

Abstract
Objectives
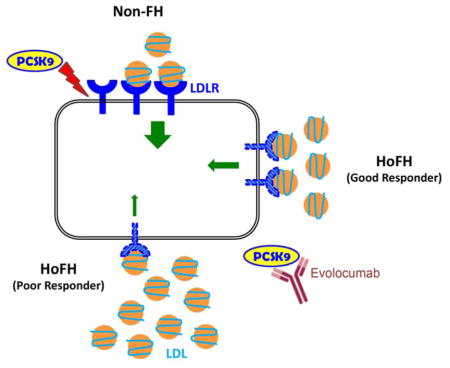
Evolocumab, a PCSK9 neutralizing antibody, lowers low-density lipoprotein cholesterol (LDL-C) in Homozygous Familial Hypercholesterolemic (HoFH) patients with reduced LDL receptor (LDLR) function. However, their individual responses are highly variable, even among carriers of identical LDLR genetic defects. We aimed to elucidate why HoFH patients variably respond to PCSK9 inhibition.
Approach and Results
Lymphocytes were isolated from 22 HoFH patients enrolled in the TAUSSIG trial. Ten patients were true homozygotes (FH1/FH1) and five identical compound heterozygotes (FH1/FH2). Lymphocytes were plated with or without mevastatin, recombinant PCSK9 (rPCSK9), or a PCSK9 neutralizing antibody. Cell surface LDLR expression was analyzed by flow cytometry. All HoFH lymphocytes had reduced cell surface LDLR expression compared with non-FH lymphocytes, for each treatment modality. Lymphocytes from FH1/FH2 patients (LDLR defective/negative) displayed the lowest LDLR expression levels followed by lymphocytes from FH1/FH1 patients (defective/defective). Mevastatin increased whereas rPCSK9 reduced LDLR expression. The PCSK9 neutralizing antibody restored LDLR expression. Lymphocytes displaying higher LDLR expression levels were those isolated from patients presenting with lowest levels of LDL-C and apolipoprotein B, prior and after 24 weeks of evolocumab treatment. These negative correlations remained significant in FH1/FH1 patients, and appeared more pronounced when patients with apolipoprotein E3/E3 genotypes were analyzed separately. Significant positive correlations were found between the levels of LDLR expression and the percentage reduction in LDL-C upon evolocumab treatment.
Conclusions
Residual LDLR expression in HoFH is a major determinant of LDL-C levels and appears to drive their individual response to evolocumab.
Subject Codes: Metabolism, Lipids and cholesterol
GRAPHIC ABSTRACT
INTRODUCTION
Homozygous familial hypercholesterolemia (HoFH) is a severe inherited disorder of lipoprotein metabolism resulting mostly from the presence of mutations on both alleles of the LDL receptor (LDLR) or in rare instances biallelic mutations of apolipoprotein B (APOB), proprotein convertase subtilisin kexin type 9 (PCSK9) or the LDLR adaptor protein (LDLRAP1) 1–3. These genetic defects sharply reduce the hepatic clearance of low-density lipoproteins (LDL). HoFH patients typically present with LDL-cholesterol (LDL-C) plasma concentrations above 500mg/dL (13mM) (although there can be considerable phenotypic variability), leading to premature and often fatal cardiovascular events in the first decades of life 1, 4.
Conventional lipid lowering treatments (LLT) currently available, statins and ezetimibe, are moderately effective in HoFH patients as they reduce LDL-C levels by only around 25%. However, even this modest LDL-C reduction has been shown to delay cardiovascular events and prolong life 4, 5. These therapies are insufficient to bring HoFH patients to therapeutic target (i.e. LDL-C < 70mg/dL (1.8mM)). Drugs that respectively block the synthesis of apoB (mipomersen) or the assembly of nascent apoB containing lipoproteins (lomitapide) reduce the endogenous production of LDL thus lowering circulating LDL-C levels independently of residual LDLR activity 1, 5. Increased hepatic fat (hepatic steatosis) is intrinsic to the mechanisms of action of mipomersen and lomitapide and may limit their use. In addition, mipomersen and lomitapide are not always well tolerated and are extremely costly. LDL apheresis is a further therapeutic option but is not available to all patients due to its cost and often is not sufficient for HoFH patients to reach LDL-C therapeutic goals.
