

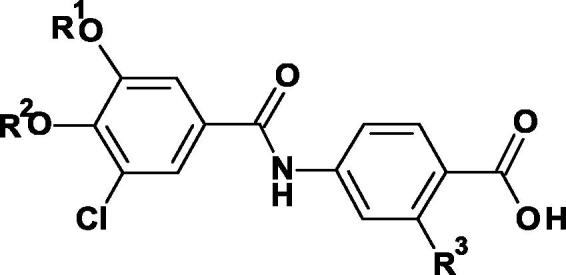
Graphical abstract

Abstract
A ligand-based virtual screening exercise examining likely bioactive conformations of AM 580 (2) and AGN 193836 (3) was used to identify the novel, less lipophilic RARα agonist 4-(3,5-dichloro-4-ethoxybenzamido)benzoic acid 5, which has good selectivity over the RARβ, and RARγ receptors. Analysis of the medicinal chemistry parameters of the 3,5-substituents of derivatives of template 5 enabled us to design a class of drug-like molecules with lower intrinsic clearance and higher oral bioavailability which led to the novel RARα agonist 4-(3-chloro-4-ethoxy-5-isopropoxybenzamido)-2-methylbenzoic acid 56 that has high RARα potency and excellent selectivity versus RARβ (2 orders of magnitude) and RARγ (4 orders of magnitude) at both the human and mouse RAR receptors with improved drug-like properties. This RARα specific agonist 56 has high oral bioavailability (>80%) in both mice and dogs with a good PK profile and was shown to be inactive in cytotoxicity and genotoxicity screens.
1. Introduction
The retinoic acid receptors (RARα, RARβ, and RARγ) are members of the nuclear receptor superfamily. Compounds which bind to and activate the RARs are termed retinoids and comprise both natural retinol (Vitamin A) metabolites and synthetic analogs. Retinoids regulate a wide variety of biological processes such as vertebrate embryonic morphogenesis and organogenesis, cell growth arrest, differentiation, and apoptosis, as well as their disorders.1
The RARα isoform is found in the majority of tissues and has been implicated in a number of diseases, most notably acute promyelocytic leukemia (APL). Selective RARα agonists have been shown to inhibit proliferation and induce apoptosis of mammary tumor oncogenesis in murine models (MMTV-neu and MMTV-wnt1 transgenic mice) relevant to human cancer,2 and to inhibit LPS-induced B-lymphocyte proliferation.3 Selective RARα agonists have also been shown to prevent neuronal cell death caused by amyloid-β and, when administered orally, can prevent amyloid-β production and Alzheimer’s disease progression in a mouse model.4 It has been shown5 that selective RARα agonists suppressed allospecific immune response and significantly prolonged the survival of mouse cardiac allografts and can ameliorate nephritis in lupus-prone mice, NZB/NZW F1.6 This supports the rationale for using RARα agonists as immunosuppressants in human organ transplantation. Thus selective RARα agonists have the therapeutic potential for the treatment of cancer, dermatological diseases, Alzheimer’s disease and immunological disorders.
Synthetic RARα, RARβ, and RARγ agonists have been developed from all-trans-retinoic acid (ATRA), and usually consist of a lipophilic ring, a linker and a carboxylic acid (Fig. 1). There has been an extensive studies on the SAR7, 8, 9, 10 of the RARα agonists based essentially on the bicyclic 5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalene derivatives which evolved from ATRA. All three agonist, AM 580 (2), AGN 193836 (3), AGN 195183 (4) and antagonist BMS 195614 (1) (Fig. 1) which contain an amide linker and a benzoic acid, arose from these studies.
Fig. 1.

RARα agonists and antagonist.
However, although AM 580 (2) and AGN 195183 (4) have moderate and good selectivity respectively for RARα, over RARβ and RARγ they are quite lipophilic (cLog P 6.3 and 7.2). In addition AM 580 (2) has been shown to be toxic,11, 12 and the more recently discovered compound AGN 195183 (4)10 which was in Phase I clinical trials for cancer has been discontinued.13 Our aim was to find a novel, potent, highly selective RARα agonist not based on the bicyclic 5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalene class that was ligand efficient, orally bioavailable and without the lipophilic obesity seen with (2), (3) and (4). We outline here how we discovered our initial hit compound 5 and how this was developed into the orally bioavailable, highly potent and selective RARα agonist 56 (Fig. 1) which exhibits promising drug-like properties.
2. Chemistry
The phenyl carboxamido-benzoic acids (Scheme 1, Scheme 3, Scheme 4, Scheme 5) and phenyl carbamoyl-benzoic acid 26 (Scheme 2) were prepared by coupling the appropriately substituted aniline with a substituted benzoic acid using a variety of standard methods for the formation of an amide bond. The 3,5-dichloro-4-alkoxy compounds 12–15 and 17–21 (Scheme 1) were prepared by alkylation of the phenolic group of methyl 3,5-dichloro-4-hydroxybenzoate 6 followed by hydrolysis of the benzoate ester. Coupling the resultant acid 7 via the acid chloride by reaction with oxalyl chloride or directly with HATU, with the appropriate methyl 4-aminobenzoate 8 followed by hydrolysis with lithium hydroxide gave the required acids 12–15 and 17–21.
Scheme 1.

4-(3,5-Dichloro-4-alkoxy-benzamido)benzoic acids. (Reagents and conditions: (i) K2CO3, R1Br, DMF, 80 °C, 3 h; (ii) LiOH, THF, H2O, room temp, 12 h; (iii) (COCl)2,CH2Cl2, DMF, 0 °C, 1 h then methyl 4-amino-2-R2-benzoate, NEt3, room temp, 12 h or HATU, DMF, DIPEA, 5 min, then methyl 4-amino-2-R2-benzoate, DMF, room temp, 18 h; (iv) R1 = Bn, R2 = Me; BCl3, CH2Cl2, 0 °C then room temp, 12 h; (v) 1,1-di-tert-butoxy-N,N-dimethylmethanamine, toluene, 80 °C, 3 h, then room temp, 12 h; (vi) 1,1-di-tert-butoxy-N,N-dimethylmethanamine, toluene, 80 °C, 3 h, then further 1,1-di-tert-butoxy-N,N-dimethylmethanamine, 2 mol, added, 80 °C, 16 h. (vii) H2, Pd/C, MeOH, room temp; (viii) R1 = Et, R2 = MeO, R = tBu; BCl3, CH2Cl2, 0 °C then room temp 2 h).
Scheme 3.

4-(3,4,5-Trialkoxybenzamido)benzoic acids. (Reagents and conditions: (i) NaHCO3, R1I, DMF, 30 °C, 72 h; (ii) K2CO3, R2Br, DMF, 50 °C, 48 h; (iii) LiOH, THF, H2O, room temp, 16 h; (iv) (COCl)2, CH2Cl2, DMF, 0 °C, 1 h then methyl 4-amino-benzoate, NEt3, room temp, 12 h).
Scheme 4.

4-(3-Chloro-4,5-dialkoxybenzamido)benzoic acids with identical alkoxy groups. (Reagents and conditions: (i) BBr3, CH2Cl2, 0 °C, 2 h; (ii) TMSCl, MeOH, 50 °C, 16 h; (iii) K2CO3, RI, DMF, 70 °C, 46 h; (iv) LiOH, THF, H2O, room temp, 18 h; (v) (COCl)2, CH2Cl2, DMF, 0 °C, 1 h then methyl 4-amino-2-R1-benzoate, NEt3, room temp, 12 h).
Scheme 5.

4-(3-Chloro-4,5-dialkoxybenzamido)benzoic acids with non-identical alkoxy groups. (Reagents and conditions: (i) BBr3, CH2Cl2, 0 °C, 2 h; (ii) TMSCl, MeOH, 50 °C, 16 h; (iii) K2CO3, BnBr, DMF, 60 °C, 0.75 h; (iv) K2CO3, R2Br, DMF, 60 °C, 2 h; (v) H2, 10% Pd/C, MeOH; (vi) K2CO3, DMF, 60 °C, 10 min, then R3I, 40 °C, 3 h; (vii) LiOH, THF, H2O, 40 °C, 1 h, then room temp, 16 h; (viii) T3P, methyl 4-amino-2-R1-benzoate, NEt3, EtOAc, 60 °C, 4 h, then room temp, 16 h. (ix) LiOH, THF, H2O, 40 °C, 16 h).
Scheme 2.

4-(3,5-Dichloro-4-ethoxyphenylcarbamoyl)benzoic acid. (Reagents and conditions: (i) (COCl)2,CH2Cl2, DMF, 0 °C, then room temp 2 h; (ii) DIPEA, CH2Cl2, room temp, 16 h, then LiOH, THF, H2O, room temp, 16 h; (iii) K2CO3, EtI, DMF, 65 °C, 18 h, then further EtI, 70 °C, 3 h; (iv) LiOH, THF, H2O, room temp, 5 h).
For compound 16 the initial alkylation of 6 was carried out with benzyl bromide, and the resulting benzyloxy compound was hydrolyzed, coupled with the aniline 8 (R2 = H) and the benzyl group was removed using boron trichloride to result in compound 11 (R2 = H). This material was then alkylated using 1,1-di-tert-butoxy-N,N-dimethylmethanamine in toluene at 80 °C. A final hydrolysis using lithium hydroxide in a mixture of tetrahydrofuran and water gave the tertiary butoxy compound 16.
A similar sequence (Scheme 2) coupling the aniline 24 and acid chloride 23 (obtained from acid 22) followed by hydrolysis gave the phenolic acid 25 which upon alkylation with ethyl iodide followed by hydrolysis gave the reverse amide analog 26.
The 3,4,5-trialkoxybenzamido-benzoic acids 31–34 were prepared in four or five steps from methyl 3,4,5-trihydroxybenzoate 27 as illustrated in Scheme 3.
For the symmetrical tri-alkoxy compound 31, treatment of 27 with sodium hydrogen carbonate and ethyl iodide gave mainly compound 28 (R1 = Et) where alkylation had only occurred in the 4-position of the substrate. After purification, this compound on treatment with potassium carbonate and 2-bromopropane gave an intermediate compound where both remaining hydroxyl groups had reacted with the alkylating reagent. Hydrolysis resulted in the fully alkylated benzoic acid 29 (R1 = Et, R2 = R3 = iPr) which was coupled via the acid chloride to give the methyl ester of compound 31. A final hydrolysis using lithium hydroxide yielded compound 31. The other tri-alkoxy derivatives 32–34 were similarly prepared (Scheme 3).
The 3-chloro-4,5-dialkoxybenzamido benzoic acids 39–45 and 49–59 were prepared as described in Scheme 4, Scheme 5. The commercially available 3-chloro-4-hydroxy-5-methoxybenzoic acid 35 was treated sequentially with boron tribromide and trimethylsilyl chloride in methanol to leave methyl 3,4-dihydroxy-5-chlorobenzoate 36.
For the derivatives 39, 40, 42, 43, and 44, (Scheme 4) where the alkoxy groups are the same, both hydroxyl groups in 36 were alkylated by using potassium carbonate and the appropriate alkyl halide in N, N-dimethylformamide heated to 70 °C.
Hydrolysis gave rise to the fully substituted benzoic acids 37 (R = iPr), 37 (R = cyclobutyl) and 37 (R = cyclopentyl. These were then coupled to the aniline 38 via the acid chloride generated by treatment of the benzoic acid with oxalyl chloride.
The di-tert-butoxy derivatives 41 and 45 were synthesized from the acid 37 (R = tBu), which was prepared by reacting the two hydroxyl groups in 36 with N, N-dimethylformamide di-tert-butyl acetal followed by hydrolysis, and then coupling the product directly with aniline 38 using HATU. A final treatment of the coupled products with lithium hydroxide in aqueous 1,4-dioxane gave the required acids.
The non-identical di-alkoxy compounds 49–57 and 59 were also prepared via methyl 3,4-dihydroxy-5-chlorobenzoate 36, while 58 was prepared from 3-chloro-4-hydroxy-5-methoxybenzoic acid 35 via benzoic acid 48 (R2 = Me, R3 = Et) (Scheme 5).
On treatment of 36 with potassium carbonate and benzyl bromide, the 4-benzyloxy methyl ester 46 was produced. For 49 this was then alkylated with isopropyl bromide and base to give the 3-isopropoxy-4-benzyloxy compound 47 (R2 = iPr) which was hydrogenated, alkylated with ethyl iodide and base and hydrolyzed to give rise to the benzoic acid 48 (R2 = iPr, R3 = Et). This benzoic acid 48 was then coupled to the aniline 38 (R1 = H) using T3P in ethyl acetate and triethylamine as a base, followed by hydrolysis with lithium hydroxide to provide the final compound 49. The other non-identical di-alkoxy compounds 50–57 and 59 were similarly prepared via their corresponding benzoic acids 48 (Scheme 5).
3. Results and discussion
A ligand-based virtual screening approach, which ranks compounds by their similarity towards known active ligands, was adopted in a search for a novel chemical series of small molecule RARα agonists. The extended electron density representation offered by the Cresset XED force-field provides a way to characterize the calculated field around a molecule.14 The subsequent molecular comparison uses four different 3D fields, positive and negative charge, steric shape and hydrophobicity, and allows a complete 3D conformational analysis of compounds to be performed.15, 16 The crystal structure of the selective RARα antagonist BMS 195614 (1) in the human RARα active site17 was overlaid with AM 580 (2), the antagonist removed and the resulting complete assembly minimized to give the putative bioactive conformation of AM 580 (2). This procedure was also performed for AGN 193836 (3) to get its bioactive conformation. Molecular fields were added to each of these bioactive conformations (Fig. 2).
