

Abstract
Objective
Several physiological stimuli activate smooth muscle cell (SMC) Gq protein-coupled receptors (GqPCRs) to cause vasoconstriction. As a protective mechanism against excessive vasoconstriction, SMC GqPCR stimulation invokes endothelial cell (EC) vasodilatory signalling. Whether Ca2+ influx in ECs contributes to the regulation of GqPCR-induced vasoconstriction remains unknown. Ca2+ influx through transient receptor potential vanilloid 4 (TRPV4) channels is a key regulator of endothelium-dependent vasodilation. We hypothesized that SMC GqPCR stimulation engages endothelial TRPV4 channels to limit vasoconstriction.
Approach and Results
Using high-speed confocal microscopy to record unitary Ca2+ influx events through TRPV4 channels (TRPV4 sparklets), we report that activation of SMC alpha1-adrenergic receptors with phenylephrine (PE) or thromboxane A2 receptors with U46619 stimulated TRPV4 sparklets in the native endothelium from mesenteric arteries. Activation of endothelial TRPV4 channels did not require an increase in Ca2+ as indicated by the lack of effect of L-type Ca2+ channel activator or chelator of intracellular Ca2+ EGTA-AM. However, gap junction communication between SMCs and ECs was required for PE- or U46619-activation of endothelial TRPV4 channels. Lowering IP3 levels with phospholipase C inhibitor or lithium chloride suppressed PE-activation of endothelial TRPV4 sparklets. Moreover, uncaging IP3 profoundly increased TRPV4 sparklet activity. In pressurized arteries, PE-induced vasoconstriction was followed by a slow, TRPV4-dependent vasodilation reflecting activation of negative regulatory mechanism. Consistent with these data, PE induced a significantly higher increase in blood pressure in TRPV4−/− mice.
Conclusion
These results demonstrate that SMC GqPCR stimulation triggers IP3-dependent activation of endothelial TRPV4 channels to limit vasoconstriction.
Keywords: Microcirculation, heterocellular communication, endothelial cells, Ca2+ signaling, myoendothelial gap junctions, TRPV4 channel
Subject Codes: Basic science research, cell signaling/signal transduction, endothelium, ion channels, mechanisms, physiology, vascular biology
INTRODUCTION
Activation of Gq protein-coupled receptors (GqPCRs) in vascular smooth muscle cells (SMCs) has emerged as a central mechanism for vasoconstriction in response to intravascular pressure and neurohumoral mediators1–6. Sympathetic nerve stimulation predominantly activates Gq-coupled alpha1-adrenergic receptors (α1ARs) in SMCs to cause vasoconstriction. Although α1AR-mediated vasoconstriction is an important component of the flight-or-fight response, excessive vasoconstriction can lower the blood flow to target tissues and elevate blood pressure to abnormal levels. In this regard, endothelial cells (ECs) play an important role in limiting the vasoconstriction induced by SMC α1ARs. SMC to EC heterocellular communication evokes endothelial vasodilatory mechanisms to regulate vasoconstriction7–10. However, the individual signaling elements of this negative regulatory mechanism remain unclear.
Recent studies reveal a crucial role for endothelial projections to SMCs, or myoendothelial projections (MEPs), in negative regulation of vasoconstriction in response to α1AR activation8–10. MEPs are specialized structures that contain key elements for endothelium-dependent vasodilation including inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) in the endoplasmic reticulum (ER) and intermediate/small conductance Ca2+-sensitive K+ (IK/SK) channels and cation channels at the cell membrane11–14. Moreover, myoendothelial gap junctions (MEGJs) at the end of the MEPs15 allow for the movement of second messengers and ionic charges between SMCs and ECs7–9, 16, 17. Localized increases in Ca2+ at the MEPs have been associated with IK/SK channel activation and endothelium-derived hyperpolarization (EDH) in small, resistance-sized arteries11, 12, 18. Activation of GqPCRs in SMCs causes vasoconstriction predominantly via two second messengers, IP3 and Ca2+. The diffusion of IP3 and Ca2+ from SMCs to ECs via MEGJs can increase Ca2+ inside the MEPs directly or via activation of IP3Rs on the ER membrane7–10, 16, 17. Whether the diffusion of second messengers from SMCs to ECs following SMC GqPCR activation also invokes Ca2+ influx pathways at the MEPs has not been elucidated.
Previous studies have established that Ca2+ influx signals through TRPV4 (transient receptor potential vanilloid 4) channels localized at MEPs regulate EDH-mediated vasodilation in resistance-sized mesenteric, cremaster, and cerebral arteries11, 14, 18, 19. TRPV4 channels are Ca2+-selective cation channels that are activated by flow/shear stress, Gq protein signaling, or changes in temperature and pressure11, 20, 21. We recently showed that local, unitary Ca2+ influx signals through endothelial TRPV4 channels, termed TRPV4 sparklets, activate IK/SK channels to dilate resistance-sized mesenteric arteries14, 18, 22. Moreover, TRPV4 channels are potentiated by Ca2+ and have a binding site for IP314, 23–27. Therefore, we hypothesized that SMC GqPCR activation engages endothelial TRPV4 channels via IP3 and Ca2+ to limit vasoconstriction.
In the current study we utilized differential kinetics and pharmacology to isolate IP3R-dependent Ca2+ signals and TRPV4 sparklets in the native endothelium. Moreover, we provide the first evidence that SMC GqPCR agonists trigger endothelial TRPV4-IK/SK signaling to negatively regulate vasoconstriction. While SMC GqPCR-induced activation of TRPV4 channels requires SMC-EC communication via gap junctions, it is independent of the movement of Ca2+ from SMCs to ECs or IP3R-mediated Ca2+ release in ECs. Our findings support a novel paradigm that IP3 is an endogenous activator of TRPV4 channels in the native endothelium and point to IP3-mediated activation of endothelial TRPV4 channels as a key negative regulatory mechanism for SMC GqPCR-induced vasoconstriction.
MATERIAL AND METHODS
Materials and Methods are available in the online-only Data Supplement.
RESULTS
Distinct kinetics and pharmacology enable the isolation of TRPV4 Ca2+ sparklets and IP3R-mediated Ca2+ signals in the native endothelium from mesenteric arteries (MAs)
SMC α1AR activation has been previously shown to stimulate IP3R-dependent Ca2+ signals in the native endothelium8, 9. To determine whether SMC α1AR activation also augments endothelial TRPV4 channel function under basal conditions, it is necessary to separate TRPV4 sparklets from IP3R-mediated Ca2+ signals. We hypothesized that IP3R Ca2+ signals and TRPV4 sparklets can be distinguished based on distinct kinetic and pharmacological properties. Ca2+ signals were recorded using high-speed spinning disk confocal microscopy in en face 3rd-order MAs from transgenic mice that express genetically encoded Ca2+ biosensor (GCaMP2) exclusively in ECs. Approximately 15 ECs were seen in a field of view measuring 110X110 μm (Figure 1A, left). Based on the fractional fluorescence (F/F0) traces, Ca2+ signals under basal conditions could be broadly categorized into: short, spike-shaped signals with one distinct peak and longer-lasting signals with square, discrete amplitudes. Consistent with previous reports on IP3R-mediated Ca2+ signals in GCaMP2 mice8, 12, the spike-shaped events had a duration of <300 ms. These events were inhibited by cyclopiazonic acid (CPA, 20 μM), an inhibitor of sarco/endoplasmic reticulum (SR/ER) Ca2+-ATPase, but not by the selective TRPV4 channel inhibitor GSK2193874 (GSK219, 100 nM) or 0 mM extracellular Ca2+ (Figure 1A, middle & right), indicating Ca2+ release from the ER. On the other hand, signals with square amplitudes lasted >300 ms and had a plateau phase with multiple data points at the peak (Figure 1B, left). A multi-Gaussian fit to the all-points histogram of square events yielded a quantal level of 0.185 ΔF/F0, consistent with the quantal level for TRPV4 sparklets14, 18 (Figure 1B, middle). Moreover, the square signals were inhibited by GSK219 and 0 mM extracellular Ca2+, but not by CPA, indicative of Ca2+ influx through TRPV4 channels at EC membranes (Figure 1B, left & right). These results confirmed the presence of two major Ca2+ signals in the native endothelium from MAs under basal conditions, TRPV4 sparklets and IP3R Ca2+ signals. Importantly, the two types of events can be isolated based on their differential kinetics and pharmacology.
Figure 1. SMC α1AR activation with phenylephrine (PE) increases both TRPV4 Ca2+ sparklets and IP3R-mediated Ca2+ signals.