Recently, the TESLA and TAUSSIG clinical trials showed that the PCSK9 inhibitor evolocumab promotes a 20–30% reduction in LDL-C in HoFH patients on top of conventional LLT 1, 6, 7. Given that PCSK9 is a circulating inhibitor of the LDLR, evolocumab does not lower LDL-C in HoFH patients totally lacking the receptor (receptor-negative)1, 6–8. However, for the vast majority of HoFH patients, the activity of the LDLR is reduced but not abrogated (receptor-defective), and evolocumab reduces LDL-C. Interestingly, the response of patients to evolocumab is extremely variable, even among homozygous carriers of identical LDLR genetic defects 1.
To elucidate why HoFH patients with similar LDLR genotypes respond variably to evolocumab, we measured the levels of LDLR expression at their lymphocyte surface. We also investigated the effects of statins, recombinant PCSK9 and a monoclonal antibody targeting PCSK9, on LDLR expression. We found an inverse association between LDLR abundance measured in vitro and the levels of LDL-C and of apoB in the plasma of HoFH patients.
MATERIAL AND METHODS
Material and Methods are available in the online only Data Supplement.
RESULTS
Lymphocytes isolated from one normolipemic control donor, one LDB HoFH patient, five HeFH patients, and 21 HoFH patients with LDLR genetic defects were incubated sequentially with increasing concentrations of mevastatin, recombinant PCSK9 (rPCSK9), and/or the PCSK9 inhibitor mAb1/31H4 (mAb1). Baseline LDLR levels measured without mevastatin, rPCSK9 and mAb1 were on average 3.5-fold lower in lymphocytes isolated from HoFH patients (ΔMFI 232±109) compared with control (ΔMFI 811±225) and LDB (ΔMFI 885±73) lymphocytes. HeFH lymphocytes displayed intermediate baseline LDLR expression levels (ΔMFI 572±159). Mevastatin treatment significantly increased the expression of the LDLR at the surface of lymphocytes up to maximal ΔMFI levels of 372±171 in HoFH, 1299±123 in HeFH, 1429±177 in control and 1392±108 in LDB (Figure 1A). In contrast, rPCSK9 significantly and dose dependently reduced LDLR cell surface expression down to ΔMFI nadirs of 73±38 in HoFH, 430±97 in HeFH, 320±65 in control, and 326±83 in LDB lymphocytes (Figure 1A). Saturating concentrations of the PCSK9 inhibitor mAb1 restored LDLR expression levels back to their maximal ΔMFI levels at 353±155 in HoFH, 1129±175 in HeFH, 1341±191 in control, and 1258±169 in LDB lymphocytes. In each experimental condition, the expression of the LDLR at the plasma membrane was on average 3 to 5-fold lower in HoFH than in control lymphocytes, and 2 to 4 fold lower in HoFH than in HeFH lymphocytes.
Figure 1. Cell surface LDLR expression in lymphocytes from one healthy donor, one HoFH patient with ApoB mutations (LDB), five HeFH with LDLR mutations, and 21 HoFH with LDLR mutations, all together (A) and for the HoFH as a function of their LDLR genotype (B).

Primary lymphocytes were plated for 24h in serum deprived medium with increasing concentrations of mevastatin and supplemented or not for the last 4h of the incubation with rPCSK9 with or without the anti-PCSK9 mAb1 prior to flow cytometry analysis. LDLR expression levels are expressed in ΔMFI. Histograms represent mean ± SD. *, p<0.05. §, p< 0.05 vs. healthy donor lymphocytes under the same experimental conditions. #, p<0.05 vs. HeFH lymphocytes under the same experimental conditions. ns, p>0.1 vs. the Meva 10μg/ml, no rPCSK9 and no anti-PCSK9 experimental condition.
When HoFH lymphocytes were analyzed with respect to the residual LDLR function associated with their genotypes (listed in Table 1), the expression of the LDLR was significantly lower at the surface of lymphocytes isolated from patients carrying one negative and one defective LDLR allele (i.e. the 5 compound heterozygotes FH1/FH2 D206E/V408M), compared with lymphocytes from patients carrying two LDLR defective alleles (i.e. the 10 true homozygotes FH1/FH1 D206E/D206E) (Figure 1B). Lymphocytes from four out of six patients carrying other mutations on both LDLR alleles and presenting with milder HoFH phenotypes, as shown by their circulating LDL-C and apoB levels at week 0 (Table 1), expressed higher baseline and maximal levels of LDLR at their surface than FH1/FH1 lymphocytes. Lymphocytes from one D206E/D154N patient (two defective LDLR alleles) and from one D206E/D461N patient (one defective and one unclassified LDLR alleles) expressed similar LDLR levels than FH1/FH1 lymphocytes (Figure 1B).