Fig. 2.
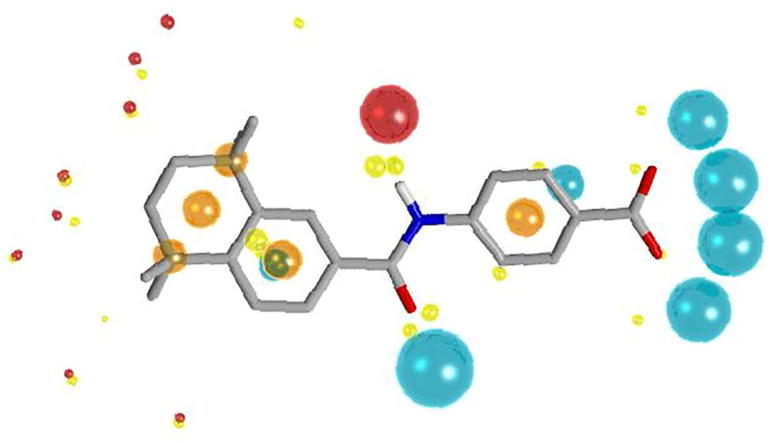
Cresset FieldScreen representation of bioactive conformation of AM580. (Blue field points (spheres) highlight energy minima for a positively charged probe, red for a negative probe. Yellow spheres represent an attractive van der Waals minima for a neutral probe and orange spheres represent hydrophobic centroids. Oxygen atoms are shown in red, nitrogen in blue. The size of the points is related to the strength of the interaction).
These unique molecular field patterns were used to search Cresset’s database of 2.5 M commercially available molecules, and the results ranked in similarity to the initial bioactive conformations (see Supplementary data for further details).
This methodology identified 3000 commercially available compounds as possible hit compounds. The 200 compounds that had the highest field overlays, Lipinski likeness, and synthetic tractability, were purchased. These were tested in transactivation assays at the RARα, β and γ receptors. Full dose-response curves were generated for each active agonist, and the potency of each compound was expressed as a ratio of its EC50 compared to that of reference ATRA EC50 value generated on each 96 well plate. This produced several potent hits, including the lead compound 5 (Table 1). The 3,5-dichloro-4-ethoxy derivative 5 was considered to be one of the better starting points for a lead optimization exercise, not only because of its potency as an RARα agonist but also because of its good selectivity over the RARβ and RARγ receptors, with moderate lipophilicity (cLog P = 4.4) compared to AGN 195183 (4) (cLog P = 7.2). In addition, it had no systematic Cyp450 liability (inactive at 25 μM at Cyp1A, 2C19, 2C9, 2D6 and 3A4 isoforms), and was not cytotoxic in COS-7 cells (i.e. showed <20% cell death @ 50 × EC50 at the RAR alpha receptors).
Table 1.

| Subtype-specific transactivationa |
|||||||
|---|---|---|---|---|---|---|---|
| Relative EC50b |
|||||||
| Compd | RO | RARα | RARβ | β/α ratioc | RARγ | γ/α ratioc | cLogPf |
| 4 | AGN195183 | 11 | 1564 | 141 | 9836 | 867 | 7.2 |
| 5 | EtO | 24 | 1917 | 79 | >3,00,000 | >12,500 | 4.4 |
| 12 | PrO | 15 | 139 | 9.5 | 1196 | 82 | 4.9 |
| 13 | BuO | 84 | 717 | 8.5 | 1477 | 18 | 4.6 |
| 14 | iPrO | 7d | 1417 | 205 | 823 | 119 | 4.7 |
| 16 | tBuO | 7 | 2927 | 426 | 6250 | 909 | 5.1 |
| 15 | 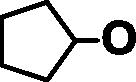 |
10 | 342 | 33 | 4703 | 452 | 5.3 |
| 26 | – | 30 | 355 | 12 | >1,08,000 | >3600 | 4.4 |
| 60 | H | 92d | 642 | 7 | 5000 | 55 | 4.2 |
| 61 | MeO | 30 | 9525 | 318 | 5850 | 195 | 3.9 |
| ATRA | 1.0(1.51 nM)e | 1.0(0.52 nM) e | 1.0(0.22 nM) e | ||||
Transactivation assays for the RAR alpha, beta and gamma receptors were performed using each of the mouse RAR ligand binding domains, Subtype-specific activity is expressed in terms of relative EC50 which is the concentration of retinoid required to produce 50% of the maximal observed response, normalised relative to that of ATRA.
Mean EC50 for each compound divided by the mean EC50 of ATRA. Values were obtained from three separate experiments. Errors in these assays are approximately 20% of the mean values.
The relative EC50 ratios of α to β and α to γ.
Compound behaves as a partial agonist relative to the amplitude of the normalizing ATRA output.
Mean of ATRA EC50 (nM).
cLog P values were calculated in ChemDraw.
Our aim was to increase the RARα potency and selectivity over RARβ while retaining the excellent selectivity over RARγ shown by 5 and achieve oral bioavailability in the rat. The target profile was RARα potency (RARα EC50/ATRA EC50 < 10) with a selectivity of 2 orders of magnitude over RARβ and 3 orders of magnitude over RARγ with an oral bioavailability of >35% in the rat.
Initial SAR showed that the three aromatic substituents in 5 seemed important for potency as the disubstituted, 3,5-dichloro derivative 60 was less potent at RARα and also less selective than the 3,5-dichloro-4-ethoxy derivative 5 at RARβ and RARγ. This helped focus our SAR on derivatives with a 3,4,5 substituted aromatic ring.
3.1. 4-Substituted derivatives
We initially concentrated on the 4-substituent (Table 1). Increasing the length of the 4-alkoxy substituent to n-propoxy 12 and n-butoxy 13 resulted in a loss of selectivity at RARβ and RARγ.
Increasing the bulk of the 4-alkoxy substituent to isopropoxy 14 and tert-butoxy 16 resulted in an increase in potency at RARα and an increase in selectivity over RARβ but a loss of selectivity at RARγ. In contrast, the cyclopentoxy compound, 15 was less selective than 5 at both RARβ and RARγ.
We also explored the reverse amide 26 of 5 which lost significant selectivity against RARβ when compared to 5 and hence further work on the reverse amides was curtailed.
We next investigated the PK profile of these 3,5-dichloro-4-alkoxy derivatives. We used intrinsic clearance figures in mouse and human microsomes as a simple in vitro screen to minimize the risk of Phase 1 metabolism, before progressing to in vivo studies. The PK profile of the 3,5-dichloro-4-alkoxy series of compounds was poor. The ethoxy 5, tert-butoxy 16 and cyclopentoxy 15 derivatives all had a high mouse, and moderate human intrinsic clearance and 15 was poorly orally absorbed with very low oral bioavailability in the rat (Table 2).
Table 2.
In vitro and in vivo PK.
| Compd | aLog D pH 7.4 | LEb | intrinsic Clintc |
rat pKd |
|||
|---|---|---|---|---|---|---|---|
| mouse (µL/min/mg protein) |
human (µL/min/mg protein) |
AUC po ng·min mL−1 |
Cl mL/kg/min |
F% | |||
| 5 | 1.7 | 0.45 | 127 | 18 | ND | ND | ND |
| 15 | 2.8 | 0.41 | 83 | 26 | 1674 | 2 | 0.3 |
| 16 | 2.6 | 0.45 | 91 | 16 | ND | ND | ND |
| 18 | 1.7 | 0.51 | 38 | 14 | 74,396 | 1.6 | 12 |
| 31 | 1.6 | 0.36 | 8 | 4 | 7,83,782 | 1 | 81 |
| 39 | 2.6 | 0.43 | 31 | – | 43,569 | 10 | 39 |
| 49 | 1.7 | 0.47 | 41 | 11 | – | – | – |
| 51 | 1.0 | 0.44 | 9 | 12 | 50,940 | 3 | 13 |
Measured by octanol/buffer shake flask method at pH 7.4 (see Supplementary data file for details).20
LE values were calculated by LE = (RT ln Kd)/N, presuming EC50 ≈ Kd.18
Intrinsic clearance Clint data for screening purposes only: Mouse and Human microsomes were incubated with the test compound at 37 °C in the presence of the co-factor, NADPH. The data is the mean of 5 separate experiments. Compound disappearance monitored over 45 min period. SEM is less than 10% of the mean values.
Rat PK (n = 4): AUC (ng·min mL−1) at 10 mg/kg, 8% Ethanol/92% PEG-400 formulation, Cl in mL min−1 kg−1. ND = not determined.
3.2. 3,5-Disubstituted derivatives
To overcome these difficulties we turned our attention to the 3,5-sustituents in 5. The patent analysis in this class of compounds showed that non-alkyl substituents in the 3,4,5-substituted aromatic ring of 5 appeared novel. With this in mind we analysed the medicinal chemistry parameters of the 3,5-substituents of our initial 4-OEt derivatives containing non-alkyl 3,5-substituents 5, 62, 63 and 3,5-dialkyl substituents 64 (Table 3). Ranking these four derivatives in terms of RARα potency against the properties of the 3,5 substituents in the second aromatic ring, such as size (MR), lipophilicity (π) and electronic resonance (σ) (Table 3), shows that potency only increases with the lipophilicity π of the 3,5-sustituents (and not with the size or resonance effects of these substituents).
Table 3.
A search of possible aromatic substituents showed that the isopropoxy group has a similar lipophilicity to a chlorine/bromine atom found in 5/63 and a similar size to a tert-butyl found in the more potent derivative 64. This suggested that the 3,5-diisopropoxy derivative 31 should be at least as active as the chloro and bromo derivatives 5 and 63, and why the 3,5-diethoxy analog 62 which is the least lipophilic, is the least active.
3.3. 3,4,5-Trialkoxy and 3,4,-dialkoxy derivatives
Encouragingly 31 proved to have good RARα potency (Table 3). In addition, 31 has high selectivity over RARβ and RARγ (Table 4), and low mouse and human intrinsic clearance with excellent oral absorption and bioavailability (81%) in the rat (Table 2), although it was shown to be only a partial RARα agonist. The close profile of 5 and 31 in terms of RARα potency, as well as RARβ and RARγ selectivity, shows that in this case, the iPrO group is a good bioisostere of the Cl group. This led the project away from the 3,5-dichloro template and enabled exploration of the alkoxy derivatives at these positions which give a lipophilic surface without the high lipophilicity of the similar sized tertiary butyl group seen in 64, making the template more drug-like. Further analogs of this trialkoxy template 31 were investigated in an attempt to increase its alpha potency while maintaining the excellent beta and gamma selectivity as well as its good PK profile. Increasing the size of the 3,5-substituents in 31 to give the di-cyclopentoxy derivative 32 or increasing the size of the 4-substituents to give 33 maintained the good RARα potency and RARβ selectivity but lost selectivity against RARγ (Table 4). Decreasing the size of both the 3- and 5-isopropoxy groups to give the 3,4,5-triethoxy derivative 62, resulted in a substantial loss of RARα potency (Table 3). In addition 31, 32 and 33 all exhibited some partial agonist activity at RARα. However a close analog the 3,4-diethoxy-5-isopropoxy derivative 34 showed that it was possible to have full RARα agonist properties with trialkoxy derivatives (Table 4).
Table 4.
| Subtype-specific transactivationa |
||||||||
|---|---|---|---|---|---|---|---|---|
| Relative EC50b |
||||||||
| Compd | R1O | R2O | R3 | RARα | RARβ |
β/α ratioc |
RARγ |
γ/α ratioc |
| 62 | EtO | EtO | EtO | 368 | 64,148 | 174 | 5882 | 16 |
| 31 | iPrO | EtO | iPrO | 26d | 4560 | 175 | 56,900 | 2190 |
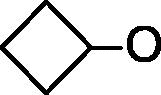
| 32 | 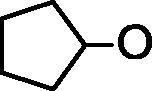 |
EtO | 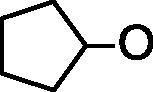 |
29d | 4200 | 145 | 550 | 19 |
| 33 | iPrO | iPrO | iPrO | 27d | 2600 | 96 | 225 | 8 |
| 34 | iPrO | EtO | EtO | 29 | 2450 | 84 | 960 | 34 |
| 49 | iPrO | EtO | Cl | 0.7d | 103 | 150 | 8083 | 11,721 |
| 50 | 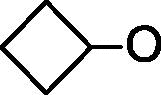 |
EtO | Cl | 1.0d | 115 | 115 | 1706 | 1706 |
| 39 | iPrO | iPrO | Cl | 1.7 | 89 | 54 | 1386 | 838 |
| 40 | 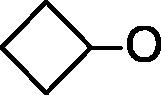 |
 |
Cl | 2.4d | 53 | 22 | 1059 | 447 |
| 41 | tBuO | tBuO | Cl | 0.9 | 38 | 44 | 162 | 189 |
| 42 | 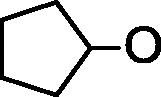 |
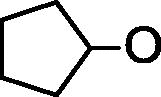 |
Cl | 1.7d | 55 | 32 | 571 | 336 |
| 51 | iPrO | MeO | Cl | 5.3 | 1500 | 283 | 10,833 | 2043 |
| 52 | EtO | iPrO | Cl | 2.1 | 7.1 | 3.1 | 1202 | 570 |
| ATRA | 1.0(1.51 nM)e | 1.0(0.52 nM)e | 1.0(0.22 nM)e | |||||
Transactivation assays were performed using the RAR alpha, beta and gamma receptors containing each of the mouse RAR ligand binding domains. Subtype-specific activity is expressed in terms of relative EC50 which is the concentration of retinoid required to produce 50% of the maximal observed response, normalised relative to that of ATRA.