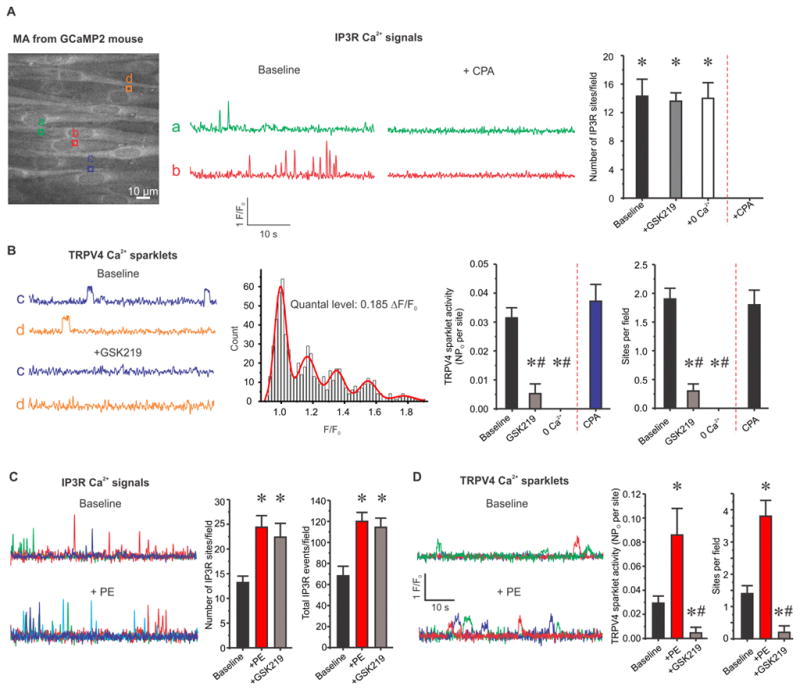
TRPV4 Ca2+ sparklets and IP3R Ca2+ signals were isolated using their distinct kinetics and pharmacological properties. (A) Left, a greyscale image of a field of view with ~15 ECs from GCaMP2 mouse. Square boxes represent the regions of interest (ROIs) placed at an event site indicating an IP3R-mediated Ca2+ signal or TRPV4 Ca2+ sparklet. Middle, representative F/F0 traces generated using the ROIs in Figure 1A indicate IP3R Ca2+ signals in the absence or presence of CPA (an inhibitor of sarco/endoplasmic reticulum (SR/ER) Ca2+-ATPase, 20 μM). Right, averaged number of IP3R Ca2+ signal sites in the absence or presence of GSK219 (a selective TRPV4 channel inhibitor, 100 nM), 0 mM extracellular Ca2+, or CPA (n=5). * p<0.05 vs. CPA. (B) Left, representative F/F0 traces generated using the ROIs in Figure 1A display TRPV4 Ca2+ sparklets under basal conditions or with GSK219 (100 nM). Middle, all-points histogram was constructed from F/F0 traces obtained from three MAs and was fit with a multi-Gaussian curve. The quantal levels (i.e. step-wise increases in amplitudes) were obtained from the peaks of the multi-Gaussian curve. Right, averaged TRPV4 sparklet activity is presented in the absence or presence of GSK219, CPA, or 0 mM extracellular Ca2+, or CPA. TRPV4 sparklet activity is expressed as NPO per site and number of sparklet sites per field. N represents the number of channels at a site and PO is the open state probability of the channels. * # p<0.05 vs. Baseline or CPA, respectively. (C) Left, representative F/F0 traces indicate IP3R Ca2+ signals under basal conditions or in the presence of PE (10 μM). Right, quantified IP3R Ca2+ signals in the absence or presence of PE alone or PE+GSK219. Ca2+ signals are presented as event sites per field and total events per field (n=5). * p<0.05 vs. Baseline. (D) Left, representative F/F0 traces show TRPV4 Ca2+ sparklet activity under basal conditions or following PE treatment (10 μM). Right, averaged TRPV4 sparklet activity is presented in the absence or presence of PE alone or PE+GSK219 (n=5). * # p<0.05 vs. Baseline or PE, respectively. Data are presented as mean ± SEM.
Activation of SMC α1ARs stimulates endothelial TRPV4 sparklets and IP3R-mediated Ca2+ signals
SMC α1AR activation generates Ca2+ and IP3 as second messengers, which can cross over to the ECs via MEGJs7–10, 16, 17. Both Ca2+ and IP3 have been shown to alter the activity of TRPV4 channels14, 23–27. Therefore, we hypothesized that SMC α1AR activation stimulates TRPV4 sparklet activity in ECs. We used the following criteria to isolate TRPV4 sparklets from IP3R-dependent Ca2+ signals: 1) durations > 300 ms and 2) inhibition by GSK219. Consistent with previous studies showing α1AR activation-induced increase in IP3R-mediated Ca2+ signals in ECs8, 9, phenylephrine (PE) stimulated the activity of IP3R-dependent Ca2+ signals in ECs from en face MAs (Figure 1C). Interestingly, SMC α1AR activation also caused a 3-fold increase in TRPV4 sparklet activity and a 2.5-fold increase in the number of sparklet sites per field (Figure 1D). GSK219 abolished the PE-induced increases in TRPV4 sparklet activity, but not the increase in IP3R Ca2+ signals (Figure 1C and D). These results confirmed that PE increases the activity of both TRPV4 sparklets and IP3R-mediated Ca2+ signals.
SMC GqPCR activation stimulates endothelial TRPV4 sparklets independent of IP3R-mediated Ca2+ release from the ER
Further mechanistic studies were performed in fluo-4 (a cell-permeable fluorescent Ca2+ dye/detector) loaded MAs (Figure 2A, left) to allow visualization of Ca2+ in EC and SMC layers from the same artery. The experiments were performed in the presence of CPA (20 μM) to exclusively record and analyze TRPV4 sparklets in absence of IP3R Ca2+ signals. CPA by itself did not alter TRPV4 sparklet activity (Figure 1B, right), suggesting that TRPV4 sparklets do not represent store-operated Ca2+ entry. An increase in SMC Ca2+ in response to PE was used as a confirmation of arterial health. Approximately 1.5 sparklet sites per field were observed in the presence of CPA alone (Figure 2B, left). PE treatment (10 μM) consistently augmented the activity of TRPV4 sparklets to a level similar to that observed in absence of CPA (Figure 2A, right; Figure 2B). The increased TRPV4 sparklet activity was substantially attenuated by GSK219 treatment (100 nM, Figure 2B, left and middle). The PE-induced increase in TRPV4 sparklet activity was eliminated in the presence of prazosin (an α1AR antagonist, 500 nM) or GSK219 pre-treatment, and in the MAs from TRPV4−/− mice (Figure 2B, right). Moreover, addition of xestospongin C (XSC, 3 μM, a potent IP3R antagonist) in the presence of CPA did not alter PE-induced increase in TRPV4 sparklet activity (Figure 2C), suggesting that TRPV4 sparklets were being activated independent of IP3R-mediated Ca2+ release. Moreover, in the presence of a membrane permeable Ca2+ chelator, EGTA-AM (5 μM), PE was able to activate TRPV4 sparklets to a similar extent (Figure 2D), thus ruling out a role for Ca2+ in PE-activation of TRPV4 sparklets. Consistent with previous studie10, 17, a strong immunostaining of α1DAR subtype was observed in SMC layer, but not in EC layer in MAs (Figure 2E). We therefore postulated that SMC α1DARs activate endothelial TRPV4 channels via SMC-EC heterocellular communication. Another SMC GqPCR agonist U46619 (a thromboxane A2 receptor agonist, 1 μM) also caused a 5-fold activation of endothelial TRPV4 sparklets (Figure 2F), supporting the idea that multiple activators of Gq-protein signaling in SMCs stimulate endothelial TRPV4 sparklets.
Figure 2. SMC GqPCR activation stimulates endothelial TRPV4 sparklets independent of IP3R-mediated Ca2+ release in MAs loaded with fluo-4.