Table 1.
HoFH patients response to evolocumab 420mg Q2W.
| PATIENTS | Plasma LDL-C, apoB and Lp(a) levels (TAUSSIG) | Maximal LDLR cell surface expression (ΔMFI) | Apo(a) polymorphism | ApoE levels & polymorphism | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Amino Acid Change | LDL-C (mM) | ApoB (g/L) | Lp(a) (nM) | KIV2 repeats (n) | Conc. (mg/dL) | Geno type | ||||||||
| Week 0 | Week 24 | Δ % | Week 0 | Week 24 | Δ % | Week 0 | Week 24 | Δ % | |||||||
| J06 | DN | 12.20 | 11.29 | −7 | 2.57 | 2.43 | −5 | 122 | 99 | −19 | 160 | 27 | 13.3 | E2/E3 | |
| J11 | DN | 13.70 | 10.64 | −22 | 2.76 | 2.39 | −13 | 76 | 92 | 21 | 173 | - | 12.6 | E3/E3 | |
| J14 | D206E/V408M | DN | 15.20 | 13.13 | −14 | 3.33 | 2.54 | −24 | 108 | 84 | −22 | 132 | 35 | 15.5 | E3/E3 |
| J20 | (FH1/FH2) | DN | 9.61 | 6.94 | −28 | 2.59 | 1.84 | −29 | 58 | 51 | −12 | 195 | 30 | 12.2 | E4/E3 |
| J23 | DN | 13.34 | 10.23 | −23 | 2.78 | 1.98 | −29 | 134 | 128 | −4 | 113 | 20 | 14.6 | E3/E3 | |
| J02 | DD | 15.20 | 14.84 | −2 | 3.05 | 3.22 | 6 | 240 | 229 | −5 | 184 | 13 | 22 | E3/E3 | |
| J10 | DD | 15.62 | 11.55 | −26 | 3.83 | 2.41 | −37 | 146 | 106 | −27 | 415 | 23 | 15 | E3/E3 | |
| J12 | DD | 10.83 | 9.69 | −11 | 2.29 | 2.16 | −6 | 79 | 96 | 22 | 468 | 6 | 12.5 | E3/E3 | |
| J13 | DD | 10.20 | 7.56 | −26 | 2.15 | 1.84 | −14 | 146 | 135 | −8 | 474 | 12 | 9.7 | E3/E3 | |
| J15* | D206E/D206E | DD | 13.65 | 12.12 | −11 | 2.87 | 2.44 | −15 | 156 | 135 | −13 | 381 | 5 | 15.5 | E3/E3 |
| J24 | (FH1/FH1) | DD | 14.81 | 13.21 | −11 | 3.37 | 2.54 | −25 | 421 | 327 | −22 | 303 | 13 | 10.1 | E3/E3 |
| C02 | DD | 12.90 | 7.23 | −44 | 2.64 | 1.79 | −32 | 17 | 14 | −18 | 418 | 10 | 8.2 | E3/E3 | |
| C03 | DD | 8.48 | 6.50 | −23 | 1.9 | 1.48 | −22 | 31 | 26 | −16 | 528 | 36 | 7.2 | E3/E3 | |
| C04 | DD | 9.78 | 5.85 | −40 | 2.22 | 1.64 | −26 | 140 | 108 | −23 | 563 | 13 | 8.5 | E3/E3 | |
| C05 | DD | 14.61 | 8.18 | −44 | 3.24 | 2.04 | −37 | 80 | 47 | −41 | 607 | 17 | 6.8 | E3/E4 | |
| J21 | C6W/C61X | UN | 3.78 | 1.30 | −66 | 1.06 | 0.43 | −59 | 71 | 39 | −45 | 511 | 6 | 7.1 | E2/E3 |
| J22 | C6W/C61X | UN | 3.99 | 2.28 | −43 | 1.13 | 0.75 | −34 | 38 | 23 | −39 | 614 | 28 | 6.7 | E2/E3 |
| J17 | D206E/D461N | DU | 5.26 | 4.20 | −20 | 1.17 | 1.1 | −6 | 86 | 83 | −3 | 393 | 13 | 7.5 | E3/E3 |
| J18 | D206E/D461N | DU | 4.66 | 4.58 | −2 | 1.07 | 1.16 | 8 | 7 | 9 | 29 | 750 | 30 | 13.4 | E3/E3 |
| J19 | D206E/D154N | DD | 9.35 | 7.85 | −16 | 1.94 | 1.65 | −15 | 5 | 5 | 0 | 333 | - | 8.9 | E2/E3 |
| J09 | P664L/P664L | UU | 7.46 | 6.84 | −8 | 1.75 | 1.67 | −5 | 133 | 140 | 5 | 475 | 15 | 11 | E3/E3 |
| D01 | R3527E/R3527E | LDB | 4.42 | 2.17 | −51 | 1.08 | 0.72 | −33 | 121 | 69 | −43 | 1392 | 5 | 2.6 | E3/E3 |
D: Defective (2–25% residual LDLR activity), N: Negative (< 2% residual LDLR activity), U: Unclassified LDLR mutation, LDB: ligand defective apoB, conc: concentration.
