

ABSTRACT
Microscopic interpretation of stained smears is one of the most operator-dependent and time-intensive activities in the clinical microbiology laboratory. Here, we investigated application of an automated image acquisition and convolutional neural network (CNN)-based approach for automated Gram stain classification. Using an automated microscopy platform, uncoverslipped slides were scanned with a 40× dry objective, generating images of sufficient resolution for interpretation. We collected 25,488 images from positive blood culture Gram stains prepared during routine clinical workup. These images were used to generate 100,213 crops containing Gram-positive cocci in clusters, Gram-positive cocci in chains/pairs, Gram-negative rods, or background (no cells). These categories were targeted for proof-of-concept development as they are associated with the majority of bloodstream infections. Our CNN model achieved a classification accuracy of 94.9% on a test set of image crops. Receiver operating characteristic (ROC) curve analysis indicated a robust ability to differentiate between categories with an area under the curve of >0.98 for each. After training and validation, we applied the classification algorithm to new images collected from 189 whole slides without human intervention. Sensitivity and specificity were 98.4% and 75.0% for Gram-positive cocci in chains and pairs, 93.2% and 97.2% for Gram-positive cocci in clusters, and 96.3% and 98.1% for Gram-negative rods. Taken together, our data support a proof of concept for a fully automated classification methodology for blood-culture Gram stains. Importantly, the algorithm was highly adept at identifying image crops with organisms and could be used to present prescreened, classified crops to technologists to accelerate smear review. This concept could potentially be extended to all Gram stain interpretive activities in the clinical laboratory.
KEYWORDS: Gram stain, blood culture, machine learning, deep learning, automated microscopy, analytics, artificial intelligence, big data, neural network
INTRODUCTION
Bloodstream infections (BSI) are rapidly progressive infections with mortality rates up to nearly 40% (1, 2). Each day of delay in institution of active antimicrobial therapy is associated with up to a ∼10% increase in mortality (3, 4). Due to relatively low bacterial burden (<10 CFU ml−1) (5), patient blood is preincubated in broth culture to detect the presence of bacteria, typically by semicontinuous measurement of CO2 production or pH with an automated blood culture instrument. If organism growth is detected, an aliquot of broth (now containing >106 CFU ml−1) is removed for Gram stain smear and subculture. The Gram stain provides the first critical piece of information that allows a clinician to tailor the appropriate therapy and to optimize the outcome (6).
Despite recent advances in automation in other stages of the BSI diagnosis process (automated blood culture incubators and Gram staining systems) (7), Gram stain interpretation remains labor and time intensive and highly operator dependent. With consolidation of hospital systems, increasing workloads, and the potential unavailability of highly trained microbiologists on site (8), automated image collection paired with computational interpretation of Gram stains to augment and complement manual testing would provide benefit. However, there has been a dearth of scientific exploration in this area, and several technical difficulties need to be overcome.
Practically, automated Gram stain interpretation requires both automated slide imaging and automated image analysis. Although automated slide scanners and microscopes are being used in anatomic pathology, for example, in telepathology (9), their application in clinical microbiology has been limited based on several technical challenges. First, Gram-stained slides are typically read using 100× objectives, greatly complicating image acquisition due to the need for addition of oil during scanning. Second, microbiology smear material can adequately be imaged only in a very narrow field of focus, a challenge for existing slide scanners. Third, Gram-stained slides exhibit ubiquitous and highly variable background staining. This background may cause autofocus algorithms to target areas that either are devoid of bacteria or miss the appropriate focal plane entirely. Image analysis to identify Gram stain characteristics presents separate hurdles. Importantly, background and staining artifacts, both fairly ubiquitous, often mimic the shape and color of bacterial cells. Therefore, algorithms relying on color intensity thresholding and shape detection provide suboptimal accuracy.
Here, we provide a proof of concept for automated, deep-learning-based Gram stain analysis. The major conceptual and technical innovations were 2-fold. First, we developed an imaging protocol using an automated slide imaging platform equipped with a 40× air objective to collect highly resolved data from Gram-stained blood culture slides. Second, image data were used to train a convolutional neural network (CNN)-based model to recognize morphologies representing the most common agents causing BSI: Gram-negative rods, Gram-positive cocci in clusters, and Gram-positive cocci in pairs or chains (1). CNNs are modeled based on the organization of neurons within the mammalian visual cortex and were applied here based on their ability to excel in image recognition tasks without requiring time-intensive selective feature extraction by humans (10). Our trained model was subsequently evaluated for accuracy in comparison to manual classification.
MATERIALS AND METHODS
Slide collection and manual slide classification.
A total of 468 deidentified Gram-stained slides from positive blood cultures were collected from the clinical microbiology laboratory at Beth Israel Deaconess Medical Center between April and July 2017 under an institutional review board (IRB)-approved protocol. Slides were prepared during the course of normal clinical workup. No preselection of organism identity, organism abundance, or staining quality was performed prior to collection. Positive blood culture broth Gram stains included those prepared from both nonlytic BD Bactec standard aerobic medium (n = 232) and lytic BD Bactec lytic anaerobic medium (n = 196) (BD, Sparks, MD).