The relative EC50 is the mean EC50 for each compound divided by the mean EC50 of ATRA. Values were obtained from three separate experiments. Errors in these assays are approximately 20% of the mean values.
The relative EC50 ratios of α to β and α to γ.
Compound behaves as a partial agonist relative to the amplitude of the normalizing ATRA output.
Mean of ATRA EC50 (nM).
This unsymmetrical derivative was further exploited by the investigation of a series of 3,4-alkoxy derivatives (Table 4). Replacing one of the isopropoxy groups in the lead 31 with a chloro atom gave the chloro-dialkoxy derivative 49 which had increased potency at RARα and also maintained the excellent selectivity at RARβ and RARγ. However, this compound was also only a partial agonist at RARα.
Increasing the size of the 3-isopropoxy in 49to 3-cyclobutyl in 50 gave no change in profile. However, increasing the 4-ethoxy group in 49 to the 4-isopropoxy in 39 gave a similar level of potency at RARα as a full agonist. The molecule was also an order of magnitude more potent than 31 at RARα while maintaining excellent selectivity at RARγ with moderate selectivity at RARβ. In addition, the di-isopropoxy derivative 39 was orally well absorbed in the rat with a bioavailability of 39% (Table 2). Thus 39 satisfied our target profile except for selectivity at RARβ. Increasing the size of the alkoxy groups to the di-cyclobutyl in 40, di-tert-butyl in 41 and di-cyclopentyl in 42 maintained potency at RARα, but decreased selectivity at RARβ and RARγ. Interestingly reducing the size of the 4-ethoxy in 49 to 4-methoxy in 51 gave a full agonist with good RARα potency and selectivity at RARβ and RARγ. However, it had a low oral bioavailability (13%) in the rat (Table 2).
3.4. Substitution of the benzoic acid ring
Ortho-fluoro substitution in the benzoic acid ring of the bicyclic 5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalene analogs which lead to AGN 195183 (4)10has been shown to increase RARα binding potency and increase selectivity over RARβ and RARγ in the transactivation assay.
Based on this precedent, a series of ortho-substituted benzoic acid derivatives of our initial lead template 5 were prepared (Table 5). While the ortho-fluoro substitution product 17 maintained potency and selectivity, the ortho-methyl substitution product, 18 improved RARα potency 20-fold and maintained good RARβ and RARγ selectivity. In addition, 18 had a lower mouse and human intrinsic clearance, as well as a somewhat improved bioavailability (12%) in the rat (Table 2), compared to the unsubstituted benzoic acid derivative 5. Compounds 19, 20 and 21 with larger substituent groups, either lost RARα potency or RARβ/RARγ selectivity compared to 5. As a result of these findings, a series ortho-methyl and ortho-fluoro substituted benzoic acid derivatives of the 3,4-dialkoxy-5-chloro template were prepared (Table 6). The initial trend from the 5 series (Table 5) was also seen in the 3,4-dialkoxy-5-chloro series (Table 6).
Table 5.
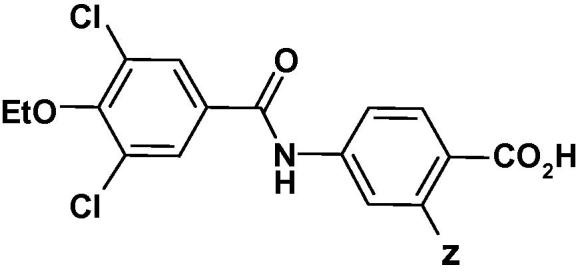
| Compd | Z | RARα rel EC50a |
β/α ratiob |
γ/α ratiob |
|---|---|---|---|---|
| 5 | H | 24 | 80 | >12,500 |
| 17 | F | 25 | 80 | >80 |
| 18 | CH3 | 0.9 | 82 | 151 |
| 19 | OH | 33c | 37 | 149 |
| 20 | Cl | 151 | 64 | >50 |
| 21 | CF3 | 1.7c | 1.5 | 11 |
a,b and c see Table 1.
Table 6.
| Potency and Selectivity |
PK |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compd | R1O | R2O | R3 | RARα rel EC50a |
β/α ratio b |
γ/α ratiob |
LogDpH 7.4d | intrinsic Clinte |
rat pof |
|||
| mouse | human | AUC | Cl | F% | ||||||||
| 53 | iPrO | MeO | F | 6.7 | 609 | 17,600 | 1.0 | 11 | 0.3 | 10,871 | 15 | 15 |
| 54 | iPrO | EtO | F | 1.1 | 487 | 6169 | 1.4 | 17.9 | 3.3 | 15,376 | 12 | 12 |
| 43 | iPrO | iPrO | F | 2.7 | 34 | 300 | 2.0 | 18.8 | 6.9 | – | – | – |
| 55 | iPrO | MeO | Me | 4.7c | 241 | 509 | 1.3 | 10.6 | 1.5 | – | – | – |
| 44 | iPrO | iPrO | Me | 0.97 | 45 | 2947 | 2.3 | 26.2 | 15 | 77,670 | 5.4 | 66 |
| 56 | iPrO | EtO | Me | 1.6 | 200 | 11,000 | 1.8 | 37.6 | 5.3 | 70,765 | 7 | 40 |
| 57 | EtO | iPrO | Me | 7 | 286 | 1675 | 1.9 | 25.7 | 8.7 | – | – | – |
| 58 | MeO | EtO | Me | 33 | 210 | >250 | – | 18.4 | – | – | – | – |
| 59 | 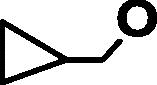 |
EtO | Me | 2.6 | 58 | 38,461 | – | 42.3 | – | – | – | – |
| 45 | tBuO | tBuO | Me | 0.64 | 13,200 | 128 | 2.9 | 36.2 | 28.7 | – | – | – |
The ortho-fluoro substituted derivatives 53, 54 and 43 (Table 6) maintained RARα potency and selectivity compared to their corresponding unsubstituted derivatives 51, 49 and 39 and, in addition, had a lower mouse and human intrinsic clearance with the latter being in single figures. However, although both 53 and 54 met our target profile in terms of high RARα potency with a selectivity of 2 orders of magnitude over RARβ and 3 orders of magnitude over RARγ, they both had low oral bioavailability (15% and 12% respectively) in the rat. The ortho-methyl substituted derivative 55 had a similar mouse and lower human intrinsic clearance (Table 6) compared to the unsubstituted derivative 51 (Table 2). However, it had only partial RARα agonist activity. Both the ortho-methyl derivatives 56 and 44 had good bioavailability (≥40%) in the rat and lower mouse intrinsic clearance (Table 6) compared to the unsubstituted derivatives 49 and 39 (Table 2), with 56 having the lowest (single figure) human intrinsic clearance of these four derivatives.
While both derivatives 56 and 44 had high RARα potency and good bioavailability 56 was superior in terms of selectivity at RARβ (2 orders of magnitude) and RARγ (4 orders of magnitude) and possessed a better overall potency, selectivity and PK profile than the other analogs 45, 57–59 shown in Table 6.
The 3-OEt, 4-OiPr geometrical isomer 57 was less potent and less selective at RARγ than 56 which is analogous to the trend seen with compounds 51 and 49 in the unsubstituted benzoic acid series. This emphasizes the need for a more lipophilic group than OEt in the 3-and 5-position in this template which was initially seen in Table 3. Thus the 3-OiPr, 4-OEt derivative 56 reached our target profile in terms of potency, selectivity, and oral bioavailability.
The excellent RARα potency, good RARβ and RARγ selectivity and PK profile of the full agonist 56 suggested further investigations to see if it had sufficient drug-like properties to be an orally bioavailable, highly potent and selective RARα agonist with therapeutic potential.
3.5. Predevelopment studies of 4-(3-chloro-4-ethoxy-5-isopropoxybenzamido)-2-methylbenzoic acid 56
3.5.1. ADME profiling
Predevelopment ADME studies revealed that 56 has a good Cyp 450 profile with no significant inhibition IC50 > 25 μM against five Cyp 450 isozymes (1A2, 2C9, 2C19, 2D6, 3A4), and has a human and mouse plasma protein binding of 93% and 91% respectively.20
Compound 56 has also been examined by CEREP in a panel of 120 other receptors, channels and enzymes. The compound at 10 μM demonstrated no significant interactions with any of the sites examined leaving a window of some 4 orders of magnitude between its actions at RAR and any non-RAR site.21 The highest inhibition of 25% was found for the 5HT2B site. To exclude potential cardiovascular side effects, compound 56 was tested in vitro on the cardiac hERG channel and did not show any significant binding to hERG up to the concentration of 10 μM.20
3.5.2. Hepatocyte stability
We initially used a microsomes assay as a screen to rank order compounds of interest in terms of their metabolic stability. As microsomes only contain phase I metabolising enzymes it was of interest to screen our lead compound 56 in a secondary screen using hepatocytes which contain the full complement of drug metabolising enzymes present in the liver.
The metabolic stability of compound 56 was tested at two concentrations (1 μM and 30 μM) in mouse, rat, dog, Cynomolgus monkey and human cryopreserved hepatocytes. The compound was shown to be stable, with a long t½ and low clearance in all species (Table 7), which correlates with the available PK data (Table 8).
Table 7.
Stability of 56 in hepatocytes.
| Conc (μM) | Species | Half-life (minutes) a | Clint (μl/min/million cells) |
|---|---|---|---|
| 1 |
Mouse | 224 | 12 |
| Rat | 357 | 4 | |
| Dog | >450 | <3 | |
| Cynomolgus monkey | >450 | <3 | |
| Human |
>450 |
<3 |
|
| 30 | Mouse | >300 | <9 |
| Rat | >450 | <3 | |
| Dog | >450 | <3 | |
| Cynomolgus monkey | >450 | <3 | |
| Human | >450 | <3 |
Data are expressed as mean values (n = 2). For assay details see Supplementary data file.
Table 8.
Pharmacokinetic Profiles of 56 in Mice and Dogs.a
| iv |
po |
|||||
|---|---|---|---|---|---|---|
| Species | Cl (mL/h/kg) | Vss (mL/kg) | t1/2 (h) | Cmax (ng/mL) | Tmax (h) | F (%) |
| miceb | 4.7 | 0.3 | 1.9 | 2007 | 0.25 | 84 |
| dogc | 2.3 | 0.66 | 9.2 | 2050 | 0.5 | 83 |
Administered at a dose of 1 mg/kg by both iv and po routes in mice. Administered at a dose of 0.5 mg/kg, iv, 1 mg/kg, po, in dogs. Vehicle = 2% DMSO in 0.05 M phosphate buffered saline, pH 7.4. Data are expressed as mean values (mice, n = 3. dogs n = 3).
C57 mice. For assay details see Supplementary data file.
Beagle dogs. For assay details see Supplementary data file.
3.5.3. PK profile in mice and dogs
The PK profile of 56 was also studied in mice and dogs (Table 8). Compound 56 showed low plasma clearance (Cl) and low volume of distribution (Vss), resulting in sustained plasma half-lives in each species (iv t1/2: mice, 1.9 h; dog, 9.2 h). In addition, oral administration of 56 exhibited high bioavailabilities >80% in both mice and dogs. These results encouraged us to investigate 56 further as a predevelopment candidate.
3.5.4. Human RAR alpha receptor
As we planned to perform PK and further in vivo evaluation in rodents, we initially used the corresponding in vitro transcriptional transactivation assays with gal4 fusion receptor constructs, created using each of the mouse RAR ligand-binding domains. Although the percentage identity of amino acid sequences between the mouse and human RAR ligand-binding domains of all three RAR types (α,β or γ) is 99–100%,23 we thought it prudent to confirm the activity and selectivity of our lead compound 56 against the human RAR ligand-binding domains in a transcriptional transactivation assay before further predevelopment studies were investigated. We also tested an earlier less active analog 15 from the 3,5-dichloro template, and AM 580 (2) for comparison (Table 9).
Table 9.

| Property | 56 |
15 |
AM580(2) |
ATRA |
||||
|---|---|---|---|---|---|---|---|---|
| mousea | humanb | mousea | humanb | mousea | humanb | mouse | human | |
| RARα rel EC50 | 1.6 | 0.59 | 10.4 | 8.1 | 0.02 | 0.13 | 1.0 (1.51 nM)c | 1.0 (1.01 nM)d |
| Selectivity β/α ratio | 200-fold | 290-fold | 33-fold | 289-fold | 1130-fold | 162-fold | 0.34-fold | 0.33-fold |
| Selectivity γ/α ratio | 11,000-fold | >13,000-fold | 452-fold | 2322-fold | 826-fold | 505-fold | 0.15-fold | 0.11-fold |
See Table 1.