Experiments were performed in the presence of CPA (20 μM). (A) Left, a greyscale image of a field of view with ~15 ECs from C57BL6/J mouse. Square boxes represent the regions of interest (ROIs) placed at TRPV4 Ca2+ sparklet sites. Right, representative F/F0 traces generated using the ROIs on the grayscale image display TRPV4 sparklets before and after the addition of PE (10 μM). (B) Left, averaged basal or PE-induced (10 μM) endothelial TRPV4 sparklet activity before or after GSK219 addition (100 nM, n=9) * # p<0.05 vs. CPA or PE, respectively. Right, summarized effect of PE on TRPV4 sparklet activity in the presence of prazosin (an α1AR antagonist, 500 nM), GSK219, or in MAs from TRPV4−/− mice (n=8). (C) Averaged TRPV4 sparklet activity in the absence or presence of XSC, +PE, or +GSK219 (n=4). XSC represents xestospongin C (a potent IP3R inhibitor, 3 μM). * # p<0.05 vs. CPA+XSC or PE, respectively. (D) Averaged TRPV4 sparklet activity in the presence of GSK101 (a TRPV4 channel agonist; 10 nM) alone, EGTA-AM (a cell permeable Ca2+ chelator; 5 μM), and PE (10 μM) (n=5). (E) Representative images for α1DAR immunostaining (left), nuclear staining with DAPI (middle), and merged staining (right) in the ECs (top) and SMCs (bottom) from an en face MA. The immunostaining image is representative of n=15 separate fields. (F) Left, representative F/F0 traces display TRPV4 sparklets before and after the addition of U46619 (1 μM). Right, averaged TRPV4 sparklet activity in the presence of CPA, +U46619, or +GSK219 (n=6). U46619 is a thromboxane receptor agonist (1 μM). * # p<0.05 vs. CPA or U46619, respectively. TRPV4 sparklet activity is expressed as NPO per site and number of sparklet sites per field (Figure 2B–D, F). N represents the number of channels at a site and PO is the open state probability of the channels. Data are presented as mean ± SEM.
α1AR activation of endothelial TRPV4 sparklets requires phospholipase C (PLC) signaling and movement of second messengers through MEGJs
IP3 and Ca2+ are the two major second messengers generated by α1AR signaling via PLC activation in SMCs. Both these second messengers can diffuse to the EC side via MEGJs7–10, 16. Therefore, we determined whether PLC signaling and MEGJ function are necessary for the activation of TRPV4 sparklets by SMC α1ARs. In the presence of U73122 (a PLC inhibitor, 3 μM), TRPV4 sparklet activity was not altered by PE (Figure 3A). Moreover, gap junction uncoupling agents 18β-glycyrrhetinic acid (18βGA, 30 μM)28, 29 or 1-heptanol (heptanol, 1 mM)30–32 also abolished PE-induced increase in TRPV4 sparklet activity (Figure 3B, Supplemental Figure IA). U73122, 18βGA, or heptanol, by itself, did not alter the activity of TRPV4 sparklets (Supplemental Figures IA, IB). These results suggested that second messengers generated by PLC signaling may diffuse to the ECs through MEGJs and activate TRPV4 sparklets. Lack of effect of PE or U46619 on TRPV4 sparklet activity in the presence of 18βGA or heptanol ruled out a direct effect of these compounds on endothelial cell TRPV4 channel function. PE and U46619 were able to increase SMC Ca2+ in the presence of CPA (Supplemental Figures IIA, IIB). Increase in SMC Ca2+ by PE or U46619 was not altered by 18βGA (Supplemental Figures IIC, IID), supporting the concept that the effect of PE or U46619 is initiated exclusively in the SMCs.
Figure 3. SMC α1AR signaling activates phospholipase C and SMC-EC coupling via MEGJs to stimulate endothelial TRPV4 channels in a Ca2+-independent manner.

Experiments were performed in the presence of CPA (20 μM). (A) Left, representative F/F0 traces for the effect of PE (10 μM) on TRPV4 sparklets in the presence of U73122 (a PLC inhibitor, 1 μM). Right, quantified TRPV4 sparklet activity in response to PE in the absence or presence of U73122 (n=7). * p<0.05 vs. CPA. (B) Left, representative F/F0 traces for the effect of PE (10 μM) on TRPV4 sparklets in the presence of 18βGA (a gap junction uncoupling agent, 30 μM). Right, averaged TRPV4 sparklet activity in response to PE in the absence or presence of 18βGA (n=6). * p<0.05 vs. CPA. (C) Quantified fluorescence intensity indicates whole-cell Ca2+ levels (arbitrary fluorescence units or AFU) in SMCs (top) and ECs (bottom) before and after BayK8644 (a voltage-dependent Ca2+ channel agonist, 1 μM, n=47–50 cells of each). * p<0.05 vs. Baseline. (D) Representative F/F0 traces (top) and quantification (bottom) of averaged TRPV4 sparklet activity in the absence or presence of BayK8644 (1 μM) or BayK8644+GSK219 (100 nM, n=6). TRPV4 Ca2+ sparklet activity is expressed as NPO per site and number of sparklet sites per field (Figure 3A, B, and D). N represents the number of channels at a site and PO is the open state probability of the channels. Data are presented as mean ± SEM.
Increase in SMC Ca2+ does not alter endothelial TRPV4 sparklet activity
PE increases SMC Ca2+ via IP3R-mediated Ca2+ release from the ER and influx of extracellular Ca2+33. Although endothelial TRPV4 channels can be potentiated by intracellular Ca2+14, 18, 25, 27, it remains unknown whether PE-induced increase in SMC Ca2+ is responsible for the activation of endothelial TRPV4 sparklets. Activation of TRPV4 sparklets by PE to a similar extent in presence or absence of CPA and XSC suggests that IP3R-mediated Ca2+ release in SMCs or ECs is not required for the activation of TRPV4 sparklets by PE (Figure 2C). To examine whether an increased Ca2+ influx in SMCs can activate TRPV4 sparklets in ECs, BayK8644 (a voltage-dependent L-type Ca2+ channel opener, 1 μM) was utilized to selectively increase Ca2+ concentration in SMCs. BayK8644 increased global Ca2+ levels in SMCs (Figure 3C, top), but did not enhance global Ca2+ levels in ECs (Figure 3C, bottom) or endothelial TRPV4 sparklet activity (Figure 3D). These results indicate that PE-induced increase in endothelial TRPV4 sparklet activity is not due to increased SMC Ca2+.
IP3 activates TRPV4 sparklets by increasing burst open times of TRPV4 channels
While PE-induced increase in TRPV4 sparklets was independent of SMC Ca2+, the PLC- and MEGJ-dependent nature of TRPV4 sparklet activation by PE suggested a role for SMC-derived IP3. In this regard, previous studies have stipulated that the movement of IP3 from SMCs to ECs can increase endothelial Ca2+8, 9, 16, 34. A recent study in expression system unravelled a binding site for IP3 on TRPV4 channel and showed that binding of IP3 to this site increases channel activity26. We, therefore, hypothesized that SMC-derived IP3 activates endothelial TRPV4 channels following α1AR activation. The presence of lithium chloride (LiCl, inositol monophosphatase inhibitor, 1 mM), which has been commonly used to lower intracellular IP3 levels35–38, inhibited PE- and U46619-induced activation of TRPV4 sparklets, suggesting an IP3-dependent nature of TRPV4 sparklet activation by PE and U46619 (Figure 4A). To determine whether IP3 can activate TRPV4 sparklets in native endothelium, we uncaged IP3 using 10 ms pulse of ultraviolet (UV) light. En face mouse MAs were simultaneously incubated with fluo-4 and caged IP3 (2 μM). CPA (20 μM) was added to prevent IP3-induced Ca2+ release from the ER. UV light alone did not alter the activity of TRPV4 sparklets (Figure 4B). Uncaging IP3 using UV light increased sparklet activity per site by 5-fold and number of sites per field by 3-fold, respectively (Figure 4C), an effect that was completely suppressed in the arteries pretreated with GSK219 (Figure 4C). Cooperative openings of TRPV4 channels in a cluster has emerged as an important regulatory mechanism for TRPV4 channel activity14. An analysis of coupling strength among TRPV4 channels in a cluster showed that IP3 did not alter the cooperative openings (κ values) of TRPV4 channels in a cluster (Figure 4D). Majority of TRPV4 sparklets represent bursts of TRPV4 channel openings39, 40. Uncaging IP3 increased the burst open times for each quantal level by 2–4-fold (Figure 4E). Taken together, these results suggest that SMC-derived IP3 activates endothelial TRPV4 channels in response to α1AR signaling by increasing channel open times.
Figure 4. IP3 augments endothelial TRPV4 sparklet activity by increasing TRPV4 channel open times, but not cooperativity.