This patient was not on evolocumab at time of blood collection. Δ: percent change from week 0
Noteworthy, LDLR cell surface expression levels measured in lymphocytes were variable, even when lymphocytes were isolated from HoFH patients with identical genetic defects. For instance, baseline LDLR expression ranged from ΔMFI levels of 74 to 103 in FH1/FH2 lymphocytes and from ΔMFI levels of 111 to 354 in FH1/FH1 lymphocytes. Maximal LDLR expression ranged from ΔMFI levels of 113 to 195 in FH1/FH2 lymphocytes and from ΔMFI levels of 184 to 607 in FH1/FH1 lymphocytes (Supplemental Figure I). Of note, mevastatin increased, whereas rPCSK9 reduced and mAb1 restored LDLR cell surface expression in all HoFH lymphocytes tested, proportionally to their baseline LDLR expression levels (r=0.976, p=0.0001 between maximal and baseline LDLR expression levels).
It is well established that PCSK9 inhibition with evolocumab promotes substantial reductions in LDL-C, apoB, as well as lipoprotein (a) [Lp(a)] in HoFH patients 1, 6, 7. This was also evident in the 21 HoFH patients included in the present study. Individual responses to treatment were however variable, even among patients with identical LDLR mutations (Table 1). To determine the molecular bases underpinning these variable responses, we performed a series of correlation analyses between (i) the maximal levels of LDLR expression measured in the lymphocytes of each patient and (ii) their levels of LDL-C, apoB and Lp(a) before (week 0, when patients are on standard LLT) or after evolocumab treatment (week 24, when patient are on standard LLT + 420mg evolocumab Q2W). We found significant negative correlations between maximal LDLR expression levels measured in patients’ lymphocytes and their circulating levels of LDL-C at week 0 (r=−0.564, p=0.007) and week 24 (r=−0.700, p=0.0004) (Figure 2A). We also found significant negative correlations between maximal LDLR expression levels of patients’ lymphocytes and their plasma apoB concentrations measured at week 0 (r=−0.564, p=0.007) and week 24 (r=−0.667, p=0.001) (Figure 2B). In contrast, the negative correlations between LDLR expression levels and circulating Lp(a) levels measured at week 0 (r=−0.336, p=0.136) and week 24 (r=−0.376, p=0.09) did not reach statistical significance (Figure 2C). Noteworthy, there was a wide distribution of apolipoprotein (a) sizes among HoFH patients, ranging from 5 to 35 kringle IV2 repeats (Table 1). We also found a significant negative correlation (r=−0.630, p=0.003) between maximal LDLR levels of primary lymphocytes and the concentrations of apoE measured by LC-MS/MS in plasma samples of HoFH patients (i.e. on standard LLT + 420mg evolocumab Q2W). The majority of patients (72%) displayed an apoE3/E3 genotype, two patients were E3/E4 and four patients were E2/E3 (Table 1). To evaluate whether variable apoE genotypes modulate LDL-C, apoB and Lp(a) levels in HoFH patients, we performed correlations analyses between LDLR expression levels and plasma lipids, separately in the subgroup of 15 patients with an apoE3/E3 genotype (Supplemental Figure II). The correlation coefficients between LDLR expression and LDL-C or apoB levels increased in this subgroup compared with those observed in the entire cohort of 21 HoFH patients (Supplemental Figure II and Figure 2). Similarly to what was observed in the entire cohort, the correlation between LDLR expression and Lp(a) did not reach statistical significance in the subgroup of 15 patients with an apoE3/E3 genotype. We did not observe any significant correlation between basal or maximal levels of LDLR expression in lymphocytes and changes in LDL-C levels induced by evolocumab in the cohort of 21 HoFH patients in TAUSSIG (r=0.361, p=0.107 and r=0.325, p=0.150, respectively). However, this correlation became significant when patient J18, the poorest responder to evolocumab, was excluded from the analysis (r=0.567, p=0.009 and r=0.546, p=0.013, respectively) (Figure 3).