Transactivation RAR human assay. For details see Supplementary data file.
Mean of ATRA EC50 (nM) mouse assay RARα.
Mean of ATRA EC50 (nM) human assay RARα.
There is a good correspondence for RARα potency between human vs mouse for 56 and 15 with the human being slightly more potent, in contrast to the RARα potency for AM 580 (2) where the human is less potent than the mouse (Table 9). Similarly, the α vs β selectivity comparison for 56 and 15, shows that the human is more selective than the mouse, while for AM 580 (2) the human is less selective than the mouse. Also, α vs γ selectivity for 56 is 4 orders of magnitude compared to AM 580 (2) where it is only 2 orders of magnitude for both human and mouse.
3.5.5. In vitro toxicology
In common with most of the other compounds in the series, the lead compound 56 showed no cytotoxicity in COS-7 cells at a 50-fold multiple of its EC50.20 When examined in a high content cell toxicity screen in HEPG2 cells (Cyprotex), 56 was found to have no effect at concentrations up to 50 μM on cell or mitochondrial viability markers.20 This is in contrast to the more lipophilic molecule AM 580 (2) which caused a significant increase in cell membrane permeability and a significant decrease in mitochondrial membrane potential at concentrations between 10 and 30 μM.
When 56 was examined for genetic toxicity, it was negative in bacterial cytotoxicity tests up to 100 μM, negative in an Ames test in three bacterial strains and in an in vitro micronucleus test in CHO-K1 cells, in all cases in both the presence and absence of S9.21 In the absence of S9 it should be noted that AM 580 (2), the reference RARα agonist has been shown by others to be a mutagen in vitro.11, 12
3.5.6. Ease of synthesis
The 4-(3-chloro-4-ethoxy-5-isopropoxybenzamido)-2-methylbenzoic acid 56 can be synthesized in 9 high yielding reaction steps from 3-chloro-4-hydroxy-5-methoxybenzoic acid (35) (Scheme 5). It is available as a stable highly crystalline, non-hygroscopic, white powder with a melting point of 186 °C, and with a solubility of >5 mg/mL, as the sodium salt in water at 35 °C.
3.5.7. Profile of lead compound 56
The 3-OiPr, 4-OEt, 5-Cl ortho methyl benzoic acid derivative 56 met our target profile in terms of high RARα agonist potency with a high degree of selectivity over RARβ (of 2 orders of magnitude) and excellent selectivity over RARγ (4 orders of magnitude) at both the mouse and human receptors. It has high levels of potency in the RARα binding assay (IC50) showing that the transactivation activity observed was being mediated through the alpha receptor (Table 10). As expected 56 was also selective vs RXR (IC50 > 10 μM in human RXR α and β binding assays).22 It also possesses good drug-like properties, a low human intrinsic clearance (5.3 µL/min/mg protein) in microsomes and a measured Log D = 1.8, which resulted in good oral exposure with low clearance and good bioavailability (40%) in the rat (Table 10). In contrast, both 15 and 2 have human intrinsic clearance in double figures and a higher Log D = 2.8, which resulted in low oral exposure in the rat with low bioavailability (0.3%) for 15. Compound 56 was also shown to be metabolically stable to hepatocytes with a long t½ and low clearance in human and 4 animal species (Table 7) together with a high bioavailability (>80%) in both mice and dogs with low plasma clearance (CL) and a sustained plasma half-live (iv t1/2: mice, 1.9 h; dog, 9.2 h) (Table 8). In addition 56 has a solubility of >5 mg/mL as the sodium salt, no systematic Cyp 450 liability against five isoforms (1A2, 2C9, 2C19, 2D6, 3A4) and demonstrated no inhibition (at 10 μM) in a binding assay for hERG channels. It was not cytotoxic in COS-7 cells and was negative for genetic toxicity in the Ames test and micronucleus test in CHO-K1 cells.
Table 10.
Comparison of the RARα Agonist Potency, selectivity versus the RARβ and RARγ Human and Mouse Receptors, Human Intrinsic Clearance and Pharmacokinetic Profile in Rat for 56 and 15.
| Binding Activity |
Agonist Potency and Selectivity |
PK |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd | RARα rel IC50a |
RARα rel EC50 bm/chu |
β/α ratio bm/chu |
γ/α ratio bm/chu |
intrinsic Clintd | rat poe |
f Log D pH 7.4 |
||
| human | AUC | Cl | F% | ||||||
| 56 | 3.6 | 1.6/0.6 | 200/298 | 11,000/>13,000 | 5.3 | 70,765 | 7 | 40 | 1.8 |
| 15 | 115 | 10.4/8.1 | 33/289 | 452/2322 | 26 | 1674 | 2 | 0.3 | 2.8 |
| AM 580(2) | 9 | 0.02/0.13 | 1130/162 | 826/505 | 15.6 | – | – | – | 2.8 |
RARα binding assay. The relative IC50 is the mean IC50 for each compound divided by the mean IC50 of ATRA (IC50 = 0.6 nM). Values were obtained from three separate experiments.
m = mouse receptor, see Table 1.
hu = human receptor, see Table 9.
Human microsomes Clint (µL/min/mg protein).
AUC po ng·min mL−1,Cl mL/kg/min.
Log D see Table 2.
4. Conclusions
We have used a ligand-based virtual screening exercise based on the bioactive conformation of AM 580 (2) and AGN 193836 (3) to identify the novel, less lipophilic RARα agonist 4-(3,5-dichloro-4-ethoxybenzamido) benzoic acid 5, which has good selectivity over the RARβ, and RARγ receptors. Analysis of the medicinal chemistry parameters of the 3,5-substituents of derivatives of template 5 showed that RARα potency is driven by the lipophilicity of these substituents. It showed that the iPrO group is a good bioisostere of the Cl group in this case and that the 4′-(3,5-diisopropoxy-4-ethoxybenzamido)benzoic acid derivative 31 has a close profile to 5 in terms of RARα potency as well as RARβ and RARγ selectivity. The low mouse and human intrinsic clearance with excellent oral absorption and bioavailability (81%) in the rat shown by 31 led to the exploration of the more drug-like branched dialkoxy derivatives, the best of which was the 4-(3-chloro-4,5-diisopropoxybenzamido)benzoic acid derivative 39 which was an order of magnitude more potent than 31 at RARα, while maintaining excellent selectivity over RARγ with moderate selectivity at RARβ and was orally well absorbed in the rat with a bioavailability of 39%. Substitution at the ortho-position of benzoic acid 5, with a range of groups, has shown that methyl groups are the best at increasing potency while maintaining good RARβ and RARγ selectivity. Methyl substitution at the ortho-position of the 4′-benzoic acid ring of a series of 4′-(3-chloro-4,5-dialkoxybenzamido)benzoic acid derivatives gave the novel RARα agonist 4-(3-chloro-4-ethoxy-5-isopropoxybenzamido)-2-methylbenzoic acid 56 as the best in terms of RARα agonist potency and selectivity versus RARβ (2 orders of magnitude) and RARγ (4 orders of magnitude) at both the human and mouse RAR receptors. This potent RARα-specific agonist with improved physicochemical properties also has high bioavailability (>80%) in both mice and dogs with a good PK profile and drug-like properties and was shown to be negative in the cytotoxicity and genotoxicity screens warranting further consideration as a potential therapeutic agent.
5. Experimental procedures
All starting materials and solvents, as well as compounds 5, 60, 61 and 62, were obtained from commercial sources. Hydrogenations were performed either on a Thales H-cube flow reactor or with a suspension of the catalyst under a balloon of hydrogen. Microwave reactions were carried out on a Personal Chemistry Smith Synthesizer Workstation with a 300 W single mode microwave cavity. Ion exchange chromatography was performed using strong cation exchange resin (SCX) cartridges purchased from Sigma-Aldrich and washed with methanol prior to use. The reaction mixture to be purified was first dissolved in methanol and then loaded directly onto the SCX and washed with methanol. The desired material was then eluted by washing with 1% NH3 in methanol. Silica gel column chromatography was performed using Silicycle pre-packed silica (230–400 mesh, 40–63 μM) cartridges. Preparative HPLC was carried out using a Gilson HPLC and an Agilent 5 µm Prep-C18 21.2 × 50 mm column. Detection was achieved using a UV detector at 254 nm. Mobile phase A: 0.1% aqueous formic acid, Mobile phase B: 0.1% formic acid in methanol. A flow rate of 40 mL/min was used and a gradient employed as follows; 0.0–0.8 min 5% B; 0.8–7.3 min 5–95% B; 7.3–8.3 min 95% B; 8.3–8.4 min 95–5% B. Analytical LCMS was performed using an Agilent 1200 HPLC and mass spectrometer system with a Scalar 5 µm C18 4.6 × 50 mm column and peaks detected by positive or negative ion electrospray ionization and a UV detector at 254 nm. All tested compounds were found to be of ≥95% purity using analytical LCMS. 1H and 13C NMR spectra were recorded using a Bruker Avance III TM 400 spectrometer at 400 and 110 MHz respectively, using either residual non-deuterated solvent or tetramethylsilane as a reference in the various solvents specified. All animal studies were ethically reviewed and carried out in accordance with the United Kingdom Animals (Scientific Procedures) Act 1986 by CXR Biosciences Ltd, James Lindsay Place, Dundee Technopole, Dundee DD 5JJ.
5.1. Chemistry
5.1.1. 4-(3,5-Dichloro-4-(cyclopentyloxy)benzamido)benzoic acid (15)
Step (i): Methyl 3,5-dichloro-4-(cyclopentyloxy)benzoate Methyl 3,5-dichloro-4-hydroxybenzoate 6 (1.00 g, 4.52 mmol) was dissolved in N, N-dimethylformamide (8 mL) and treated with bromocyclopentane (534 µL, 4.98 mmol), followed by potassium carbonate (937 mg, 6.79 mmol). The mixture was stirred at 80 °C for 3 h and then partitioned between ethyl acetate (100 mL) and water (100 mL). The aqueous phase was extracted with ethyl acetate (50 mL) and the combined organic phases washed successively with water (5 × 50 mL) and brine (50 mL), then dried over magnesium sulfate and filtered. The solvent was removed in vacuo to afford methyl 3,5-dichloro-4-(cyclopentyloxy) benzoate (1.10 g, 84% yield). 1H NMR (400 MHz, CDCl3) δ 7.97 (2H, s), 5.04 (1H, m), 3.90 (3H, s), 2.04–1.91 (4H, m), 1.82–1.75 (2H, m), 1.69–1.60 (2H, m).
Step (ii): 3,5-Dichloro-4-(cyclopentyloxy)benzoicacid (7: R1 = cyclopentyloxy). Methyl 3,5-dichloro-4-(cyclopentyloxy)benzoate (1.05 g, 3.63 mmol) and lithium hydroxide (174 mg, 7.26 mmol) were combined in tetrahydrofuran (10 mL), and water (1.5 mL) was added dropwise until a solution formed. The resultant mixture was stirred at room temperature for 12 h. The tetrahydrofuran was removed in vacuo and the residue acidified using aqueous 1 M hydrochloric acid. The resultant precipitate was filtered to afford 3,5-dichloro-4-(cyclopentyloxy)benzoic acid (7: R1 = cyclopentyloxy), (820 mg, 82% yield). 1H NMR (400 MHz, DMSO‑d6) δ 8.10 (2H, s), 5.03 (1H, m), 2.04–1.91 (4H, m), 1.82–1.75 (2H, m), 1.69–1.60 (2H, m).
Step (iii): Methyl 4-[3,5-dichloro-4-(cyclopentyloxy)benzamido]benzoate (10: R1 = cyclopentyloxy, R2 = H, R = Me). A solution of (7: R1 = cyclopentyloxy) (100 mg, 363 µmol) in dichloromethane (5 mL), cooled to 0 °C, was treated with oxalyl chloride (63.6 µL, 727 µmol), followed by a drop of N, N-dimethylformamide. The resultant mixture was stirred for 1 h at room temperature. The solvent was evaporated in vacuo and the residue dissolved in dichloromethane (5 mL) and then treated with a solution of methyl 4-aminobenzoate (8: R2 = H, R = Me) (54.9 mg, 363 µmol) and di-isopropylethylamine (190 µL, 1.09 mmol) in dichloromethane (5 mL). The reaction mixture was stirred for12 h at room temperature and then partitioned between dichloromethane (20 mL) and aqueous 1 M hydrochloric acid (20 mL). The phases were separated, and the organic phase was washed successively with water (2 × 20 mL), and brine (20 mL), dried over magnesium sulfate, filtered and then the solvent was removed in vacuo. The residue was purified by silica gel chromatography (12 g, 0–100% ethyl acetate/isohexane) to afford methyl 4-[3,5-dichloro-4-(cyclopentyloxy)benzamido]benzoate (10: R1 = cyclopentyloxy, R2 = H, R = Me), (30 mg, 20% yield). 1H NMR (400 MHz, CDCl3) δ 8.06 (2H, d, J = 8.8 Hz,), 7.85 (1H, br s), 7.82 (2H, s), 7.71 (2H, d, J = 8.8 Hz), 5.07–5.03 (1H, m), 3.92 (3H, s), 2.10–1.90 (4H, m), 1.85–1.70 (2H, m), 1.70–1.60 (2H, m).