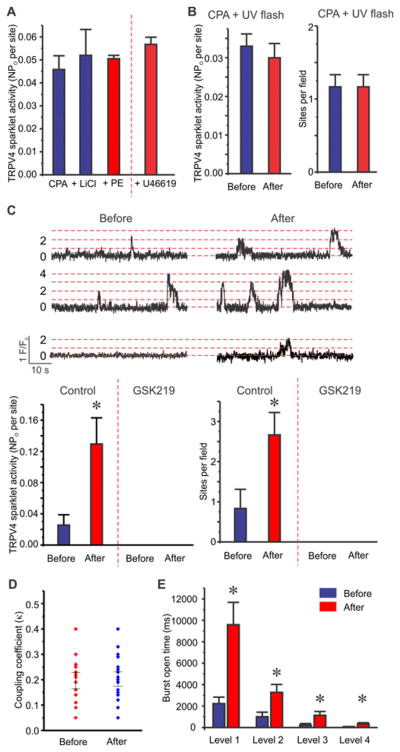
Experiments were performed in the presence of CPA (20 μM). (A) Averaged TRPV4 sparklet activity in response to PE (10 μM) or U46619 (1 μM) in the presence of LiCl (1 mM, n=7). (B) Averaged TRPV4 sparklet activity before and after UV light exposure in the presence of CPA (n=6). (C) Top, representative F/F0 traces for TRPV4 sparklets before (left) and after (right) IP3 uncaging in the presence of CPA. Dotted red lines indicate the quantal levels (i.e. stepwise openings of 0–4 TRPV4 channels at a site) derived from all-points histograms (Figure 1B). Bottom, averaged TRPV4 sparklet activity before and after IP3 uncaging in the absence or presence of GSK219 (100 nM, n=6). * p<0.05 vs. Before IP3 uncaging. TRPV4 sparklet activity is expressed as NPO per site or number of sparklet sites per field (Figure 4A and 4B). N represents the number of channels at a site and PO is the open state probability of the channels. (D) Summarized coupling coefficient (κ) values indicating the effect of IP3 on cooperative openings of TRPV4 channels at a site (n=14–20 events). Coupling coefficient (κ) values vary from 0 (no coupling or independent gating) to 1 (maximum coupling). (E) Mean burst open times (ms) for each quantal level of TRPV4 sparklets before and after IP3 uncaging (n=6). * p<0.05 vs. Before IP3 uncaging. Data are presented as mean ± SEM.
SMC α1AR signaling activates TRPV4 sparklets exclusively at MEPs
Since MEGJs between endothelial and SMCs form at the ends of MEPs41–43, we postulated that PE or U46619-induced increase in TRPV4 sparklet activity occurs exclusively at the MEPs. The MEPs are indicated by black holes in the internal elastic lamina (IEL, Figure 5A and 5C). Sparklet sites within 5 μm from the center of a hole in the IEL were considered as MEP sparklet sites14. PE-induced a 3.5-fold increase in TRPV4 sparklet activity at the MEPs but did not affect the sparklet activity at other sites (Figure 5B). Similarly, U46619 increased sparklet activity at MEPs by 5.5-fold and did not alter sparklet activity at non-MEP sites (Figure 5D). Collectively, these data support the concept that SMC-derived IP3 locally diffuses through MEGJs and selectively activates TRPV4 channels at the MEPs.
Figure 5. SMC GqPCR signaling induced an increase in TRPV4 sparklet activity exclusively at the myoendothelial projections (MEPs).
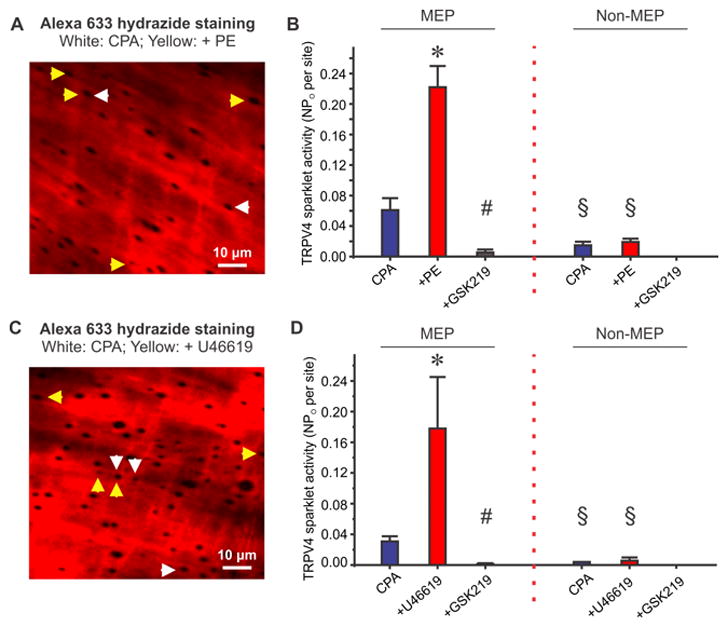
Experiments were performed in the presence of CPA (20 μM). (A) Representative images of an en face MA loaded with fluo-4 and counter-stained with Alexa Fluor 633 hydrazide (10 μM) displays TRPV4 sparklet sites at the holes in internal elastic lamina (IEL), indicative of the MEPs, in the absence (white arrows) or presence (yellow arrows) of PE (10 μM). White and yellow arrows represent TRPV4 sparklet sites that overlapped with MEPs. (B) Summarized data for TRPV4 sparklet activity at the MEP or non-MEP sites in the absence of presence of PE or PE+GSK219 (n=9). * # p<0.05 vs. CPA or PE, respectively. § p<0.05 vs. corresponding value at MEP. (C) Representative Alexa Fluor 633 hydrazide images in the absence (white arrows) or presence (yellow arrows) of U46619 (1 μM). White and yellow arrows represent TRPV4 sparklet sites that overlapped with MEPs. (D) Quantified TRPV4 sparklet activity at the MEPs or non-MEPs in the absence or presence of U46619 or U46619+GSK219 (n=6). * # p<0.05 vs. CPA or PE, respectively. § p<0.05 vs. corresponding value at MEP. TRPV4 sparklet activity is expressed as NPO per site (Figure 5B and 5D). N represents the number of channels at a site and PO is the open state probability of the channels. Data are presented as mean ± SEM.
Endothelial TRPV4 channels negatively regulate PE-induced vasoconstriction
Activation of endothelial TRPV4 sparklets promotes endothelium-dependent vasodilation in small MAs14, 18. Therefore, we hypothesized that PE-induced activation of endothelial TRPV4 channels contributes to the feedback regulation of vasoconstriction. Vascular reactivity was assessed in MAs constricted by intravascular pressure of 80 mmHg. PE-induced vasoconstriction peaked at ~2 minutes after addition of PE and was followed by a slow vasodilation (Figure 6A, PE alone, Supplemental Table 1). The vasodilation following PE-induced vasoconstriction has been attributed to negative feedback regulation by endothelium7, 44, 45. The peak vasodilation following PE was significantly attenuated in the presence of TRPV4 inhibitor GSK219, in endothelium-denuded arteries, and in the MAs from TRPV4−/− mice (Figure 6A–C), indicating that endothelial TRPV4 channels are responsible for the vasodilation following PE-induced vasoconstriction.
Figure 6. TRPV4-IK/SK signaling mediates endothelium-dependent vasodilation following PE-induced vasoconstriction.

(A) Representative diameter traces for a biphasic response to PE (1–10 μM) showing an immediate vasoconstriction followed by a subsequent, slow vasodilation in the absence or presence of GSK219 (100 nM) or Tram-34 (1 μM)+apamin (300 nM), or from EC-denuded arteries. (B) Representative area under curves (AUCs, grey color) or diameter responses (dotted lines) illustrating the secondary vasodilation following PE (1–10 μM)-evoked vasoconstriction in the absence or presence of GSK219 (100 nM) or Tram-34 (1 μM)+apamin (300 nM). (C & D) Averaged AUCs (Figure 6C) or diameter responses (% vasodilation, Figure 6D) to PE (1–10 μM) in the absence or presence of GSK219 (100 nM), Tram-34+apamin, 18βGA (30 μM), Tram-34+apamin+L-NNA (100 μM), in MAs from TRPV4−/− mice, in EC-denuded MAs, and in the presence of 20 μM CPA (n=4–7). * p<0.05 vs. Control. Data are presented as mean ± SEM. (E) Left, mean arterial pressure in control and TRPV4−/− mice averaged over three days and three nights. Right, increase in mean arterial pressure after a single bolus injection of PE at noon (0.1 mg/kg, intraperitoneal), n =4. * p<0.05 vs. Control. Data are presented as mean ± SEM.
In systemic arteries, TRPV4 sparklets activate Ca2+-sensitive IK/SK channels to cause vasodilation18. Consistent with these prior findings, vasodilation after PE treatment was significantly reduced in the presence of Tram-34 (1 μM) /apamin (300 nM) (IK/SK channel inhibitors, Figure 6A–D). Addition of nitric oxide synthase (NOS) inhibitor L-NNA (L-NG-Nitroarginine; NG-nitro-L-Arginine, 100 μM) in the presence of IK/SK channel inhibitors abolished the remaining vasodilation after PE (Figure 6B–6D), supporting a minor role for NOS in opposing PE-induced constriction. Moreover, vasodilation after PE application was markedly attenuated in the presence of MEGJ uncoupling agent (18βGA, 30 μM, Figure 6C, 6D), indicating that SMC α1AR initiates SMC-EC gap junction communication that evokes endothelial TRPV4-IK/SK signaling to cause vasodilation following PE-induced vasoconstriction. In the presence of CPA, to eliminate IP3R-mediated Ca2+ release in ECs and SMCs, PE-induced vasoconstriction was followed by a vasodilation similar to that in the absence of CPA (Figure 6C, 6D, Supplemental Figure III). Moreover, the vasodilation was almost entirely inhibited by GSK219 (Supplemental Figure III). Thus, TRPV4 channel-dependent vasodilation following PE addition in the presence of CPA was consistent with that observed in the absence of CPA.
