SH, Elmore SW. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 14.Vandenberg CJ, Cory S. ABT-199, a new Bcl-2-specific BH3 mimetic, has in vivo efficacy against aggressive Myc-driven mouse lymphomas without provokingthrombocytopenia. Blood. 2013;121:2285–8. doi: 10.1182/blood-2013-01-475855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–8. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 16.Peirs S, Matthijssens F, Goossens S, Van de Walle I, Ruggero K, de Bock CE, Degryse S, Canté-Barrett K, Briot D, Clappier E, Lammens T, De Moerloose B, Benoit Y, Poppe B, Meijerink JP, Cools J, Soulier J, Rabbitts TH, Taghon T, Speleman F, Van Vlierberghe P. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood. 2014;124:3738–47. doi: 10.1182/blood-2014-05-574566. [DOI] [PubMed] [Google Scholar]
- 17.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discovery. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 18.Cosulich SC, Worrall V, Hedge PJ, Green S, Clarke PR. Regulation of apoptosis by BH3 domains in a cell-free system. Curr Biol. 1997;7:913–20. doi: 10.1016/s0960-9822(06)00410-6. [DOI] [PubMed] [Google Scholar]
- 19.Moreau C, Cartron PF, Hunt A, Meflah K, Green DR, Evan G, Vallette FM, Juin P. Minimal BH3 peptides promote cell death by antagonizing anti-apoptotic proteins. J Biol Chem. 2003;278:19426–35. doi: 10.1074/jbc.M209472200. [DOI] [PubMed] [Google Scholar]
- 20.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 21.Delgado-Soler L, Pinto M, Tanaka-Gil K, Rubio-Martinez J. Molecular determinants of Bim(BH3) peptide binding to pro-survival proteins. J Chem Inf Model. 2012;52:2107–18. doi: 10.1021/ci3001666. [DOI] [PubMed] [Google Scholar]
- 22.LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C, Fisher JK, Godes M, Pitter K, Kung AL, Walensky LD. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. 2012;122:2018–31. doi: 10.1172/JCI46231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kashiwagi H, McDunn JE, Goedegebuure PS, Gaffney MC, Chang K, Trinkaus K, Piwnica-Worms D, Hotchkiss RS, Hawkins WG. TAT-Bim induces extensive apoptosis in cancer cells. Ann Surg Oncol. 2007;14:1763–71. doi: 10.1245/s10434-006-9298-z. [DOI] [PubMed] [Google Scholar]
- 24.Procko E, Berguig GY, Shen BW, Song Y, Frayo S, Convertine AJ, Margineantu D, Booth G, Correia BE, Cheng Y, Schief WR, Hockenbery DM, Press OW, Stoddard BL, Stayton PS, Baker D. A computationally designed inhibitor of an Epstein-Barr viral Bcl-2 protein induces apoptosis in infected cells. Cell. 2014;157:1644–56. doi: 10.1016/j.cell.2014.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keller S, Wilson JT, Patilea GI, Kern HB, Convertine AJ, Stayton PS. Neutral polymer micelle carriers with pH-responsive, endosome-releasing activity modulate antigen trafficking to enhance CD8(+) T cell responses. J Controlled Release. 2014;191:24–33. doi: 10.1016/j.jconrel.2014.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson JT, Keller S, Manganiello MJ, Cheng C, Lee CC, Opara C, Convertine A, Stayton PS. pH-Responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS Nano. 2013;7:3912–25. doi: 10.1021/nn305466z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lundy BB, Convertine A, Miteva M, Stayton PS. Neutral polymeric micelles for RNA delivery. Bioconjugate Chem. 2013;24:398–407. doi: 10.1021/bc300486k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manganiello MJ, Cheng C, Convertine AJ, Bryers JD, Stayton PS. Diblock copolymers with tunable pH transitions for gene delivery. Biomaterials. 2012;33:2301–9. doi: 10.1016/j.biomaterials.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng C, Convertine AJ, Stayton PS, Bryers JD. Multifunctional triblock copolymers for intracellular messenger RNA delivery. Biomaterials. 2012;33:6868–76. doi: 10.1016/j.biomaterials.