Step (iv): 4-[3,5-Dichloro-4-(cyclopentyloxy)benzamido]benzoicacid (15). Compound (10: R1 = cyclopentyloxy, R2 = H, R = Me), (30.0 mg, 73 µmol) and lithium hydroxide (3.5 mg, 0.147 mmol) were combined in tetrahydrofuran (3 mL) and water was added dropwise until a solution formed. The resultant mixture was stirred at room temperature for 16 h. The tetrahydrofuran was removed in vacuo and the residue acidified using aqueous 1 M hydrochloric acid. The resultant precipitate was filtered to afford 4-[3,5-dichloro-4-(cyclopentyloxy)benzamido]benzoic acid 15 (15.0 mg, 51% yield) as a white solid. 1H NMR (400 MHz, DMSO‑d6) δ 12.77 (1H, s), 10.58 (1H, s), 8.07 (2H, s), 7.93 (2H, d, J = 8.8 Hz,), 7.88 (2H, d, J = 8.8 Hz), 5.06–5.01 (1H, m), 1.90–1.60 (8H, m). m/z 392 (M−H)− (ES−).
The compounds 12–14, 17, 20, 21, 63 and 64 were similarly prepared as 15: see Supplementary data for experimental and spectroscopic details.
5.1.2. 4-(4-(tert-Butoxy)-3,5-dichlorobenzamido)benzoic acid (16)
Step (i): Methyl 4-(benzyloxy)-3,5-dichlorobenzoate. Crude methyl 4-(benzyloxy)-3,5-dichlorobenzoate (16.9 g) was prepared from methyl 3,5-dichloro-4-hydroxybenzoate (6) (10 g, 45.2 mmol) and benzyl bromide (15.5 g, 90 mmol) using a procedure essentially the same as in step (i) for 15, except that the mixture was stirred at room temperature for 18 h. The crude product was partially purified by silica gel chromatography (330 g, 0–10% EtOAc/isohexane) to afford a white solid. The material was used in the next step without further purification.
Step (ii): 4-(Benzyloxy)-3,5-dichlorobenzoicacid (7: R1 = CH2Ph). 4-(Benzyloxy)-3,5-dichlorobenzoic acid (7: R1 = CH2Ph) (12.8 g, 96% over 2 steps) was prepared from crude 4-(benzyloxy)-3,5-dichlorobenzoate (16.9 g) using a procedure essentially the same as in step (iv) for 15: 1H NMR (400 MHz, DMSO‑d6) δ 7.88 (2H, s), 7.56–7.48 (2H, m), 7.44–7.37 (3H, m), 5.05 (2H, s). m/z 295 (M−H)− (ES−).
Step (iii): Methyl 4-(4-(benzyloxy)-3,5-dichlorobenzamido)benzoate (10: R1 = CH2Ph, R2 = H, R = Me). Methyl 4-(4-(benzyloxy)-3,5-dichlorobenzamido)benzoate (10: R1 = CH2Ph, R2 = H, R = Me) (9.81 g, 51%) was prepared from 4-(benzyloxy)-3,5-dichlorobenzoic acid (7: R1 = CH2Ph) (12.8 g, 43.2 mmol) using a procedure essentially the same as in step (iii) for 15, except the crude product was crystallized from isohexane/EtOAc to afford the product as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.07 (2H, d, J = 8.8 Hz), 7.84 (2H, s), 7.73 (2H, d, J = 8.8 Hz), 7.59–7.52 (2H, m), 7.44–7.36 (3H, m), 5.13 (2H, s), 3.92 (3H, s). m/z 428 (M−H)− (ES−).
Step (iv): Methyl 4-(3,5-dichloro-4-hydroxybenzamido)benzoate(11: R2=H). A solution of methyl 4-(4-(benzyloxy)-3,5-dichlorobenzamido)benzoate (10: R1 = CH2Ph, R2 = H, R = Me) (8.8 g, 20.5 mmol) in DCM (500 mL) was cooled to 0 °C and treated dropwise with boron trichloride (20.5 mL, 20.5 mmol, 1 M in DCM). The mixture was then allowed to stir at room temperature for 12 h. The mixture was cooled in an ice bath then quenched by addition of water (150 mL). The resultant mixture was partitioned between EtOAc (200 mL) and H2O (100 mL). The aqueous phase was extracted with EtOAc (2 × 75 mL) and the combined organic phases washed successively with water (50 mL) and brine (50 mL), then dried over MgSO4 and filtered. The solvent was removed in vacuo. The residue was crystallized from isohexane/EtOAc to afford methyl 4-(3,5-dichloro-4-hydroxybenzamido)benzoate (11: R2 = H) (5.81 g, 84%): 1H NMR (400 MHz, DMSO‑d6) δ 11.06 (1H, s), 10.52 (1H, s), 8.06 (2H, s), 8.04–7.91 (4H, m), 3.88 (3H, s). m/z 338 (M−H)− (ES−).
Step (v): Methyl 4-(4-(tert-butoxy)-3,5-dichlorobenzamido)benzoate. A stirred suspension of methyl 4-(3,5-dichloro-4-hydroxybenzamido)benzoate (11: R2 = H) (100 mg, 294 µmol) in toluene (2 mL) was heated at 80 °C until homogenous. The resultant solution was treated with 1,1-di-tert-butoxy-N,N-dimethylmethanamine (141 µL, 588 µmol) and the mixture heated at 80 °C for 3 h, and then at room temperature for 18 h. Additional 1,1-di-tert-butoxy-N,N-dimethylmethanamine (141 µL, 588 µmol) was added and the mixture was heated at 80 °C for 5 h. The reaction mixture was cooled to room temperature and the solvent was removed in vacuo. The residue was diluted with water and extracted with Et2O. The organic layer was dried over MgSO4, filtered and concentrated in vacuo. The residue was partially purified by silica gel chromatography (12 g, 0–50% EtOAc in isohexane) to afford methyl 4-(4-(tert-butoxy)-3,5-dichlorobenzamido)benzoate (82 mg, 71%). The material was used in the next step without further purification.
Step (vi): 4-(4-(tert-Butoxy)-3,5-dichlorobenzamido)benzoicacid (16). 4-(4-(tert-Butoxy)-3,5-dichlorobenzamido)benzoic acid 16 (39 mg, 51%) was prepared as a white solid from methyl 4-(4-(tert-butoxy)-3,5-dichlorobenzamido)benzoate (82 mg, 294 µmol) using a procedure essentially the same as in step (ii) for 15: 1H NMR (400 MHz, DMSO‑d6) δ 12.79 (1H, s), 10.60 (1H, s), 8.06 (2H, s), 7.94 (2H, d, J = 8.1 Hz), 7.87 (2H d, J = 8.1 Hz), 1.49 (9H, s). m/z 380 [M−H]− (ES−).
5.1.3. 4-(3,5-Dichloro-4-ethoxybenzamido)-2-methylbenzoic acid (18)
Step (iii): Methyl 4-(3,5-dichloro-4-ethoxybenzamido)-2-methylbenzoate (10: R1 = Et, R2 = Me). A solution of 3,5-dichloro-4-ethoxybenzoic acid (7: R1 = Et) (285 mg, 1.21 mmol) and DIPEA (1.05 mL, 6.05 mmol) in DMF (2.5 mL) was added to HATU (690 mg, 1.82 mmol) and the orange mixture was stirred for 5 min prior to the addition of methyl 4-amino-2-methylbenzoate (8: R = R2 = Me) (200 mg, 1.21 mmol) in DMF (1 mL). The resulting dark orange solution was stirred for 18 h. 2 M HCl (10 mL) was added and stirring continued for 10 min, and then the mixture was extracted with diethyl ether. The organic layer was washed with water (3 × 15 mL), dried over MgSO4, filtered and the solvent was evaporated in vacuo. The yellow residue was purified by silica gel chromatography (40 g, 0–100% EtOAc in isohexane) to afford methyl 4-(3,5-dichloro-4-ethoxybenzamido)-2-methylbenzoate (10: R1 = Et, R = R2 = Me) (267 mg, 56%): 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.5 Hz, 1H), 7.81 (2H, s), 7.83–7.77 (1H, m), 7.59–7.48 (2H, m), 4.18 (2H, q, J = 7.0 Hz), 3.89 (3H, s), 2.63 (3H, s), 1.49 (3H, t, J = 7.0 Hz). m/z 380 (M−H)− (ES−).
Step (ii): 4-(3,5-Dichloro-4-ethoxybenzamido)-2-methylbenzoicacid (18). Lithium hydroxide (60 mg, 2.51 mmol) in water (1 mL) was added dropwise to a stirring solution of methyl 4-(3,5-dichloro-4-ethoxybenzamido)-2-methylbenzoate (10: R1 = Et, R = R2 = Me) (267 mg, 56%) (240 mg, 0.628 mmol) in THF (5 mL) and the resulting yellow solution was stirred for 5 days at room temperature. The solvent was evaporated in vacuo and dissolved in water (5 mL), then acidified with 2 M HCl. The resultant mixture was extracted with EtOAc. The organic layer was washed with water, dried over MgSO4 and filtered and pre-adsorbed on silica. Silica gel chromatography (40 g, 0–10% IPA in DCM) provided 4-(3,5-dichloro-4-ethoxybenzamido)-2-methylbenzoic acid 18 (52 mg, 22%) as a white solid: 1H NMR (400 MHz, DMSO‑d6) δ 12.66 (1H, s), 10.49 (1H, s), 8.08 (2H, s), 7.94–7.84 (1H, m), 7.75–7.65 (2H, m), 4.14 (2H, q, J = 7.0 Hz), 2.54 (3H, s), 1.40 (3H, t, J = 7.0 Hz). m/z 366 (M−H)− (ES−).
The compound 58 was similarly prepared as 18: see Supplementary data for experimental and spectroscopic details.
5.1.4. 4-(3,5-Dichloro-4-ethoxybenzamido)-2-hydroxybenzoic acid (19)
Steps (vi) and (vii): tert-Butyl 4-amino-2-methoxybenzoate (8: R = tBu, R2 = OMe). 1,1-di-tert-Butoxy-N,N-dimethylmethanamine (608 µL, 2.54 mmol) was added dropwise to a solution of 2-methoxy-4-nitrobenzoic acid 9 (250 mg, 1.27 mmol) in toluene (7.5 mL) at 80 °C. The reaction mixture was heated at 80 °C for 3 h, then a further quantity of 1,1-di-tert-butoxy-N,N-dimethylmethanamine (608 µL, 2.54 mmol) was added. The reaction mixture was heated at 80 °C for 16 h, then diluted with water (10 mL) and extracted with Et2O (3 × 10 mL). The combined organic phases were washed with brine (30 mL), dried over MgSO4, filtered and then concentrated in vacuo to afford tert-butyl 2-methoxy-4-nitrobenzoate (271 mg, 78%) as a pale yellow solid. The material was used in the next step without further purification. tert-Butyl 2-methoxy-4-nitrobenzoate (271 mg, 1.07 mmol) was dissolved in MeOH (270 mL) and passed through a Thales ‘H-cube’ cartridge (10% Pd/C) at a flow rate of 1 mL/min at 25 °C under full H2 mode. The solvent was removed in vacuo to afford tert-butyl 4-amino-2-methoxybenzoate (8: R = tBu, R2 = OMe) (234 mg, 92%) as a pale yellow solid: 1H NMR (400 MHz, DMSO‑d6) δ 7.41 (1H, d, J = 8.5 Hz), 6.16 (1H, d, J = 2.0 Hz), 6.09 (1H, dd, J = 8.5, 2.0 Hz), 5.82 (2H, br s), 3.68 (3H, s), 1.45 (9H, s). m/z 222 [M−H]− (ES−).
Step (iii): tert-Butyl 4-(3,5-dichloro-4-ethoxybenzamido)-2-methoxybenzoate (10: R1 = Et, R2 = OMe, R = tBu). 3,5-Dichloro-4-ethoxybenzoic acid (7: R1 = Et) (75 mg, 0.32 mmol) in DCM (5 mL) was treated with oxalyl chloride (56 μL, 0.64 mmol) dropwise, followed by a drop of DMF. The reaction mixture was stirred at room temperature for 1 h, and then the solvent was removed in vacuo. The residue was dissolved in DCM (5 mL) and TEA (133 μL, 957 μmol) was added. The mixture was added to tert-butyl 4-amino-2-methoxybenzoate (8: R = tBu, R2 = OMe) (71 mg, 0.32 mmol) and stirred at room temperature for 16 h. The mixture was sequentially washed with sat. aq. NaHCO3 (5 mL) and 1 M HCl (5 mL), and the organic phase was concentrated in vacuo. The residue was purified by silica gel chromatography (12 g, 0–100% EtOAc in isohexane) to afford tert-butyl 4-(3,5-dichloro-4-ethoxybenzamido)-2-methoxybenzoate (10: R1 = Et, R2 = OMe, R = tBu) (59 mg, 42%) as a white solid: 1H NMR (400 MHz, DMSO‑d6) δ 10.51 (1H, s), 8.09 (2H, s), 7.63 (1H, d, J = 8.5 Hz), 7.60 (1H, d, J = 1.9 Hz), 7.43 (1H, dd, J = 8.5, 1.9 Hz), 4.15 (2H, q, J = 7.0 Hz), 3.81 (3H, s), 1.51 (9H, s), 1.41 (3H, t, J = 7.0 Hz). m/z 384 [M−tBu+2H]+ (ES+).