Figure 2. Association between the maximal levels of LDLR cell surface expression in lymphocytes and the concentrations of (A) LDL-C, (B) ApoB and (C) Lp(a) measured in the plasma of HoFH patients before (week 0 – left panels) and after (week 24 – right panels) treatment with evolocumab 420mg Q2W.

Spearman correlation coefficients (r) and significance (p) values are indicated.
Figure 3. Association between the basal (top panels) and maximal (bottom panels) levels of LDLR expression in lymphocytes and the reduction in LDL-C levels in 21 HoFH patients (A & B) and in ten FH1-FH1 HoFH patients (C & D) after treatment with evolocumab 420mg Q2W.

Pearson correlation coefficients (r) and significance (p) values as well as J18 patient are indicated.
When the subgroup of ten HoFH patients with identical LDLR defects (FH1/FH1) was analysed separately, we observed a significant negative correlation between the maximal levels of LDLR expression measured in patients lymphocytes and their circulating LDL-C levels at week 0 (r=−0.648, p=0.049) and week 24 (r=−0.8303, p=0.0047). The correlation coefficients between LDLR expression and LDL-C levels at week 0 (r=−0.883, p=0.003) and week 24 (r=−0.950, p=0.0004) were further increased in the subgroup of nine FH1/FH1 patients with an apoE3/E3 genotype. The negative correlation between LDLR expression in lymphocytes and plasma apoB at week 0 (r=−0.833, p=0.008) and week 24 (r=−0.933, p=0.0007) also reached statistical significance. The association between LDLR expression and Lp(a) levels was significant at week 0 (r=−0.711, p=0.034) but did not reach statistical significance at week 24 (r=−0.569, p=0.11). As in the entire cohort, the negative association between LDLR expression and apoE levels was significant in FH1/FH1 (r=−0.818, p=0.007). The changes in LDL-C induced by evolocumab treatment in TAUSSIG were positively correlated with basal and maximal LDLR expression of FH1/FH1 lymphocytes (r=0.775, p=0.008 and r=0.737, p=0.015, respectively) (Figure 3).
DISCUSSION
To understand why HoFH patients, even those with similar LDLR genetic defects, variably respond to evolocumab in clinical trials, we investigated their ability to express the LDLR ex vivo under a wide set of conditions. We first showed that LDLR expression levels were quite variable in primary lymphocytes isolated from HoFH patients with distinct as well as similar LDLR mutations. We also showed that the levels of LDLR expression correlated negatively with the circulating levels of LDL-C of patients before and after treatment with evolocumab, demonstrating that residual LDLR functionality and expression are important determinants of LDL clearance in HoFH.
We comprehensively investigated LDLR expression at the surface of lymphocytes isolated from patients enrolled in TAUSSIG. As anticipated, LDLR expression was sharply reduced in HoFH lymphocytes compared with non-FH, HeFH and LDB cells. Not surprisingly, LDLR expression varied widely between lymphocytes isolated from HoFH patients carrying different LDLR mutations. Lymphocytes from FH1/FH2 patients (one negative and one defective LDLR alleles) displayed reduced cell surface LDLR expression compared with lymphocytes from FH1/FH1 patients (two identical defective LDLR alleles), in line with previous observations made in primary fibroblasts from carriers of those mutations 8, 9. Even among lymphocytes isolated from patients with identical genetic defects (FH1/FH1), cell surface expression of the LDLR appeared quite variable, as previously observed in primary fibroblasts 8. The reasons for these variations within FH1/FH1 lymphocytes are unclear. Given that no neutralizing antibodies were seen in TAUSSIG, it is very unlikely that the variability in response to PCSK9 inhibition might be due to a reduced efficacy of evolocumab in some patients. The variations observed may rather result from the presence of epigenetic modifications on the LDLR gene or on the genes controlling LDLR expression such as the SREBPs transcription factors in some patients 10. These modifications could also promote a differential response to statin treatment.