PE induced a larger increase in blood pressure in TRPV4 knockout mice
Radiotelemetric recording of blood pressure showed that the systolic and diastolic blood pressures, and heart rate were not different in global TRPV4 knockout mice when compared to the wild-type control mice (Supplemental Figure IV). However, PE injection (0.1 mg/kg, intraperitoneal) elevated the mean arterial pressure (Figure 6E), as well as both systolic and diastolic blood pressures (Supplemental Figure IV), to a significantly greater extent in TRPV4 knockout mice when compared to wild-type control mice. The heart rate following PE injections was not different between TRPV4 knockout and control mice (Supplemental Figure IV). These results support an important physiological role for TRPV4 channels in negatively regulating α1-adrenergic signaling-induced increase in blood pressure.
DISCUSSION
Heterocellular communication between SMCs and ECs is crucial for the regulation of blood pressure and blood flow on a moment-to-moment basis. One EC is structurally surrounded by several SMCs in the arteriolar wall46, thus uniquely positioned to receive signals from SMCs to modulate their contractile state. While the role of endothelium in moderating nerve stimulation-induced vasoconstriction is well-established7–10, 17, 34, the individual signaling elements involved in this negative feedback regulation of vasoconstriction remain unclear. In this study we provide the first evidence that SMC Gq protein signaling activates Ca2+ influx through endothelial TRPV4 channels to negatively regulate vasoconstriction. Moreover, activation of endothelial TRPV4 channels by PE appears to be independent of increases in SMC Ca2+ and may represent direct activation of TRPV4 channels by IP3 transfer from SMCs to ECs via MEGJs. SMC GqPCR activation has emerged as an important mechanism for vasoconstriction in response to intravascular pressure and neurohumoral mediators1–6. Activation of TRPV4 channels in ECs represents an immediate feedback mechanism for the regulation of vasoconstriction caused by SMC GqPCR activation. The negative feedback regulation of vasoconstriction mainly involves activation of endothelial TRPV4-IK/SK signaling. Several vascular disorders including hypertension are characterized by sympathetic overactivation and endothelial dysfunction47, 48. Moreover, the function of endothelial TRPV4 channels is impaired in angiotensin II-induced hypertension14. An impairment of TRPV4 channel-dependent feedback regulation of vasoconstriction may result in higher vasoconstriction and exacerbate the effects of endothelial dysfunction in vascular disorders.
Intracellular Ca2+ plays a pivotal role in regulating diverse functions of endothelium11, 12, 18, 49, 50. Previous studies of negative feedback regulation of vasoconstriction have mostly focused on IP3R-mediated Ca2+ release in ECs. While endothelial TRPV4 channels have been shown to regulate endothelium-dependent vasodilation in different vascular beds11, 14, 18, 19, their role in negative feedback regulation of vasoconstriction remains unknown. Our results represent the first evidence for IP3R and TRPV4 Ca2+ signals as two main Ca2+ signals in native endothelium under basal conditions. Distinct kinetic and pharmacological properties of these two signals can be utilized to obtain greater mechanistic details on how Ca2+ regulates endothelial function under normal and disease conditions. Moreover, an ability to simultaneously record the two signals enables the studies on how they are modulated by physiological stimuli.
MEGJs, an essential component of heterocellular coupling between ECs and SMCs, allow the transfer of small signaling molecules (i.e. Ca2+ and IP3; below 1 KDa of molecular mass)41–43. Previous studies have postulated that Ca2+ and IP3 produced by SMC GqPCR signaling diffuse to ECs through MEGJs and alter endothelial Ca2+ signaling7, 8, 10, 16, 17, 34. A recent study by Garland et al.10 suggested that Ca2+ influx through SMC voltage-dependent L-type Ca2+ channels (VDCCs) increases EC Ca2+ signals. While the current study does not exclude the possibility of the Ca2+ diffusing across MEGJs, an increase in SMC VDCC activity was unable to stimulate endothelial TRPV4 sparklets (Figure 3D). Moreover, a similar activation of TRPV4 sparklets by PE or U46619 in the presence or absence of VDCC blocker nifedipine (Supplemental Figure V) rules out a role for SMC VDCCs in PE- or U46619-activation of TRPV4 sparklets. Garland et al.10 demonstrated that nifedipine completely inhibited the effect of 0.3 μM PE, but not 3 μM PE, on endothelial Ca2+ signals, suggesting VDCC-independent mechanism for the activation of endothelial Ca2+ signals at higher PE concentration (3 μM). Consistent with these data, we find that PE does not activate endothelial TRPV4 sparklets at 0.3 μM (Supplemental Figure VI), and TRPV4 sparklet activation by 1–10 μM PE is not dependent on VDCCs. Thus, it is possible that the activation of endothelial TRPV4 channels by SMC GqPCR signaling is dependent on the concentration of GqPCR agonist, as seen with 1–10 μM PE or 1 μM U46619.
Additionally, lithium chloride (inositol monophosphatase inhibitor, Figure 4A), PLC inhibition (Figure 3A), and uncoupling of gap junctions (Figure 3B, Supplemental Figure I) attenuated the activation of TRPV4 sparklets by PE, suggesting that IP3 generated by SMC GqPCR signaling may cross over to the EC side and activate TRPV4 sparklets. This interpretation is consistent with previous studies demonstrating that IP3 is the key molecule delivered to ECs after α1AR stimulation and results in localized Ca2+ signals-Ca2+ pulsars or wavelets-in ECs8, 9, 51. In conjunction with the data that IP3 activated TRPV4 sparklets, these results support the idea that IP3 crossing over to the EC side not only activates IP3R-mediated Ca2+ signals but also stimulates TRPV4 channels. In this regard, glycyrrhetinic acids (GAs, 18αGA and 18βGA) have been used to disrupt gap junction coupling between SMCs and ECs in arteries52–54. In particular, the efficacy and potency of 18βGA for blocking vascular gap junctions were found to be much higher than that of 18αGA28; therefore, 18βGA was employed in this study to examine whether the delivery of SMC-derived second messengers evokes endothelial TRPV4 channel activity. Heptanol has also been used extensively to suppress gap junction communication30–32. While 18βGA and heptanol (Supplemental Figure I) did not alter basal TRPV4 sparklet activity or SMC Ca2+, both the compounds abolished PE-induced activation of TRPV4 sparklets, supporting their effectiveness as MEGJ uncoupling agents.
Our results provide the first evidence of IP3 as an endogenous activator for TRPV4 channel function in native endothelium. The IP3-induced increase in TRPV4 sparklet activity was independent of IP3R-mediated Ca2+ release from the ER (Figure 1D, Figure 2). How IP3 activates TRPV4 channels remains unknown. A recent study delineated that the ankyrin repeat domain (ARD) of TRPV4 channels has an IP3 binding sites and binding of IP3 to this site increases TRPV4 channel activity26. In addition, IP3 application considerably increased whole-cell currents evoked by 4α-PDD (an activator of TRPV4 channels) in HEK293 cells26. TRPV4 sparklets represent bursts of openings of TRPV4 channels in a cluster39, 40. Our results indicate that IP3 increases the mean burst open times for TRPV4 channels, but does not alter the cooperative openings among the channels in a cluster. We, therefore, postulate that SMC-derived IP3 stimulates endothelial TRPV4 channels via a direct interaction that increases the channel open times.