Step (viii): 4-(3,5-Dichloro-4-ethoxybenzamido)-2-hydroxybenzoicacid (19). A solution of tert-butyl 4-(3,5-dichloro-4-ethoxybenzamido)-2-methoxybenzoate (10: R1 = Et, R2 = OMe, R = tBu) (55 mg, 0.13 mmol) in DCM (5 mL) was cooled to 0 °C and treated dropwise with a solution of 1 M boron trichloride in DCM (349 μL, 349 μmol). The reaction mixture was stirred at 0 °C for 1 h and then at room temperature for 2 h. The reaction mixture was cooled to 0 °C and water (0.5 mL) and sat. aq. NaHCO3 (2 mL) were added. The resulting white precipitate was collected by filtration and washed with water (2 mL). The solid was dried, then purified by capture and release on SAX, eluting with 5% AcOH in THF to afford 4-(3,5-dichloro-4-ethoxybenzamido)-2-hydroxybenzoic acid 19 (11 mg, 24%) as a white solid: 1H NMR (400 MHz, DMSO‑d6) δ 10.51 (1H, s), 8.06 (2H, s), 7.76 (1H, d, J = 8.7 Hz), 7.48 (1H, d, J = 2.0 Hz), 7.32–7.25 (1H, m), 4.14 (2H, q, J = 7.0 Hz), 1.91 (1H, s), 1.40 (3H, t, J = 7.0 Hz), 1.35 (1H, s). m/z 370 [M+H]+ (ES+), 368 [M−H]− (ES−).
5.1.5. 4-(3,5-Dichloro-4-ethoxyphenylcarbamoyl)benzoic acid (26)
Steps (i) and (ii): 4-(3,5-Dichloro-4-hydroxyphenylcarbamoyl)benzoicacid (25). A mixture of 4-(chlorocarbonyl)benzoic acid methyl ester 23 (600 mg, ca. 3.02 mmol) contaminated with 4-(methoxycarbonyl)benzoic acid 22 was suspended in DCM (5 mL) and cooled to 0 °C. The mixture was treated with oxalyl chloride (529 µL, 6.04 mmol) and DMF (1 drop). The resultant mixture was warmed to room temperature, stirred for 2 h, and then concentrated in vacuo. The residue was dissolved in DCM (3 mL) and a suspension of 4-amino-2,6-dichlorophenol 24 (511 mg, 2.9 mmol) in DCM (18 mL) was added. The resultant suspension was treated with DIPEA (1.58 mL, 9.06 mmol) and was stirred at room temperature for 16 h. The solvent was removed in vacuo and the residue partitioned between EtOAc/DCM and aqueous HCl (1 M). The layers were separated and the organic layer was washed with water and brine. The organic layer was dried over MgSO4, filtered and then the solvent evaporated in vacuo to afford a pale brown solid (930 mg), which was triturated in hot acetonitrile/methanol (9:1) and filtered. The precipitate and filtrate were recombined, the solvent was evaporated in vacuo and then the residue was dissolved in THF (40 mL). Water (10 mL) was added and the mixture treated with lithium hydroxide (340 mg, 14.2 mmol). The mixture was stirred for 16 h and then partitioned between EtOAc and aqueous HCl (1 M). The organic layer was washed successively with water (2 × 50 mL), brine, dried over MgSO4, filtered and then concentrated in vacuo to afford crude 4-(3,5-dichloro-4-hydroxyphenylcarbamoyl)benzoic acid 25 as a pale brown solid. This material was used in the subsequent reaction step without purification.
Step (iii): Ethyl 4-(3,5-dichloro-4-ethoxyphenylcarbamoyl)benzoate. Crude 4-(3,5-dichloro-4-hydroxyphenylcarbamoyl)benzoic acid 25 (450 mg) was dissolved in DMF (15 mL) and treated with potassium carbonate (829 mg, 6.00 mmol) and iodoethane (436 µL, 5.4 mmol). The mixture was stirred at 65 °C for 16 h. Iodoethane (200 µL, 2.48 mmol) was added and the reaction mixture stirred at 70 °C for 3 h. The mixture was partitioned between EtOAc (150 mL) and aqueous HCl (100 mL, 1 M). The layers were separated and the organic layer was washed successively with saturated aqueous NaHCO3 and water. The organic layer was dried over MgSO4, filtered and the solvent evaporated in vacuo. The residue was purified by silica gel chromatography (10–25% EtOAc/isohexane) to afford ethyl 4-(3,5-dichloro-4-ethoxyphenylcarbamoyl)benzoate (500 mg, 75% over 2 steps) as a pale pink solid: m/z 380 (M−H)+ (ES−).
Step (iv): 4-(3,5-Dichloro-4-ethoxyphenylcarbamoyl)benzoicacid (26). Ethyl 4-(3,5-dichloro-4-ethoxyphenylcarbamoyl)benzoate (109 mg, 285 µmol) in THF (5 mL) was treated with aqueous lithium hydroxide (1.43 mL, 1 M, 1.43 mmol) and the mixture was stirred at room temperature for 5 h. The reaction mixture was partitioned between EtOAc and aqueous HCl (1 M). The organic layer was separated and washed successively with water and brine. The organic layer was dried over MgSO4, filtered and then concentrated in vacuo to afford 4-(3,5-dichloro-4-ethoxyphenylcarbamoyl)benzoic acid 26 (89 mg, 88%) as a pale lilac solid: 1H NMR (400 MHz, DMSO‑d6) δ 13.30 (1H, s), 10.58 (1H, s), 8.13–7.99 (4H, m), 7.94 (2H, s), 4.04 (2H, q, J = 7.0 Hz), 1.37 (3H, t, J = 7.0 Hz). m/z 352 [M−H]− (ES−).
5.1.6. 4-(4-Ethoxy-3,5-diisopropoxybenzamido)benzoic acid (31)
Step (i): Methyl 3,5-dihydroxy-4-ethoxybenzoate(28: R1 = Et). A mixture of methyl 3,4,5-trihydroxybenzoate 27 (5g, 27.2 mmol), iodoethane (2.194 mL, 27.2 mmol) and sodium hydrogen carbonate (9.12 g, 109 mmol) was stirred in N,N-dimethylformamide (50 mL) at 30 °C for 72 h. Water (50 mL) was added and the mixture was extracted with ethyl acetate (2 × 50 mL). The organic layer was then washed with water (50 mL), brine (50 mL), dried over magnesium sulfate, filtered and concentrated in vacuo. The product was then purified by silica gel chromatography (80 g, 0–20% hexane/ethyl acetate) to leave 28 (2.90 g, 50% yield). 1H NMR (400 MHz, CDCl3) δ 7.24 (2H, s), 5.68 (2H, s), 4.21 (2H, q, J = 7.1 Hz), 3.89 (3H, s), 1.42 (3H, t, J = 7.0 Hz).
Step (ii): 3,5-Diisopropoxy-4-ethoxybenzoicacid (29: R1 = Et, R2 = R3 = iPr). Compound 28 (500 mg, 2.36 mmol) was combined with 2-bromopropane (885 μL, 9.43 mmol) and potassium carbonate (651 mg, 4.71 mmol) in N,N-dimethylformamide (5 mL). The resulting suspension was stirred at 50 °C for 48 h. Water (5 mL) was added and the mixture was extracted with ethyl acetate (2 × 5 mL). The organic layer was then washed with water (5 mL), brine (5 mL), dried over magnesium sulfate, filtered and concentrated in vacuo. The crude product was then purified by silica gel chromatography (40 g, 0–50% hexane/ethyl acetate) to leave methyl 3,5-diisopropoxy-4-ethoxybenzoate (530 mg, 76% yield). 1H NMR (400 MHz, CDCl3) δ 7.27 (2H, s), 4.61–4.55 (2H, m), 4.10 (2H, q, J = 7.1 Hz), 3.88 (3H, s), 1.38–1.33 (15H, m).
Step (iii): Methyl 3,5-diisopropoxy-4-ethoxybenzoatewas converted to compound (29: R1 = Et, R2 = R3 = iPr) in 57% yield using lithium hydroxide in the procedure described for compound 15. 1H NMR (400 MHz, CDCl3) δ 7.34 (2H, s), 4.61–4.55 (2H, m), 4.10 (2H, q, J = 7.1 Hz), 1.40–1.33 (15H, m).
Step (iv): 4-(4-Ethoxy-3,5-diisopropoxybenzamido)benzoicacid (31). Compound (29: R1 = Et, R2 = R3 = iPr) and 30 were coupled and hydrolyzed to the title compound 31 (288 mg, 57% for final step) as a white solid, using the procedures described for the preparation of 15. 1H NMR (400 MHz, CDCl3) δ 8.02 (2H, d, J = 8.7 Hz), 7.85 (1H, br s), 7.75 (2H, d, J = 8.8 Hz), 7.09 (2H, s), 4.66–4.58 (2H, m), 4.10 (2H, q, J = 7.1 Hz), 1.39–1.33 (15H, m). m/z 400 (M−H)− (ES−), 402 (M+H)+ (ES+).
The compounds 32–34 were similarly prepared as 31: see Supplementary data for experimental and spectroscopic details.
5.1.7. 4-[3-Chloro-4,5-bis(cyclopentyloxy)benzamido]benzoic acid (42)
Step (i): Methyl 3-chloro-4,5-dihydroxybenzoate (36). Tribromoborane (7.86 mL, 82 mmol) was added dropwise to a stirring mixture of 3-chloro-4-hydroxy-5-methoxybenzoic acid 35 (6.61 g, 32.6 mmol) in dichloromethane (50 mL) under nitrogen at 0 °C. The resulting orange mixture was stirred at the same temperature for 2 h then poured portion wise onto ice/brine (250 mL). The aqueous phase was extracted with ethyl acetate (2 × 150 mL) and the combined organic extracts were dried over magnesium sulfate and filtered. The solvent was removed in vacuo to give 3-chloro-4,5-dihydroxybenzoic acid (5.11 g, 79% yield). 1H NMR (400 MHz, DMSO‑d6) δ 12.69 (1H, br s), 10.14 (2H, br s), 7.35 (1H, d, J = 2.0 Hz), 7.32 (1H, d, J = 2.0 Hz). m/z 187 [M−H]− (ES).
Step (ii): A solution of 3-chloro-4,5-dihydroxybenzoic acid (3.16 g, 16.76 mmol) and chlorotrimethylsilane (6.36 mL, 50.3 mmol) in methanol (50 mL) was stirred at 50 °C, for 16 h, under an atmosphere of nitrogen. The solvent was removed in vacuo and the residue was partitioned between brine (75 mL) and ethyl acetate (75 mL). The organic layer was washed with brine (75 mL), dried over magnesium sulfate and filtered. The solvent was removed in vacuo to give methyl 3-chloro-4,5-dihydroxybenzoate 36 (3.26 g, 82% yield). 1H NMR (400 MHz, DMSO‑d6) δ 10.17 (2H, br s), 7.38 (1H, d, J = 2.0 Hz), 7.35 (1H, d, J = 2.0 Hz), 3.78 (3H, s). m/z 201 [M−H]− (ES−).
Step (iii): 3-Chloro-4,5-bis(cyclopentyloxy)benzoicacid (37, R = cyclopentyl). A mixture of methyl 3-chloro-4,5-dihydroxybenzoate 36 (300 mg, 1.48 mmol), iodocyclopentane (558 µL, 4.44 mmol) and potassium carbonate (614 mg, 4.44 mmol) in DMF (10 mL) was stirred at 70 °C for 46 h. The reaction mixture was cooled to room temperature and then partitioned between 1 M hydrochloric acid (75 mL) and ethyl acetate (100 mL). The phases were separated and the organic phase was washed with brine (2 × 75 mL) then dried over magnesium sulfate and filtered. The solvent was removed in vacuo and the residue was purified by silica gel chromatography (40 g, 0–100% EtOAc and isohexane) to give methyl 3-chloro-4,5-bis(cyclopentyloxy)benzoate (427 mg, 85% yield). 1H NMR (400 MHz, CDCl3) δ 7.66 (1H, d, J = 2.0 Hz), 7.45 (1H, d, J = 2.0 Hz), 5.05–4.98 (1H, m), 4.87–4.83 (1H, m), 3.89 (3H, s), 1.95–1.55 (16H, m). m/z 339 [M+H]+ (ES+).
Step (iv): Methyl 3-chloro-4,5-bis(cyclopentyloxy)benzoate (400 mg, 1.18 mmol) was dissolved in a mixture of 1,4-dioxane (10 mL) and water (5 mL) and lithium hydroxide (226 mg, 9.44 mmol) was added. After stirring for 18 h at room temperature, the mixture was partitioned between 1 M hydrochloric acid (20 mL) and ethyl acetate (25 mL). The phases were separated and the organic phase was washed with water (20 mL) then dried over magnesium sulfate and filtered. The solvent was removed in vacuo to give the title compound (37: R = cyclopentyl) (380 mg, 99% yield). 1H NMR (400 MHz, DMSO‑d6) δ 13.07 (1H, br s), 7.52 (1H, d, J = 2.0 Hz), 7.45 (1H, d, J = 2.0 Hz), 4.97–4.91 (2H, m), 1.99–1.90 (2H, m), 1.70–1.57 (14H, m). m/z 323 [M−H]− (ES−).