The significant negative correlations between the expression of the LDLR measured in lymphocytes and the levels of LDL-C and apoB in the plasma of HoFH patients, in particular in the FH1/FH1 subgroup, underpins the important role of residual LDLR activity for the clearance of LDL particles in those individuals. Interestingly, these correlations appeared more pronounced with plasma LDL-C and apoB levels measured after evolocumab treatment. A potential explanation is that the expression and/or the activity of PCSK9 might greatly vary between individual HoFH subjects, a parameter blunted by evolocumab treatment, since a dose of 420mg every two weeks inhibits 94–100% of circulating PCSK91.
Besides the residual LDLRs, other receptors that contribute to apoB containing lipoproteins clearance may also function variably in HoFH patients. Since these receptors are LDLR family members that are also regulated by PCSK9 11, 12 [e.g. the LDLR related protein], their variable functionality could account in part for the variability observed in the LDL-C lowering effects of evolocumab. The presence of distinct apoE isoforms that bind the LDLR with variable affinities 13, could also explain to some extent the substantial range of LDL-C levels and the variability in response to evolocumab observed in HoFH patients with identical LDLR genetic defects 13, 14. Thus, the correlations coefficients between LDLR expression and LDL-C or apoB100 levels increased when patients with an apoE genotype other than E3/E3 were excluded from our analyses.
Unlike the correlations between LDLR expression in lymphocytes and LDL-C or apoB levels in patients pre and post evolocumab treatment that reached significance in the whole group as well as in the FH1/FH1 subgroup, the association between LDLR expression and Lp(a) levels reached significance only in the subgroup of FH1/FH1 patients at a single time point (week 0). This suggests that the residual LDLR activity is probably not a significant determinant of Lp(a) clearance in HoFH, which is further evidenced by the fact that evolocumab reduced Lp(a) but not LDL-C in two HoFH patients totally lacking the receptor7. The role of the LDLR in Lp(a) clearance remains a controversial issue with recent in vitro studies showing that Lp(a) catabolism is mediated to some extent by the LDLR (when LDLR expression is very high) and other studies showing that the LDLR is not involved in Lp(a) uptake 15–17.
Our study has several limitations. The number of HoFH patients with the same LDLR genotypes included was small. Given the limited amount of lymphocytes available from each patient, we were only able to measure LDLR expression but not LDLR activity (i.e. fluorescent LDL uptake). This would have provided valuable information to the study. We have however previously shown that LDLR expression and LDLR activity parallel nicely in primary human lymphocytes3. In addition, we used lymphocytes to assess LDLR expression ex vivo as a proxy for hepatocytes3, a more relevant cell type for LDL and Lp(a) plasma clearance studies. We also assumed that the maximal ability of primary lymphocytes to express the LDLR at their plasma membrane reflects what happens in vivo in patients’ hepatocytes upon treatment with high doses of statins and evolocumab. Nevertheless, our data clearly indicate that residual activity of the LDLR expression is a major determinant of LDL-C circulating levels in HoFH patients. Enhancing this molecular pathway with evolocumab substantially lowers plasma lipids in these difficult–to-treat patients, and will undoubtedly improve their cardiovascular health and their life expectancy.
Supplementary Material
HIGHLIGHTS.
Residual expression of the LDL receptor in homozygous familial hypercholesterolemia patients with identical LDL receptor genetic defects is highly variable.
It negatively correlates their circulating levels of LDL cholesterol before and after treatment with evolocumab on top of maximal statin therapy.
Ex-vivo LDL receptor expression measurement is a useful approach to dissect the individual response of homozygous FH patients to lipid lowering treatments.
Acknowledgments
We thank the Cytometry Facility Cytocell from Nantes for expert technical assistance.
SOURCES OF FUNDING
This study was funded in part by AMGEN. Gilles Lambert is the recipient of an Allocation de Recherche Chaire Mixte (Inserm-Université), and a Program Grant ANR-16-RHUS-0007 CHOPIN (Agence Nationale de la Recherche).