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berguig GY, Convertine AJ, Frayo S, Kern HB, Procko E, Roy D, Srinivasan S, Margineantu DH, Booth G, Palanca-Wessels MC, Baker D, Hockenbery D, Press OW, Stayton PS. Intracellular Delivery System for Antibody-Peptide Drug Conjugates. Mol Ther. 2015;23:907–917. doi: 10.1038/mt.2015.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palanca-Wessels MC, Convertine AJ, Cutler-Strom R, Booth GC, Lee F, Berguig GY, Stayton PS, Press OW. Anti-CD22 antibody targeting of pH-responsive micelles enhances small interfering RNA delivery and gene silencing in lymphoma cells. Mol Ther. 2011;19:1529–37. doi: 10.1038/mt.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Convertine AJ, Benoit DS, Duvall CL, Hoffman AS, Stayton PS. Development of a novel endosomolytic diblock copolymer for siRNA delivery. J Controlled Release. 2009;133:221–9. doi: 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duvall CL, Convertine AJ, Benoit DS, Hoffman AS, Stayton PS. Intracellular delivery of a proapoptotic peptide via conjugation to a RAFT synthesized endosomolytic polymer. Mol Pharmaceutics. 2010;7:468–76. doi: 10.1021/mp9002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berguig GY, Convertine AJ, Frayo S, Kern HB, Procko E, Roy D, Srinivasan S, Margineantu DH, Booth G, Palanca-Wessels MC, Baker D, Hockenbery D, Press OW, Stayton PS. Intracellular delivery system for antibody-peptide drug conjugates. Mol Ther. 2015;23:907–17. doi: 10.1038/mt.2015.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 36.Thorpe PE, Wallace PM, Knowles PP, Relf MG, Brown AN, Watson GJ, Knyba RE, Wawrzynczak EJ, Blakey DC. New coupling agents for the synthesis of immunotoxins containing a hindered disulfide bond with improved stability in vivo. Cancer Res. 1987;47:5924–31. [PubMed] [Google Scholar]
- 37.Thorpe PE, Wallace PM, Knowles PP, Relf MG, Brown AN, Watson GJ, Blakey DC, Newell DR. Improved antitumor effects of immunotoxins prepared with deglycosylated ricin A-chain and hindered disulfide linkages. Cancer Res. 1988;48:6396–403. [PubMed] [Google Scholar]
- 38.Xie H, Audette C, Hoffee M, Lambert JM, Blättler WA. Pharmacokinetics and biodistribution of the antitumor immunoconjugate, cantuzumab mertansine (huC242-DM1), and its two components in mice. J Pharmacol Exp Ther. 2004;308:1073–82. doi: 10.1124/jpet.103.060533. [DOI] [PubMed] [Google Scholar]
- 39.Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ, Senter PD. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjugate Chem. 2008;19:759–65. doi: 10.1021/bc7004329. [DOI] [PubMed] [Google Scholar]
- 40.Roberts R. Lysosomal cysteine proteases: structure, function and inhibition of cathepsins. Drug News Perspect. 2005;18:605–14. doi: 10.1358/dnp.2005.18.10.949485. [DOI] [PubMed] [Google Scholar]
- 41.Authier F, Métioui M, Bell AW, Mort JS. Negative regulation of epidermal growth factor signaling by selective proteolytic mechanisms in the endosome mediated by cathepsin B. J Biol Chem. 1999;274:33723–31. doi: 10.1074/jbc.274.47.33723. [DOI] [PubMed] [Google Scholar]
- 42.Blum JS, Fiani ML, Stahl PD. Proteolytic cleavage of ricin A chain in endosomal vesicles. Evidence for the action of endosomal proteases at both neutral and acidic pH. J Biol Chem. 1991;266:22091–5. [PubMed] [Google Scholar]
- 43.Lautwein A, Kraus M, Reich M, Burster T, Brandenburg J, Overkleeft HS, Schwarz G, Kammer W, Weber E, Kalbacher H, Nordheim A, Driessen C. Human B lymphoblastoid cells contain distinct patterns of cathepsin activity in endocytic compartments and regulate MHC class II transport in a cathepsin S-independent manner. J Leukocyte Biol. 2004;75:844–55. doi: 10.1189/jlb.0803367. [DOI] [PubMed] [Google Scholar]
- 44.Dubowchik GM, Firestone RA, Padilla L, Willner D, Hofstead SJ, Mosure K, Knipe JO, Lasch SJ, Trail PA. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjugate Chem. 2002;13:855–69. doi: 10.1021/bc025536j. [DOI] [PubMed] [Google Scholar]