Some of the previous studies have proposed a direct movement of free Ca2+ from SMCs to ECs as a mechanism for myoendothelial feedback regulation of vasoconstriction7, 10. In the current study an increase in SMC Ca2+ did not increase endothelial TRPV4 sparklet activity, a result that is in agreement with previous investigations suggesting that an increase in SMC Ca2+ does not alter EC Ca2+ 8, 34. Although there is evidence suggesting that SMC Ca2+ diffuses to EC7, 10, any possible diffusion of Ca2+ appears to be insufficient to stimulate endothelial TRPV4 channels. Furthermore, a similar activation of endothelial TRPV4 sparklets by PE in the absence or presence of CPA (Figure 1D, Figure 2B), EGTA-AM (Figure 2D), and nifedipine (Supplemental Figure V), and a similar vasodilation to PE in the presence of CPA rule out the role of increased intracellular Ca2+ in PE-activation of TRPV4 sparklets. Ca2+-independent heterocellular communication between SMCs and ECs could be explained by the following possibilities: 1) The mobility of IP3 is significantly higher than Ca2+ because of vastly different diffusion coefficients of IP3 and Ca2+ (i.e. ~280 vs. 38 μm2/s)55. Free Ca2+ promptly interacts with Ca2+-binding proteins including calmodulin, thereby limiting the mobility of Ca2+ 55. While Ca2+-calmodulin complex is too large to be diffused across narrow pores of the MEGJs, SMC-derived IP3 can freely move to the ECs and participate in myoendothelial feedback regulation. Notably, a recent study showed that the diffusion constant of IP3 is less than 10 μm2/s in SH-SY5Y neuroblastoma cells56. However, further studies and computational modeling are required to confirm the 30-fold slower IP3 diffusion rate since it could be affected by IP3R buffering capacity, intracellular IP3 concentration, photorelease of IP3, or cell size/subcellular structure57. 2) MEGJs are mainly comprised of connexin 37 or 4058, 59. Connexin composition of MEGJs is a key determinant of selective permeability of the MEGJs to IP3 and Ca2+60–62. It is plausible that the MEGJs in mouse MAs are selectively permeable to IP3. Taken together, these results suggest that an increase in SMC Ca2+ is not an essential component for TRPV4 channel activation by PE.
PE-induced vasoconstriction is followed by slow vasodilation7, 44, 45, a response that is attributed to myoendothelial feedback regulation. Specifically, PE induces an increase in endothelial Ca2+ that activates vasodilatory mechanisms including NO or IK/SK channel-mediated EDH8–10, 17. While PE treatment (1–10 μM) consistently induced the biphasic vasomotor responses in pressurized MAs, the vasodilatory component was abolished in the presence of IK/SK channel inhibitors. It is well-documented that endothelium-dependent vasodilation of resistance-sized arteries is primarily controlled by EDH, whereas NO and/or prostacyclin are the prevailing vasodilators in larger, conduit arteries63, 64. The number of MEPs increases inversely with vessel size15, supporting the notion that MEP-localized EDH signaling plays a crucial role in resistance-sized arteries. MEPs serve as the cellular platforms that house the biological apparatus including IK/SK channels and MEGJs necessary for EDH-mediated vasodilation11–14. While TRPV4 sparklets can occur at MEP and non-MEP sites14, our results that PE activated TRPV4 sparklets selectively at the MEPs support SMC-EC communication at the MEPs and subsequent activation of EDH signaling. It is likely that IP3 generated by SMC GqPCR signaling locally diffuses to ECs via MEGJs and initiates EDH signaling by activating TRPV4 channels at the MEPs. Additional inhibition of vasodilation by NOS inhibitor in the presence of IK/SK inhibitors may be an indication for a minor role of NO in myoendothelial feedback regulation in MAs (Figure 6). Compared to the TRPV4 channel inhibitor, IK/SK channel inhibitors in the absence or presence of L-NNA had a larger effect on the vasodilatory component following PE, suggesting that the IP3R Ca2+ signal activated by PE may couple with IK/SK channels and eNOS (Figure 6).
Several limitations of the present investigation need to be acknowledged: 1) There is no tracking of IP3 as it moves from SMCs to ECs. While this will be an important area for future investigation, the currently available tools do not allow for accurate tracking of IP3 in intact arteries. This will require development of reliable IP3 probes or isotope labeling of SMC IP3. 2) Despite the many advantages of en face preparation (more ECs per field of view, higher signal/noise at high imaging speed), it is likely that SMCs and ECs experience different stretch levels in pressurized and en face preparations, and therefore, have different resting membrane potentials. It was observed that the membrane potential of endothelial cells in en face arteries is ~ −45 mV12. The SMC membrane potential in en face preparation, and EC membrane potential in pressurized arteries are not known. Although the results in en face preparation are consistent with those from diameter studies in pressurized arteries, minor differences in signaling mechanisms due to differences in membrane potentials cannot be ruled out. 3) The Ca2+ images represent snapshots of 1 minute. In this duration, some cells show Ca2+ signals, while others are quiescent. Whether this is due to endothelial cell heterogeneity or is a function of probability of finding active sites during the recording duration remains unknown. Definitively addressing this possibility may require data acquisition for several hours. The high imaging rates required for recording TRPV4 sparklets and IP3 receptor events and the possibility of bleaching the fluorophores or damaging the endothelium limit our ability to record Ca2+ signals for several hours. 4) Endothelium-specific TRPV4 knockout mice will provide the most direct evidence for the role of TRPV4 channels in negatively regulating PE-induced vasoconstriction and increase in blood pressure. However, such mice have not been generated. 5) The number of TRPV4 sparklet sites activated by PE and U46619 appear to be different. This could be due to differences in the number of sparklet sites under baseline conditions. Indeed, the number of TRPV4 sparklet sites varies from one field to another, while the sparklet activity/site remains unchanged.
In conclusion, our studies reveal that distinct endothelial Ca2+ signals could be isolated and analyzed under physiological conditions using differential kinetics and pharmacology. Moreover, the study also provides first evidence for a novel endothelial mechanism that acts as a feedback regulator for vasoconstriction induced by SMC GqPCR activation. In agreement with previous studies, this mechanism involves heterocellular communication and transfer of second messengers from SMCs to ECs. This is followed by the activation of EC TRPV4 channels, possibly through the movement of IP3 from SMCs to ECs and a direct interaction of IP3 with TRPV4 channels. GqPCR activation is an important mechanism for SMC contraction. Indeed, numerous physiological vasoconstrictors including intravascular pressure (myogenic tone), α1AR activation, TXA2R activation, and angiotensin II receptor activation involve Gq protein signaling in SMCs. Activation of EC TRPV4 channels may be a key mechanism that negatively regulates vasoconstriction to these stimuli, thus preventing excessive vasoconstriction. An abnormal TRPV4 channel-dependent negative feedback regulation of vasoconstriction may result in elevated blood pressure, and therefore is a potential mechanism that could be targeted for treating hypertension.
Supplementary Material
HIGHLIGHTS.
Ca2+ influx signals through TRPV4 channels can be distinguished from IP3R-mediated Ca2+ release from the ER in the native endothelium from resistance-sized arteries using distinct kinetics and pharmacology of the two signals.
Activation of SMC GqPCRs initiates SMC-EC communication that stimulates endothelial TRPV4 channel function.
SMC-derived IP3, rather than Ca2+, activates endothelial TRPV4 sparklets in response to SMC GqPCR activation.
Endothelial TRPV4 sparklets negatively regulate vasoconstriction caused by the activation of SMC GqPCRs via TRPV4-IK/SK channel signalling. This signalling pathway represents a novel mechanism for regulation of blood pressure in response to SMC GqPCR activation.
Acknowledgments
We thank Drs. Michael Kotlikoff (Cornell University), Adrian Bonev (University of Vermont), and Avril Somlyo for help with GCaMP2 mice, software program for image analysis, and comments on the manuscript, respectively. We also thank GlaxoSmithKline for providing TRPV4−/− breeder mice.
SOURCES OF FUNDING
Studies were supported by grants from the National Institutes of Health to S.K.S. (HL121484, HL138496), B.E.I. (HL088554), C.M. (T32 Training Grant HL007284), UVA School of Medicine and Robert M. Berne Cardiovascular Research Center startup funds (S.K.S.), and American Heart Association Predoctoral Fellowship to C.M. (17PRE33660762).
Abbreviations
- α1ARs
alpha1-adrenergic receptors
- ECs
endothelial cells
- EDH
endothelium-derived hyperpolarization
- ER
endoplasmic reticulum
- GqPCRs
Gq protein-coupled receptors
- IK/SK
intermediate/small conductance Ca2+-sensitive K+ channels
- IP3
inositol 1,4,5-trisphosphate
- IP3Rs
IP3 receptors
- MEGJs
Myoendothelial gap junctions
- MEPs
Myoendothelial projections
- NOS
nitric oxide synthase
- PE
phenylephrine
- PLC
phospholipase C
- SMCs
vascular smooth muscle cells
- TRPV4
transient receptor potential vanilloid 4
- TXA2R
thromboxane A2 receptors
- VDCCs
voltage-dependent L-type Ca2+ channels
Footnotes
DISCLOSURES
None.