Step (v) and (iv): 4-[3-Chloro-4,5-bis(cyclopentyloxy)benzamido]benzoicacid (42). A mixture of 3-chloro-4,5-bis(cyclopentyloxy)benzoic acid (37: R = cyclopentyl) and methyl 4-aminobenzoate (38: R1 = H) was converted to the methyl 4-[3-chloro-4,5-bis(cyclopentyloxy)benzamido]benzoate in 49% yield using the procedure in step (iii) described for compound 15, 1H NMR (400 MHz, CDCl3) δ 8.06 (2H, d, J = 8.8H), 7.84 (1H, br s), 7.72 (2H, d, J = 8.8 Hz), 7.42–7.35 (2H, m), 5.08–4.98 (1H, m), 4.90–4.86 (1H, m), 3.92 (3H, s), 1.95–1.63 (16H, m). m/z 458 [M+H]+ (ES+), 456 [M−H]− (ES−). Hydrolysis of methyl-4-[3-chloro-4,5-bis(cyclopentyloxy)benzamido]benzoate using the procedure in step (iv) described in the preparation of compound (37: R = cyclopentyl) gave 4-[3-Chloro-4,5-bis(cyclopentyloxy)benzamido]benzoic acid 42 in 72% yield as white solid. 1H NMR (400 MHz, DMSO‑d6) δ 12.74 (1H, br s), 10.45 (1H, s), 7.98–7.84 (4H, m), 7.69 (1H, d, J = 2.0 Hz), 7.52 (1H, d, J = 2.0 Hz), 5.01–4.95 (2H, m), 1.99–1.93 (2H, m), 1.73–1.48 (14H, m). m/z 442 [M−H]− (ES−).
The compounds 39, 40, 43, 44 were similarly prepared as 42: see Supplementary data for experimental and spectroscopic details.
5.1.8. 4-(3,4-Di-tert-butoxy-5-chlorobenzamido)benzoic acid (41)
Step (ix): 3,4-Di-tert-butoxy-5-chlorobenzoicacid (37: R = tBu). N,N-Dimethylformamide di-tert-butyl acetal (5.92 mL, 24.7 mmol) was added to a solution of methyl 3-chloro-4,5-dihydroxybenzoate 36 (500 mg, 2.47 mmol) in toluene (10 mL) and the reaction mixture was stirred at RT under nitrogen for 21 h. The solvent was removed in vacuo and the residue was purified by silica gel chromatography (40 g, 0–20% EtOAc in iso-hexane) to give the bis-alkylated intermediate, which was dissolved in 1,4-dioxane/water (20 mL, 1:1) and treated with lithium hydroxide (591 mg, 24.7 mmol). The mixture was stirred 18 h at room temperature. The mixture was poured into 10% aqueous citric acid (100 mL) and the precipitate was collected by filtration. The solid was washed with water and dried to give 3,4-di-tert-butoxy-5-chlorobenzoic acid (37: R = tBu) (534 mg, 70%). 1H NMR (400 MHz, DMSO‑d6) δ 13.13 (1H, br s), 7.67 (1H, s), 7.53 (1H, s), 1.39 (9H, s), 1.32 (9H, s). m/z 299 [M−H]− (ES−).
Step (x): 4-(3,4-Di-tert-butoxy-5-chlorobenzamido)benzoic acid (41).
A mixture of 3,4-di-tert-butoxy-5-chlorobenzoic acid (37: R = tBu) (250 mg, 0.831 mmol) and methyl 4-aminobenzoate (38: R1 = H) was converted to themethyl 4-(3,4-di-tert-butoxy-5-chlorobenzamido)benzoate (185 mg, 50%) using the procedure described for compound 18. 1H NMR (400 MHz, DMSO‑d6) δ:10.54 (1H, s), 7.96 (2H, d), 7.91 (2H, d), 7.86 (1H, d), 7.60 (1H, d), 3.84 (3H, s), 1.41 (9H, s), 1.32 (9H, s). m/z 432 [M−H]− (ES−). Hydrolysis of methyl 4-(3,4-di-tert-butoxy-5-chlorobenzamido)benzoate (175 mg, 0.403 mmol) using the procedure described in the preparation of compound (37: R = cyclopentyl) step (vii). gave 4-(3,4-di-tert-butoxy-5-chlorobenzamido)benzoic acid 41 (110 mg, 64%) as a white solid: 1H NMR (400 MHz, DMSO‑d6) δ 12.78 (1H, br s). 10.49 (1H, s), 7.97–7.86 (3H, m), 7.85 (2H, d, J = 2.3 Hz), 7.59 (1H, d, J = 2.2 Hz), 1.40 (9H, s), 1.34 (9H, s). m/z 418 [M−H]− (ES−).
The compound 45 was similarly prepared as 41: see Supplementary data for experimental and spectroscopic details.
5.1.9. 4-(3-Chloro-4-ethoxy-5-isopropoxybenzamido)-2-methylbenzoic acid (56)
Step (iii): Methyl 4-benzyloxy-3-chloro-5-hydroxybenzoate (46). Methyl 3-chloro-4,5-dihydroxybenzoate 36 (14.19 g, 70 mmol) was dissolved in N,N-dimethylformamide (210 mL) and treated with potassium carbonate (8.71 g, 63 mmol). After stirring for 5 min, benzyl bromide (8.32 mL, 70 mmol) was added and the mixture was heated to 60 °C for 0.75 h. The reaction mixture was diluted with diethyl ether (500 mL) and washed successively with 1 M hydrochloric acid (500 mL) and with brine (2 × 500 mL). The aqueous phase was re-extracted with diethyl ether (500 mL) and the combined organic layers were washed with brine (2 × 500 mL) and dried with magnesium sulfate. Filtration and evaporation left the crude product which was purified by silica gel chromatography (330 g, 0–100% ethyl acetate/isohexane) to leave methyl 4-(benzyloxy)-3-chloro-5-hydroxybenzoate 46 as an off-white solid (9.84 g, 48% yield). 1H NMR (400 MHz, DMSO‑d6) δ 10.50 (1H, s), 7.57–7.53 (2H, m), 7.52 (1H, d, J = 2.1 Hz), 7.47 (1H, d, J = 2.1 Hz), 7.46–7.37 (3H, m), 5.14 (2H, s), 3.82 (3H, s). (m/z 293.3 [M+H]+ (ES+), 291.2 [M−H]− (ES−).
Step (iv): Methyl 3-chloro-4-benzyloxy-5-isopropoxybenzoate (47: R2 = iPr). Methyl 4-(benzyloxy)-3-chloro-5-hydroxybenzoate 46 (7.5 g, 25.6 mmol) was combined with potassium carbonate (7.08 g, 51.2 mmol) in N,N-dimethylformamide (25 mL). The mixture was stirred at RT for 5 min. 2-Bromopropane (4.81 mL, 51.2 mmol) was added and the mixture stirred at 60 °C for 2 h. Water (25 mL) was added and the mixture was extracted with ethyl acetate (3 × 50 mL). The combined organic phase was washed with brine (2 × 50 mL) and then dried over magnesium sulfate, filtered and concentrated in vacuo to leave a crude mixture which was purified by silica gel chromatography (120 g, 0–100% ethyl acetate/isohexane) to afford methyl 3-chloro-4-benzyloxy-5-isopropoxybenzoate (47: R2 = iPr), as a clear oil (4.91 g, 57% yield). 1H NMR (400 MHz, DMSO‑d6) δ 7.68 (1H, d, J = 2.0 Hz), 7.56–7.47 (3H, m), 7.43–7.28 (3H, m), 5.12 (2H, s), 4.77–4.72 (1H, m), 3.90 (3H, s), 1.38 (6H, d, J = 6.1 Hz). m/z 335 [M+H]+ (ES+).
3-Chloro-4-ethoxy5-isopropoxybenzoicacid (48: R2 = iPr, R3 = Et). Steps (v), (vi) and (vii).
Step (v): Methyl 3-chloro-4-hydroxy-5-isopropoxybenzoate. Methyl 3-chloro-4-(benzyloxy)-5-isopropoxybenzoate (47: R2 = iPr) (4.91 g, 14.7 mmol) was dissolved in a mixture of methanol (160 mL), dichloromethane (16 mL) and acetic acid (0.16 mL) and the solution was passed through a Thales ‘H-cube’ cartridge (10% Pd/C) at a flow rate of 1 mL/min at 25 °C under an atmosphere of hydrogen (full H2 mode). The solvents were removed in vacuo to afford methyl 3-chloro-4-hydroxy-5-isopropoxybenzoate (3.42 g, 85%). 1H NMR (400 MHz, DMSO‑d6) δ 10.07 (1H, s), 7.53 (1H, d, J = 2.0 Hz), 7.43 (1H, d, J = 2.0 Hz), 4.70–4.63 (1H, m), 3.82 (3H, s), 1.30 (6H, d, J = 6.0 Hz). m/z 245 [M+H]+ (ES+), 243 [M−H]− (ES−).
Step (vi): Methyl 3-chloro-4-ethoxy-5-isopropoxybenzoate. Methyl 3-chloro-4-hydroxy-5-isopropoxybenzoate (3.42 g, 14 mmol) was combined with potassium carbonate (3.86 g, 28 mmol) in N,N-dimethylformamide (5 mL) and the mixture heated at 60 °C for 10 min. Iodoethane (2.26 mL, 28 mmol) was added dropwise whereupon the mixture was stirred at 40 °C for 3 h. A further aliquot of iodoethane was added and heating and stirring was continued for 16 h. Water (50 mL) was added and the mixture was extracted with ethyl acetate (3 × 100 mL). The combined organic phase was washed with brine (3 × 50 mL) and then dried over magnesium sulfate, filtered and concentrated in vacuo to leave a crude mixture which was purified by silica gel chromatography (120 g, 0–100% ethyl acetate/isohexane) to afford methyl 3-chloro-4-ethoxy-5-isopropoxybenzoate as a white solid (3.17 g, 82% yield). 1H NMR (400 MHz, DMSO‑d6) δ 7.67 (1H, d, J = 2.0 Hz), 7.48 (1H, d, J = 2.5 Hz), 4.66–4.59 (1H, m), 4.16 (2H, q, J = 7.1 Hz), 3.90 (3H, s), 1.40 (3H, t, J = 7.0 Hz), 1.37 (6H, d, J = 6.0 Hz). m/z 245 [M+H]+ (ES+), 243 [M−H]− (ES−).
Step (vii): 3-Chloro-4-ethoxy5-isopropoxybenzoicacid (48: R2 = iPr, R3 = Et). Methyl 3-chloro-4-ethoxy-5-isopropoxybenzoate (3.17 g, 11.6 mmol) was dissolved in tetrahydrofuran (226 mL) and treated with 1 M aqueous lithium hydroxide solution (23.25 mL, 23.25 mmol). Methanol (5 mL) was added so that a solution formed and this was heated at 40 °C for 1 h. After stirring for a further 16 h at room temperature, the reaction mixture was acidified with 1 M hydrochloric acid and extracted with diethyl ether (3 × 100 mL). The organic layer was dried over magnesium sulfate, filtered and concentrated in vacuo to leave 3-chloro-4-ethoxy 5-isopropoxybenzoic acid(48: R2 = iPr, R3 = Et) as a white solid (2.83 g, 94% yield). 1H NMR (400 MHz, DMSO‑d6) δ 13.21 (1H, s), 7.54 (1H, d, J = 1.9 Hz), 7.48 (1H, d J = 1.9 Hz,), 4.74–4.78 (1H, m), 4.11 (2H, q, J = 7.0 Hz), 1.37–1.21 (9H, m). m/z 257 [M−H]− (ES−).
5.1.10. 4-(3-Chloro-4-ethoxy-5-isopropoxybenzamido)-2-methylbenzoic acid (56)
Step (viii): Methyl 4-(3-chloro-4-ethoxy-5-isopropoxybenzamido)-2-methylbenzoate. A suspension of 3-chloro-4-ethoxy 5-isopropoxybenzoic acid (48: R2 = iPr, R3 = Et) (2.82 g, 10.9 mmol) and methyl 4-amino-2-methylbenzoate (38: R1 = Me) (2.16 g, 13.1 mmol) in ethyl acetate (33 mL) was treated with triethylamine (4.56 mL, 32.7 mmol) followed by T3P (50 wt% in ethyl acetate) (17.34 mL, 27.3 mmol) and the mixture was heated at 60 °C for 4 h and allowed to cool to room temperature for 16 h. The reaction mixture was stirred vigorously with an aqueous solution of sodium hydrogencarbonate (50 mL) for 10 min and separated. The aqueous layer was extracted with dichloromethane (3 × 100 mL) and the combined organic phases were dried (magnesium sulfate), filtered concentrated in vacuo and the residue purified by silica gel chromatography (40 g, 0:50:50 to 20:40:40 ethyl acetate:dichloromethane:isohexane) to produce methyl 4-(3-chloro-4-ethoxy-5-isopropoxybenzamido)-2-methylbenzoate as a beige solid (3.24 g, 70% yield). 1H NMR (400 MHz, CDCl3) δ 8.0 (1H, d, J = 8.4 Hz), 7.8 (1H, s), 7.59–7.50 (2H, m), 7.43–7.36 (2H, m), 4.69–4.73 (1H, m), 4.18 (2H, q, J = 7.1 Hz), 3.92 (3H, s), 2.6 (3H, s), 1.46–1.37 (9H, m). m/z 406 [M+H]+ (ES+), 404 [M−H]− (ES−).