- 45.Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, Francisco JA, Wahl AF, Meyer DL, Senter PD. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol. 2003;21:778–84. doi: 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- 46.Shao LH, Liu SP, Hou JX, Zhang YH, Peng CW, Zhong YJ, Liu X, Liu XL, Hong YP, Firestone RA, Li Y. Cathepsin B cleavable novel prodrug Ac-Phe-Lys-PABC-ADM enhances efficacy at reduced toxicity in treating gastric cancer peritoneal carcinomatosis: an experimental study. Cancer. 2012;118:2986–96. doi: 10.1002/cncr.26596. [DOI] [PubMed] [Google Scholar]
- 47.Chu DS, Johnson RN, Pun SH. Cathepsin B-sensitive polymers for compartment-specific degradation and nucleic acid release. J Controlled Release. 2012;157:445–54. doi: 10.1016/j.jconrel.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cezari MH, Puzer L, Juliano MA, Carmona AK, Juliano L. Cathepsin B carboxydipeptidase specificity analysis using internally quenched fluorescent peptides. Biochem J. 2002;368(Pt 1):365–9. doi: 10.1042/BJ20020840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cotrin SS, Puzer L, de Souza Judice WA, Juliano L, Carmona AK, Juliano MA. Positional-scanning combinatorial libraries of fluorescence resonance energy transfer peptides to define substrate specificity of carboxydipeptidases: assays with human cathepsin B. Anal Biochem. 2004;335:244–52. doi: 10.1016/j.ab.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 50.Hasnain S, Hirama T, Tam A, Mort JS. Characterization of recombinant rat cathepsin B and nonglycosylated mutants expressed in yeast. New insights into the pH dependence of cathepsin B-catalyzed hydrolyses. J Biol Chem. 1992;267:4713–21. [PubMed] [Google Scholar]
- 51.Dubowchik GM, Firestone RA. Cathepsin B-sensitive dipeptide prodrugs. 1. A model study of structural requirements for efficient release of doxorubicin. Bioorg Med Chem Lett. 1998;8:3341–6. doi: 10.1016/s0960-894x(98)00609-x. [DOI] [PubMed] [Google Scholar]
- 52.Stachowiak K, Tokmina M, Karpinśka A, Sosnowska R, Wiczk W. Fluorogenic peptide substrates for carboxydipeptidase activity of cathepsin B. Acta Biochim Polym. 2004;51:81–92. [PubMed] [Google Scholar]
- 53.Huang C, Neoh KG, Xu L, Kang ET, Chiong E. Polymeric nanoparticles with encapsulated superparamagnetic iron oxide and conjugated cisplatin for potential bladder cancer therapy. Biomacromolecules. 2012;13:2513–20. doi: 10.1021/bm300739w. [DOI] [PubMed] [Google Scholar]
- 54.Lutz JF. Polymerization of oligo(ethylene glycol) (meth)-acrylates: Toward new generations of smart biocompatible materials. J Polym Sci, Part A: Polym Chem. 2008;46:3459–3470. [Google Scholar]
- 55.Pohlit H, Bellinghausen I, Schömer M, Heydenreich B, Saloga J, Frey H. Biodegradable pH-Sensitive Poly(ethylene glycol) Nanocarriers for Allergen Encapsulation and Controlled Release. Biomacromolecules. 2015;16:3103–11. doi: 10.1021/acs.biomac.5b00458. [DOI] [PubMed] [Google Scholar]
- 56.Cohen N, Binyamin L, Levi-Kalisman Y, Berguig GY, Convertine A, Stayton P, Yerushalmi Rozen R. pH and Salt Effects on Surface Activity and Self-Assembly of Copolymers Containing a Weak Polybase. Langmuir. 2016;32(36):9286–92. doi: 10.1021/acs.langmuir.6b02452. [DOI] [PubMed] [Google Scholar]
- 57.Mahamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer. 2006;6:764–75. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- 58.Satchi-Fainaro R, Puder M, Davies JW, Tran HT, Sampson DA, Greene AK, Corfas G, Folkman J. Targeting angiogenesis with a conjugate of HPMA copolymer and TNP-470. Nat Med. 2004;10:255–61. doi: 10.1038/nm1002. [DOI] [PubMed] [Google Scholar]
- 59.Vicent MJ, Duncan R. Polymer conjugates: nanosized medicines for treating cancer. Trends Biotechnol. 2006;24:39–47. doi: 10.1016/j.tibtech.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 60.Ducry L, Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjugate Chem. 2010;21:5–13. doi: 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.