References
- 1.Gebremedhin D, Lange AR, Narayanan J, Aebly MR, Jacobs ER, Harder DR. Cat cerebral arterial smooth muscle cells express cytochrome P450 4A2 enzyme and produce the vasoconstrictor 20-HETE which enhances L-type Ca2+ current. J Physiol. 1998;507:771–781. doi: 10.1111/j.1469-7793.1998.771bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hong K, Zhao G, Hong Z, Sun Z, Yang Y, Clifford PS, Davis MJ, Meininger GA, Hill MA. Mechanical activation of angiotensin II type 1 receptors causes actin remodelling and myogenic responsiveness in skeletal muscle arterioles. J Physiol. 2016;594:7027–7047. doi: 10.1113/JP272834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mederos y Schnitzler M, Storch U, Meibers S, Nurwakagari P, Breit A, Essin K, Gollasch M, Gudermann T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. The EMBO journal. 2008;27:3092–3103. doi: 10.1038/emboj.2008.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pires PW, Ko EA, Pritchard HAT, Rudokas M, Yamasaki E, Earley S. The angiotensin II receptor type 1b is the primary sensor of intraluminal pressure in cerebral artery smooth muscle cells. J Physiol. 2017;595:4735–4753. doi: 10.1113/JP274310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schleifenbaum J, Kassmann M, Szijarto IA, Hercule HC, Tano JY, Weinert S, Heidenreich M, Pathan AR, Anistan YM, Alenina N, Rusch NJ, Bader M, Jentsch TJ, Gollasch M. Stretch-activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circ Res. 2014;115:263–272. doi: 10.1161/CIRCRESAHA.115.302882. [DOI] [PubMed] [Google Scholar]
- 6.Storch U, Blodow S, Gudermann T, Mederos YSM. Cysteinyl leukotriene 1 receptors as novel mechanosensors mediating myogenic tone together with angiotensin II type 1 receptors-brief report. Arterioscler Thromb Vasc Biol. 2015;35:121–126. doi: 10.1161/ATVBAHA.114.304844. [DOI] [PubMed] [Google Scholar]
- 7.Dora KA, Doyle MP, Duling BR. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc Natl Acad Sci U S A. 1997;94:6529–6534. doi: 10.1073/pnas.94.12.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nausch LW, Bonev AD, Heppner TJ, Tallini Y, Kotlikoff MI, Nelson MT. Sympathetic nerve stimulation induces local endothelial Ca2+ signals to oppose vasoconstriction of mouse mesenteric arteries. Am J Physiol Heart Circ Physiol. 2012;302:H594–H602. doi: 10.1152/ajpheart.00773.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran CH, Taylor MS, Plane F, Nagaraja S, Tsoukias NM, Solodushko V, Vigmond EJ, Furstenhaupt T, Brigdan M, Welsh DG. Endothelial Ca2+ wavelets and the induction of myoendothelial feedback. Am J Physiol Cell Physiol. 2012;302:C1226–1242. doi: 10.1152/ajpcell.00418.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garland CJ, Bagher P, Powell C, Ye X, Lemmey HAL, Borysova L, Dora KA. Voltage-dependent Ca(2+) entry into smooth muscle during contraction promotes endothelium-mediated feedback vasodilation in arterioles. Sci Signal. 2017:10. doi: 10.1126/scisignal.aal3806. [DOI] [PubMed] [Google Scholar]
- 11.Bagher P, Beleznai T, Kansui Y, Mitchell R, Garland CJ, Dora KA. Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca2+ events, and IKCa channels, reducing arteriolar tone. Proc Natl Acad Sci U S A. 2012;109:18174–18179. doi: 10.1073/pnas.1211946109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI, Nelson MT. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci U S A. 2008;105:9627–9632. doi: 10.1073/pnas.0801963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandow SL, Neylon CB, Chen MX, Garland CJ. Spatial separation of endothelial small- and intermediate-conductance calcium-activated potassium channels (K(Ca)) and connexins: possible relationship to vasodilator function? J Anat. 2006;209:689–698. doi: 10.1111/j.1469-7580.2006.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sonkusare SK, Dalsgaard T, Bonev AD, Hill-Eubanks DC, Kotlikoff MI, Scott JD, Santana LF, Nelson MT. AKAP150-dependent cooperative TRPV4 channel gating is central to endothelium-dependent vasodilation and is disrupted in hypertension. Sci Signal. 2014;7:ra66. doi: 10.1126/scisignal.2005052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sandow SL, Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ Res. 2000;86:341–346. doi: 10.1161/01.res.86.3.341. [DOI] [PubMed] [Google Scholar]
- 16.Isakson BE, Ramos SI, Duling BR. Ca2+ and inositol 1,4,5-trisphosphate-mediated signaling across the myoendothelial junction. Circ Res. 2007;100:246–254. doi: 10.1161/01.RES.0000257744.23795.93. [DOI] [PubMed] [Google Scholar]
- 17.Jackson WF, Boerman EM, Lange EJ, Lundback SS, Cohen KD. Smooth muscle alpha1D-adrenoceptors mediate phenylephrine-induced vasoconstriction and increases in endothelial cell Ca2+ in hamster cremaster arterioles. Br J Pharmacol. 2008;155:514–524. doi: 10.1038/bjp.2008.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336:597–601. doi: 10.1126/science.1216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L, Papadopoulos P, Hamel E. Endothelial TRPV4 channels mediate dilation of cerebral arteries: impairment and recovery in cerebrovascular pathologies related to Alzheimer’s disease. Br J Pharmacol. 2013;170:661–670. doi: 10.1111/bph.12315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M. Heat-evoked activation of the ion channel, TRPV4. J Neurosci. 2002;22:6408–6414. doi: 10.1523/JNEUROSCI.22-15-06408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watanabe H, Vriens J, Suh SH, Benham CD, Droogmans G, Nilius B. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J Biol Chem. 2002;277:47044–47051. doi: 10.1074/jbc.M208277200. [DOI] [PubMed] [Google Scholar]
- 22.Sonkusare SK, Dalsgaard T, Bonev AD, Nelson MT. Inward rectifier potassium (Kir2.1) channels as end-stage boosters of endothelium-dependent vasodilators. J Physiol. 2016;594:3271–3285. doi: 10.1113/JP271652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandes J, Lorenzo IM, Andrade YN, Garcia-Elias A, Serra SA, Fernandez-Fernandez JM, Valverde MA. IP3 sensitizes TRPV4 channel to the mechano- and osmotransducing messenger 5′-6′-epoxyeicosatrienoic acid. J Cell Biol. 2008;181:143–155. doi: 10.1083/jcb.200712058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Elias A, Lorenzo IM, Vicente R, Valverde MA. IP3 receptor binds to and sensitizes TRPV4 channel to osmotic stimuli via a calmodulin-binding site. J Biol Chem. 2008;283:31284–31288. doi: 10.1074/jbc.C800184200. [DOI] [PubMed] [Google Scholar]
- 25.Strotmann R, Schultz G, Plant TD. Ca2+-dependent potentiation of the nonselective cation channel TRPV4 is mediated by a C-terminal calmodulin binding site. J Biol Chem. 2003;278:26541–26549. doi: 10.1074/jbc.M302590200. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi N, Hamada-Nakahara S, Itoh Y, et al. TRPV4 channel activity is modulated by direct interaction of the ankyrin domain to PI(4,5)P(2) Nat Commun. 2014;5:4994. doi: 10.1038/ncomms5994. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe H, Vriens J, Janssens A, Wondergem R, Droogmans G, Nilius B. Modulation of TRPV4 gating by intra- and extracellular Ca2+ Cell Calcium. 2003;33:489–495. doi: 10.1016/s0143-4160(03)00064-2. [DOI] [PubMed] [Google Scholar]
- 28.Guan BC, Si JQ, Jiang ZG. Blockade of gap junction coupling by glycyrrhetinic acids in guinea pig cochlear artery: a whole-cell voltage- and current-clamp study. Br J Pharmacol. 2007;151:1049–1060. doi: 10.1038/sj.bjp.0707244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guan X, Wilson S, Schlender KK, Ruch RJ. Gap-junction disassembly and connexin 43 dephosphorylation induced by 18 beta-glycyrrhetinic acid. Mol Carcinog. 1996;16:157–164. doi: 10.1002/(SICI)1098-2744(199607)16:3<157::AID-MC6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 30.Georgescu A, Alexandru N, Constantinescu E, Popov D. Effect of gap junction uncoupler heptanol on resistance arteries reactivity in experimental models of diabetes, hyperlipemia and hyperlipemia-diabetes. Vascul Pharmacol. 2006;44:513–518. doi: 10.1016/j.vph.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 31.Sahu G, Sukumaran S, Bera AK. Pannexins form gap junctions with electrophysiological and pharmacological properties distinct from connexins. Sci Rep. 2014;4:4955. doi: 10.1038/srep04955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takens-Kwak BR, Jongsma HJ, Rook MB, Van Ginneken AC. Mechanism of heptanol-induced uncoupling of cardiac gap junctions: a perforated patch-clamp study. Am J Physiol. 1992;262:C1531–C1538. doi: 10.1152/ajpcell.1992.262.6.C1531. [DOI] [PubMed] [Google Scholar]