Non Standard Abbreviations
- PCSK9
Proprotein Convertase Subtilisin Kexin Type 9
- rPCSK9
recombinant PCSK9
- LDL
low density lipoprotein
- LDL-C
LDL-Cholesterol
- LDLR
LDL Receptor
- FH
Familial Hypercholesterolemia
- HeFH
Heterozougous FH
- HoFH
Homozygous FH
- Lp(a)
Lipoprotein (a)
- ΔMFI
Specific Mean Fluorescence Intensity
- Apo(a)
apolipoprotein (a)
- ApoE
apolipoprotein E
- ApoB
apolipoprotein B
- LDLRAP1
LDLR adaptor protein
- LLT
Lipid lowering treatment
- PBMC
Peripheral Blood Mononuclear Cells
- LC-MS/MS
Liquid chromatography tandem mass spectrometry
- Q2W
Every 2 weeks
- LDB
Ligand defective apoB
- mAb1
PCSK9 inhibitor mAb1/31H4
Footnotes
DISCLOSURES
LT is a fulltime employee of Amgen. GL has received research funding, consulting fees and/or honoraria from Amgen, Sanofi-Regeneron, and Pfizer. MF received grants, consulting fees and/or honoraria from Abbott, Akcea/Ionis, Amgen, AstraZeneca, Eli Lilly, Kowa, Merck, Mylan, Pfizer, Roche, Sanofi-Regeneron and Servier. FJR has received reimbursement for conducting clinical trials from Amgen, Regeneron Pharmaceuticals, Inc., and Sanofi Aventis; modest speaker fees or honoraria for lectures from AstraZeneca, Pfizer, Merck, Amgen, and Sanofi; and consultant/advisory board fees from Amgen, Sanofi Aventis and Regeneron Pharmaceuticals, Inc. DJB has received research support from Amgen and Aegerion (both significant), honoraria for lectures from MSD, AstraZeneca, Pfizer, Sanofi-Aventis, Amgen, and Servier (all modest), and has served as a consultant/advisory board member for AstraZeneca, Sanofi, MSD, Aegerion, Gemphire, and Amgen (all modest). DB has received reimbursement for conducting clinical trials from Amgen, Regeneron Pharmaceuticals, Ionis, Eli Lilly and AstraZeneca. BC has received research funding and personal fees from Sanofi and Regeneron Pharmaceuticals, Inc.; research funding from Pfizer; and honoraria from Amgen, AstraZeneca, Pierre Fabre, Eli Lilly and Company, MSD Merck & Co., Novo Nordisk, and Sanofi,
References
- 1.Raal FJ, Hovingh GK, Blom D, Santos RD, Harada-Shiba M, Bruckert E, Couture P, Soran H, Watts GF, Kurtz C, Honarpour N, Tang L, Kasichayanula S, Wasserman SM, Stein EA. Long-term treatment with evolocumab added to conventional drug therapy, with or without apheresis, in patients with homozygous familial hypercholesterolaemia: an interim subset analysis of the open-label TAUSSIG study. Lancet Diabetes Endocrinol. 2017;5:280–290. doi: 10.1016/S2213-8587(17)30044-X. [DOI] [PubMed] [Google Scholar]
- 2.Alves AC, Etxebarria A, Medeiros AM, Benito-Vicente A, Thedrez A, Passard M, Croyal M, Martin C, Lambert G, Bourbon M. Characterization of the first PCSK9 gain of function homozygote. Journal of the American College of Cardiology. 2015;66:2152–4. doi: 10.1016/j.jacc.2015.08.871. [DOI] [PubMed] [Google Scholar]
- 3.Thedrez A, Sjouke B, Passard M, Prampart-Fauvet S, Guedon A, Croyal M, Dallinga-Thie G, Peter J, Blom D, Ciccarese M, Cefalu AB, Pisciotta L, Santos RD, Averna M, Raal F, Pintus P, Cossu M, Hovingh K, Lambert G. Proprotein Convertase Subtilisin Kexin Type 9 Inhibition for Autosomal Recessive Hypercholesterolemia. Arteriosclerosis, thrombosis, and vascular biology. 2016;36:1647–50. doi: 10.1161/ATVBAHA.116.307493. [DOI] [PubMed] [Google Scholar]
- 4.Raal FJ, Pilcher GJ, Panz VR, van Deventer HE, Brice BC, Blom DJ, Marais AD. Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy. Circulation. 2011;124:2202–7. doi: 10.1161/CIRCULATIONAHA.111.042523. [DOI] [PubMed] [Google Scholar]
- 5.Cuchel M, Bruckert E, Ginsberg HN, Raal FJ, Santos RD, Hegele RA, Kuivenhoven JA, Nordestgaard BG, Descamps OS, Steinhagen-Thiessen E, Tybjaerg-Hansen A, Watts GF, Averna M, Boileau C, Boren J, Catapano AL, Defesche JC, Hovingh GK, Humphries SE, Kovanen PT, Masana L, Pajukanta P, Parhofer KG, Ray KK, Stalenhoef AF, Stroes E, Taskinen MR, Wiegman A, Wiklund O, Chapman MJ European Atherosclerosis Society Consensus Panel on Familial H. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35:2146–57. doi: 10.1093/eurheartj/ehu274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raal FJ, Honarpour N, Blom DJ, Hovingh GK, Xu F, Scott R, Wasserman SM, Stein EA Investigators T. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:341–50. doi: 10.1016/S0140-6736(14)61374-X. [DOI] [PubMed] [Google Scholar]