Step (ix):1 M Lithium hydroxide solution (15.97 mL, 15.97 mmol) was added to a solution of methyl 4-(3-chloro-4-ethoxy-5-isopropoxybenzamido)-2-methylbenzoate (3.24 g, 7.98 mmol) in tetrahydrofuran (32 mL). Methanol (5 mL) was added and the mixture stirred at 40 °C for 16 h. A further aliquot of lithium hydroxide solution (7.98 mL, 7.98 mmol) in methanol (5 mL) was added and stirring at 40 °C was continued for 3 h. The reaction mixture was partitioned between water (50 mL) and diethyl ether (100 mL). The layers were separated and the aqueous layer was acidified with 1 M hydrochloric acid solution. A precipitate evolved which was filtered and washed with water (3 × 10 mL) and diethyl ether (3 × 10 mL). After drying, this left 4-(3-chloro-4-ethoxy-5-isopropoxybenzamido)-2-methylbenzoic acid 56 as a white solid (2.55 g, 81%). Recrystallisation from dioxane/water (82:18) gave white crystals mp 186 °C. 1H NMR (400 MHz, DMSO‑d6) δ 12.64 (1H, br s), 10.34 (1H, s), 7.86 (1H, d, J = 8.5 Hz,), 7.74–7.63 (3H, m), 7.55 (1H, d, J = 2.0 Hz), 4.79–4.73 (1H, m), 4.10 (2H, q, J = 7.1 Hz), 2.52 (3H, s), 1.37–1.26 (9H, m). 13C NMR (101 MHz, DMSO‑d6) δ 168.5, 164.4, 151.9, 148.0, 142.4, 140.9, 132.0, 130.9, 127.9, 125.5, 123.0, 121.4, 117.7, 114.2, 71.6, 69.3, 22.3, 22.2, 15.9. m/z 392 [M+H]+ (ES+), 390 [M−H]− (ES−). HRMS: C20H23ClNO5 requires (M+H)+ 392.1265, found 392.1249 (error −4.0 ppm).
The compounds 49–55, 57 and 59 were similarly prepared as 56: see Supplementary data for experimental and spectroscopic details.
5.2. Biological and ADME assays
5.2.1. Transactivation assays for mouse RAR alpha, beta and gamma receptors
Transcriptional transactivation assays have been performed with gal4 fusion receptor constructs, created using each of the mouse RAR ligand-binding domains, co-transfected with the pFR-luc (Stratagene) reporter construct in COS-7 cells. Thus, transfected cells will constitutively express the gal4-RAR fusion protein which in turn may be transactivated by ATRA to induce the expression of the luciferase that is driven by a gal4UAS. Briefly, on day one, 96 well plates were seeded with 8000 cells per well then left to recover overnight. On day two, the cells were co-transfected with 100 ng of reporter plasmid and 10 ng of the appropriate receptor plasmid per well using lipofectamine (Invitrogen). On day three, the lipofectamine containing media was replaced by a DMEM without phenol red, followed by the addition of novel compounds dissolved in 1 µL of DMSO to each well’s 100 µL total volume. Finally, on day four, the cells were lysed and their luciferase substrate was provided by the BrightGlo reagent (Promega), the plates were then read on the MicroBeta TriLux (Perkin Elmer). In each experiment, an 8 point dose response curve of ATRA was run in duplicate, and the various compounds tested were compared to these values.
5.2.2. FlashPlate® Scintillation Proximity Binding Assay (SPA)
In the FlashPlate® Scintillation Proximity Assay (SPA) wells of a 96-well plate are coated with scintillant and capture antibody (or similar) for tagged proteins. This requires just 100 ng of RAR proteins and 2 nM [3H]-retinoic acid per well. This enables competition of specifically bound [3H]-retinoic acid by unlabelled retinoid compounds. As only radioligand specifically bound to the captured protein is sufficiently close to the scintillant to produce a signal, separation of bound and free radioactivity is not required. Binding of the tritiated retinoid to biotinylated RARα is specific, saturable, time dependent and reversible. We have successfully applied our assay to a screen of known retinoid standards and novel compounds and it is both rapid and reproducible (see Supplementary data file for details).
5.2.3. Intrinsic clearance Clint
In this in vitro model of hepatic clearance mouse or human liver microsomes were incubated with the test compound at 37 °C in the presence of the co-factor, NADPH, which initiates the reaction. The reaction is terminated by the addition of methanol. Following centrifugation, the supernatant is analyzed on the LC-MS/MS. The disappearance of the test compound is monitored over a 45 min time period. The data is the mean on 5 separate experiments. SEM is less than 10% of the mean values.
The ln peak area ratio (compound peak area/internal standard peak area) is plotted against time and the gradient of the line determined.
The elimination rate constant (k) = (−gradient), the Half life (t1/2)(min) = 0.693/k and V(µL/mg) = volume of incubation (µL)/protein in the incubation (mg).
Intrinsic Clearance = (CLint)(µL/min/mg/protein) = V × 0.693/k. (see Supplementary data file for details).
5.2.4. PK studies in rats
Test compounds were administered orally and intravenously to groups of 4 male Sprague-Dawley rats. Oral dosing solutions of each Test Item were prepared at a concentration of 1 mg/mL in 8% ethanol and 92% PEG-400. The Test Items were orally administered at a dose of 10 mg/kg and a dosing volume of 10 mL/kg. Intravenous dosing solutions of each Test Item were prepared at a concentration of 0.25 mg/mL in 8% ethanol, 92% PEG-400. The Test Items were intravenously administered at a dose of 0.5 mg/kg and a dosing volume of 2 mL/kg. Approximately eight blood samples were collected from each animal at appropriate intervals up to 6 h post dosing for the iv groups and up to 24 h for the oral groups. Whole blood concentrations of the Test Items were measured using LC-MS/MS and selected pharmacokinetic parameters calculated using Pharsight WinNonLin software. For furthers details see Supplementary data file.
Acknowledgements
We thank the Wellcome Trust for their financial support (Grant 084286).
Footnotes
Supplementary data including experimental and spectroscopic details for similarly prepared compounds 12–14, 17, 20, 21, 32–34, 39, 40, 43–45, 49–55, 57–59, 63 and 64. ADME, biological assays, and virtual screening details associated with this article can be found, in the online version, at https://doi.org/10.1016/j.bmc.2017.12.015.
A. Supplementary data
References
- 1.Dollé P., Neiderreither K., editors. The Retinoids: Biology, Biochemistry, and Disease. John Wiley Sons Inc; Hoboken, NJ: 2015. [Google Scholar]
- 2.Lu Y., Bertran S., Samuels T.A., Mira-y-Lopez R., Farias E.F. Mechanism of inhibition of MMTV-neu and MMTV-wnt1 induced mammary oncogenesis by RAR[alpha] agonist AM 580. Oncogene. 2010;29:3665–3676. doi: 10.1038/onc.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshimura H., Kikuchi K., Hibi S. Discovery of novel and potent retinoic acid receptor α agonists: synthesis and evaluation of benzofuranyl-pyrrole and benzothiophenyl-pyrrole derivatives. J Med Chem. 2000;43:2929–2937. doi: 10.1021/jm000098s. [DOI] [PubMed] [Google Scholar]
- 4.Jarvis C.I., Goncalves M.B., Clarke E. Retinoic acid receptor-α signalling antagonizes both intracellular and extracellular amyloid-β production and prevents neuronal cell death caused by amyloid-β. Eur J Neurosci. 2010;32:1246–1255. doi: 10.1111/j.1460-9568.2010.07426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seino K.I., Yamauchi T., Shikata K. Prevention of acute and chronic allograft rejection by a novel retinoic acid receptor-α-selective agonist. Int Immunol. 2004;16:665–673. doi: 10.1093/intimm/dxh066. [DOI] [PubMed] [Google Scholar]
- 6.Yamauchi T., Ishibashi A., Shikata K. Effect of E6060 [4-{5-[7-fluoro-4-(trifluoromethyl)benzo[b]furan-2-yl]-1H- 2-pyrrolyl}benzoic acid], a novel subtype-selective retinoid, on lupus-like nephritis in female (NZBxNZW)F1 mice. J Pharmacol Exp Ther. 2005;312:938–944. doi: 10.1124/jpet.104.075598. [DOI] [PubMed] [Google Scholar]
- 7.Shudo K., Kagechika H. Structural evolution of retinoids. Adv Drug Res. 1993;24:81–119. [Google Scholar]
- 8.Teng M., Duong T.T., Klein E.S., Pino M.E., Chandraratna R.A. Identification of a retinoic acid receptor α subtype specific agonist. J Med Chem. 1996;39:3035–3038. doi: 10.1021/jm9603532. [DOI] [PubMed] [Google Scholar]
- 9.Kagechika H., Kawachi E., Hashimoto Y., Himi T., Shudo K. Retinobenzoic acids. 1. Structure-activity relationships of aromatic amides with retinoidal activity. J Med Chem. 1988;31:2182–2192. doi: 10.1021/jm00119a021. [DOI] [PubMed] [Google Scholar]
- 10.Beard R.L., Duong T.T., Teng M., Klein E.S., Standevan A.M., Chandraratna R.A. Synthesis and biological activity of retinoic acid receptor-alpha specific amides. Bioorg Med Chem Lett. 2002;12:3145–3148. doi: 10.1016/s0960-894x(02)00647-9. [DOI] [PubMed] [Google Scholar]
- 11.Arafa H.M., Elmazar M.M., Hamada F.M., Reichert U., Shroot B., Nau H. Selective agonists of retinoic acid receptors: comparative toxicokinetics and embryonic exposure. Arch Toxicol. 2000;73:547–556. doi: 10.1007/s002040050007. [DOI] [PubMed] [Google Scholar]
- 12.Elmazar M.M., Reichert U., Shroot B., Nau H. Pattern of retinoid-induced teratogenic effects: possible relationship with relative selectivity for nuclear retinoid receptors RAR alpha, RAR beta, and RAR gamma. Teratology. 1996;53:158–167. doi: 10.1002/(SICI)1096-9926(199603)53:3<158::AID-TERA3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 13.Altucci L., Leibowitz M.D., Ogilvie K.M., de Lera A.R., Gronemeyer H. RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov. 2007;6:793–810. doi: 10.1038/nrd2397. [DOI] [PubMed] [Google Scholar]
- 14.Vinter J.G. Extended electron distributions applied to the molecular mechanics of some intermolecular interactions. J Comput Aided Mol Des. 1994;8:653–668. doi: 10.1007/BF00124013. [DOI] [PubMed] [Google Scholar]
- 15.(a) Cheeseright T., Mackey M., Rose S., Vinter A. Molecular field extrema as descriptors of biological activity: definition and validation. J Chem Inf Model. 2006;46:665–676. doi: 10.1021/ci050357s. [DOI] [PubMed] [Google Scholar]; (b) Cheeseright T.J., Mackey M.D., Melville J.L., Vinter J.G. FieldScreen: virtual screening using molecular fields. Application to the DUD data set. J Chem Inf Model. 2008;48:2108–2117. doi: 10.1021/ci800110p. [DOI] [PubMed] [Google Scholar]
- 16.Vinter J.G., Trollope K.I. Multiconformational composite molecular-potential fields in the analysis of drug-action. 1. Methodology and first evaluation using 5-HT and histamine action as examples. J Comput Aided Mol Des. 1995;9:297–307. doi: 10.1007/BF00125171. [DOI] [PubMed] [Google Scholar]
- 17.Bourguet W., Vivat V., Wurtz J.M., Chambon P., Gronemeyer H., Moras D. Crystal structure of a heterodimeric complex of RAR and RXR ligand-binding domains. Mol Cell. 2000;5:289–298. doi: 10.1016/s1097-2765(00)80424-4. [DOI] [PubMed] [Google Scholar]
- 18.Hopkins A.L., Groom C.R., Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 19.Mannhold R., Krogsgaard-Larsen P., Timmerman H. Vol. 1. John Wiley & Sons; 2008. QSAR: Hansch analysis and related approaches. [Google Scholar]
- 20.ADME assays: LogD octanol/water pH7.4, Cyp450 activity, and human plasma protein binding and the Toxicity assays: High-Content Cell Toxicity Screen in HEPG2 cells, Cytotoxicity Assay in COS-7 cells were carried out by the CRO Cyprotex (see Supplementary data file for details).
- 21.Genetic Toxicity assays: Bacterial Cytotoxicity, Ames Tests, in vitro Micronucleus test and the in vitro Pharmacology: Binding and Enzyme Assays: including the CEREP panel of 120 other receptors, channels and enzymes, and the hERG Binding Assay were carried out by the CRO Cerep (see Supplementary data file for details).
- 22.The Retinoid X Receptor RXRα and RXRβ radioligand binding assays using [3H] 9-cis-retinoic acid and the respective human recombinant receptors, were carried out by the CRO Panlabs Eurofins Pharma Discovery Services UK Limited, Gemini Crescent, Dundee Technology Park, Dundee, DD2 1SW.
- 23.Leid M., Kastner P., Chambon P. Multiplicity generates diversity in the retinoic acid signalling pathways. Trends Biochem Sci. 1992;17:427–433. doi: 10.1016/0968-0004(92)90014-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.