- 33.Utz J, Eckert R, Trautwein W. Changes of intracellular calcium concentrations by phenylephrine in renal arterial smooth muscle cells. Pflugers Arch. 1999;438:725–731. doi: 10.1007/s004249900091. [DOI] [PubMed] [Google Scholar]
- 34.Lamboley M, Pittet P, Koenigsberger M, Sauser R, Beny JL, Meister JJ. Evidence for signaling via gap junctions from smooth muscle to endothelial cells in rat mesenteric arteries: possible implication of a second messenger. Cell Calcium. 2005;37:311–320. doi: 10.1016/j.ceca.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 35.Hallcher LM, Sherman WR. The effects of lithium ion and other agents on the activity of myo-inositol-1-phosphatase from bovine brain. J Biol Chem. 1980;255:10896–10901. [PubMed] [Google Scholar]
- 36.Kennedy ED, Challiss RA, Ragan CI, Nahorski SR. Reduced inositol polyphosphate accumulation and inositol supply induced by lithium in stimulated cerebral cortex slices. Biochem J. 1990;267:781–786. doi: 10.1042/bj2670781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phiel CJ, Klein PS. Molecular targets of lithium action. Annu Rev Pharmacol Toxicol. 2001;41:789–813. doi: 10.1146/annurev.pharmtox.41.1.789. [DOI] [PubMed] [Google Scholar]
- 38.Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170:1101–1111. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loukin S, Su Z, Zhou X, Kung C. Forward genetic analysis reveals multiple gating mechanisms of TRPV4. J Biol Chem. 2010;285:19884–19890. doi: 10.1074/jbc.M110.113936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mah W, Sonkusare SK, Wang T, Azeddine B, Pupavac M, Carrot-Zhang J, Hong K, Majewski J, Harvey EJ, Russell L, Chalk C, Rosenblatt DS, Nelson MT, Seguin C. Gain-of-function mutation in TRPV4 identified in patients with osteonecrosis of the femoral head. J Med Genet. 2016;53:705–709. doi: 10.1136/jmedgenet-2016-103829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Figueroa XF, Duling BR. Gap junctions in the control of vascular function. Antioxid Redox Signal. 2009;11:251–266. doi: 10.1089/ars.2008.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McSherry IN, Sandow SL, Campbell WB, Falck JR, Hill MA, Dora KA. A role for heterocellular coupling and EETs in dilation of rat cremaster arteries. Microcirculation. 2006;13:119–130. doi: 10.1080/10739680500466400. [DOI] [PubMed] [Google Scholar]
- 43.Rummery NM, Hill CE. Vascular gap junctions and implications for hypertension. Clin Exp Pharmacol Physiol. 2004;31:659–667. doi: 10.1111/j.1440-1681.2004.04071.x. [DOI] [PubMed] [Google Scholar]
- 44.Dora KA, Hinton JM, Walker SD, Garland CJ. An indirect influence of phenylephrine on the release of endothelium-derived vasodilators in rat small mesenteric artery. Br J Pharmacol. 2000;129:381–387. doi: 10.1038/sj.bjp.0703052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yashiro Y, Duling BR. Integrated Ca(2+) signaling between smooth muscle and endothelium of resistance vessels. Circ Res. 2000;87:1048–1054. doi: 10.1161/01.res.87.11.1048. [DOI] [PubMed] [Google Scholar]
- 46.Haas TL, Duling BR. Morphology favors an endothelial cell pathway for longitudinal conduction within arterioles. Microvasc Res. 1997;53:113–120. doi: 10.1006/mvre.1996.1999. [DOI] [PubMed] [Google Scholar]
- 47.Bruno RM, Ghiadoni L, Seravalle G, Dell’oro R, Taddei S, Grassi G. Sympathetic regulation of vascular function in health and disease. Front Physiol. 2012;3:284. doi: 10.3389/fphys.2012.00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vanhoutte PM, Shimokawa H, Tang EH, Feletou M. Endothelial dysfunction and vascular disease. Acta Physiol (Oxf) 2009;196:193–222. doi: 10.1111/j.1748-1716.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- 49.Francis M, Waldrup JR, Qian X, Solodushko V, Meriwether J, Taylor MS. Functional Tuning of Intrinsic Endothelial Ca2+ Dynamics in Swine Coronary Arteries. Circ Res. 2016;118:1078–1090. doi: 10.1161/CIRCRESAHA.115.308141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tiruppathi C, Minshall RD, Paria BC, Vogel SM, Malik AB. Role of Ca2+ signaling in the regulation of endothelial permeability. Vascul Pharmacol. 2002;39:173–185. doi: 10.1016/s1537-1891(03)00007-7. [DOI] [PubMed] [Google Scholar]
- 51.Nagaraja S, Kapela A, Tran CH, Welsh DG, Tsoukias NM. Role of microprojections in myoendothelial feedback--a theoretical study. J Physiol. 2013;591:2795–2812. doi: 10.1113/jphysiol.2012.248948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coleman HA, Tare M, Parkington HC. K+ currents underlying the action of endothelium-derived hyperpolarizing factor in guinea-pig, rat and human blood vessels. J Physiol. 2001;531:359–373. doi: 10.1111/j.1469-7793.2001.0359i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiang ZG, Nuttall AL, Zhao H, Dai CF, Guan BC, Si JQ, Yang YQ. Electrical coupling and release of K+ from endothelial cells co-mediate ACh-induced smooth muscle hyperpolarization in guinea-pig inner ear artery. J Physiol. 2005;564:475–487. doi: 10.1113/jphysiol.2004.080960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamamoto Y, Klemm MF, Edwards FR, Suzuki H. Intercellular electrical communication among smooth muscle and endothelial cells in guinea-pig mesenteric arterioles. J Physiol. 2001;535:181–195. doi: 10.1111/j.1469-7793.2001.00181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Allbritton NL, Meyer T, Stryer L. Range of messenger action of calcium ion and inositol 1,4,5-trisphosphate. Science. 1992;258:1812–1815. doi: 10.1126/science.1465619. [DOI] [PubMed] [Google Scholar]
- 56.Dickinson GD, Ellefsen KL, Dawson SP, Pearson JE, Parker I. Hindered cytoplasmic diffusion of inositol trisphosphate restricts its cellular range of action. Sci Signal. 2016;9:ra108. doi: 10.1126/scisignal.aag1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leybaert L. IP3, still on the move but now in the slow lane. Sci Signal. 2016;9:fs17. doi: 10.1126/scisignal.aal1929. [DOI] [PubMed] [Google Scholar]
- 58.Haddock RE, Grayson TH, Brackenbury TD, Meaney KR, Neylon CB, Sandow SL, Hill CE. Endothelial coordination of cerebral vasomotion via myoendothelial gap junctions containing connexins 37 and 40. Am J Physiol Heart Circ Physiol. 2006;291:H2047–H2056. doi: 10.1152/ajpheart.00484.2006. [DOI] [PubMed] [Google Scholar]
- 59.Mather S, Dora KA, Sandow SL, Winter P, Garland CJ. Rapid endothelial cell-selective loading of connexin 40 antibody blocks endothelium-derived hyperpolarizing factor dilation in rat small mesenteric arteries. Circ Res. 2005;97:399–407. doi: 10.1161/01.RES.0000178008.46759.d0. [DOI] [PubMed] [Google Scholar]
- 60.Bedner P, Niessen H, Odermatt B, Kretz M, Willecke K, Harz H. Selective permeability of different connexin channels to the second messenger cyclic AMP. J Biol Chem. 2006;281:6673–6681. doi: 10.1074/jbc.M511235200. [DOI] [PubMed] [Google Scholar]
- 61.Locke D, Stein T, Davies C, Morris J, Harris AL, Evans WH, Monaghan P, Gusterson B. Altered permeability and modulatory character of connexin channels during mammary gland development. Exp Cell Res. 2004;298:643–660. doi: 10.1016/j.yexcr.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 62.Niessen H, Harz H, Bedner P, Kramer K, Willecke K. Selective permeability of different connexin channels to the second messenger inositol 1,4,5-trisphosphate. J Cell Sci. 2000;113(Pt 8):1365–1372. doi: 10.1242/jcs.113.8.1365. [DOI] [PubMed] [Google Scholar]
- 63.Hill CE, Phillips JK, Sandow SL. Heterogeneous control of blood flow amongst different vascular beds. Med Res Rev. 2001;21:1–60. doi: 10.1002/1098-1128(200101)21:1<1::aid-med1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 64.Shimokawa H, Godo S. Diverse Functions of Endothelial NO Synthases System: NO and EDH. J Cardiovasc Pharmacol. 2016;67:361–366. doi: 10.1097/FJC.0000000000000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.