- 7.Stein EA, Honarpour N, Wasserman SM, Xu F, Scott R, Raal FJ. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128:2113–20. doi: 10.1161/CIRCULATIONAHA.113.004678. [DOI] [PubMed] [Google Scholar]
- 8.Lambert G, Chatelais M, Petrides F, Passard M, Thedrez A, Rye KA, Schwahn U, Gusarova V, Blom DJ, Sasiela W, Marais AD. Normalization of low-density lipoprotein receptor expression in receptor defective homozygous familial hypercholesterolemia by inhibition of PCSK9 with alirocumab. Journal of the American College of Cardiology. 2014;64:2299–300. doi: 10.1016/j.jacc.2014.07.995. [DOI] [PubMed] [Google Scholar]
- 9.Lambert G, Petrides F, Chatelais M, Blom DJ, Choque B, Tabet F, Wong G, Rye KA, Hooper AJ, Burnett JR, Barter PJ, Marais AD. Elevated plasma PCSK9 level is equally detrimental for patients with nonfamilial hypercholesterolemia and heterozygous familial hypercholesterolemia, irrespective of low-density lipoprotein receptor defects. Journal of the American College of Cardiology. 2014;63:2365–73. doi: 10.1016/j.jacc.2014.02.538. [DOI] [PubMed] [Google Scholar]
- 10.Adaikalakoteswari A, Finer S, Voyias PD, McCarthy CM, Vatish M, Moore J, Smart-Halajko M, Bawazeer N, Al-Daghri NM, McTernan PG, Kumar S, Hitman GA, Saravanan P, Tripathi G. Vitamin B12 insufficiency induces cholesterol biosynthesis by limiting s-adenosylmethionine and modulating the methylation of SREBF1 and LDLR genes. Clin Epigenetics. 2015;7:14. doi: 10.1186/s13148-015-0046-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roubtsova A, Munkonda MN, Awan Z, Marcinkiewicz J, Chamberland A, Lazure C, Cianflone K, Seidah NG, Prat A. Circulating proprotein convertase subtilisin/kexin 9 (PCSK9) regulates VLDLR protein and triglyceride accumulation in visceral adipose tissue. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:785–91. doi: 10.1161/ATVBAHA.110.220988. [DOI] [PubMed] [Google Scholar]
- 12.Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, Mayer H, Nimpf J, Prat A, Seidah NG. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. The Journal of biological chemistry. 2008;283:2363–72. doi: 10.1074/jbc.M708098200. [DOI] [PubMed] [Google Scholar]
- 13.Mahley RW. Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J Mol Med (Berl) 2016;94:739–46. doi: 10.1007/s00109-016-1427-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moriarty PM, Varvel SA, Gordts PL, McConnell JP, Tsimikas S. Lipoprotein(a) Mass Levels Increase Significantly According to APOE Genotype: An Analysis of 431 239 Patients. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:580–588. doi: 10.1161/ATVBAHA.116.308704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romagnuolo R, Scipione CA, Boffa MB, Marcovina SM, Seidah NG, Koschinsky ML. Lipoprotein(a) catabolism is regulated by proprotein convertase subtilisin/kexin type 9 through the low density lipoprotein receptor. The Journal of biological chemistry. 2015;290:11649–62. doi: 10.1074/jbc.M114.611988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villard EF, Thedrez A, Blankenstein J, Croyal M, Tran TTT, Poirier B, LeBail JC, Illiano S, Nobécourt E, Krempf M, Blom DJ, Marais AD, Janiak P, Muslin AJ, Guillot E, Lambert G. PCSK9 modulates the secretion but not the cellular uptake of Lipoprotein (a) ex vivo: an effect blunted by alirocumab. J Am Coll Cardiol Basic Transl Sci. 2016;1:419–427. doi: 10.1016/j.jacbts.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma M, Redpath GM, Williams MJ, McCormick SP. Recycling of Apolipoprotein(a) After PlgRKT-Mediated Endocytosis of Lipoprotein(a) Circulation research. 2017;120:1091–1102. doi: 10.1161/CIRCRESAHA.116.310272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.