All slides were imaged without coverslips using a MetaFer Slide Scanning and Imaging platform (MetaSystems Group, Inc., Newton, MA) with a 140-slide-capacity automated slide loader equipped with a ×40 magnification Plan-Neofluar objective (Zeiss, Oberkochen, Germany) (0.75 numerical aperture). For each slide, 54 images were collected from defined positions spanning the entirety of the slide. The first 279 slides collected were used in training, validation, and evaluation of our deep-learning model. The remaining 189 slides were classified manually as Gram-negative rods, Gram-positive chains/pairs, or Gram-positive clusters using a Nikon Labophot 2 microscope (Nikon Inc., Tokyo, Japan) equipped with a 100× oil objective. Results were recorded for later use in evaluation of our whole-slide classification algorithm.
Training a deep convolutional neural network.
A training data set consisting of 146-by-146-pixel image crops was generated manually with the assistance of a custom Python script. The script allowed crop selection, classification, and file archiving with a single mouse click, allowing large numbers of annotated crops to be saved in a short period of time in a manner directly accessible to the deep-learning training program. Each crop was assigned to one of four classifications: Gram-positive cocci in pairs or chains, Gram-positive cocci in clusters, Gram-negative rods, or background (no cells). Prior to training, the data set was randomly divided into three subsets: 70% of image crops were used to train the model, 10% were reserved for hold-out validation during model training, and 20% were reserved for testing to evaluate model performance after completion of training. We used a transfer learning technique based on the Inception v3 convolutional neural network (CNN) architecture pretrained on the ImageNet Large Scale Visual Recognition Competition (ILSVRC) 2012 image database (11). We used the Python language (version 3.5) and the TensorFlow library (12) (version 1.0.1) to retrain the final layer of the model using a custom graphical user interface (GUI) controlling a modified script (“retrain.py”) found in the TensorFlow GitHub repository (12, 13). Training was performed using mini-batch gradient descent (batch size, 200) with Nesterov momentum (momentum = 0.9) (14) and cross entropy as the loss function (15). The initial learning rate was 0.001 and decayed exponentially at a rate of 0.99 per epoch. The output layer was a 4-way softmax classification which assigned probabilities to each of the four categories described above.
Analysis of model performance on a per-crop basis.
Using our trained CNN, we evaluated model performance on a per-image-crop basis using an evaluation set of 1,000 manually selected crops from each class (total crops = 4,000), all of which were independent of the training, validation, and testing data sets. For each category, true positives were defined as crops correctly classified as the category of interest; false positives were defined as crops that were incorrectly classified as the category of interest; true negatives were defined as crops correctly classified as a category other than the category of interest; and false negatives were defined as crops incorrectly classified as a category other than the category of interest. Sensitivity and specificity were modeled as receiver operating characteristic (ROC) curves for each classification label by adjusting the softmax classification thresholds required for positivity. Sensitivity was defined as true positive/(true positive + false negative). Specificity was defined as true negative/(true negative + false positive). Values for area under the ROC curve (AUC) were calculated for each label using the trapezium rule as implemented in the scipy library (16). ROC curves were visualized using the matplotlib library (17).
Development of whole-slide classification algorithm.
False-positive rates for automatically cropped images containing only background were determined by analysis of 350 whole images from 40 different slides. Images contained no visible cells and were independent of the training, validation, testing, and evaluation data sets. Each image was automatically segmented into 192 nonoverlapping crops of 146 by 146 pixels using a custom Python script (total crops = 67,200) and classified with our trained CNN using a stringent cutoff for positivity (cutoff = 0.99). If no label achieved a probability greater than or equal to the cutoff, the associated crop was called background. False-positive rates were recorded for each classification label.
Whole-slide classification.
Using the automated imaging protocol outlined in the “Automated image collection” section, we evaluated whole-slide classification accuracy using images collected from 189 slides which were previously manually classified (outlined in the “slide collection and manual slide classification” section). For each slide, a custom Python script was employed to automatically divide each image among the 54 images collected from predefined locations into 192 crops of 146 by 146 pixels. Each crop was evaluated by our trained deep-learning model, and probabilities were assigned to each category (Gram-negative rods, Gram-positive chains/pairs, Gram-positive clusters, or background) with a stringent cutoff for classification (cutoff = 0.99). If no label met the classification cutoff, the crop was classified as background.
After classification of all crops from a slide, the category corresponding to the greatest number of predicted crops was selected; however, the section was performed only if the number of crops in the selected category exceeded the number of expected false positives (calculated as described in the “Development of whole-slide classification algorithm” section). If none of the three label categories representing organisms were selected based on these criteria, the slide was classified as background. All results were recorded and used to construct a confusion matrix tabulation per convention in the deep-learning field (18). Whole-slide sensitivity and specificity were defined and calculated as described in the “Analysis of model performance on a per-crop basis” section. Classification accuracy levels for slides from aerobic or anaerobic bottles were compared using Fisher's exact test, with significance defined as a P value of <0.05 (JMP Pro version 13.0).
RESULTS
Slide collection and manual classification.
Blood culture Gram stain slides prepared manually during the course of normal laboratory operation were used for analysis. Slides were selected based on the presence of any of the three most common morphotypes observed in bloodstream infection: Gram-positive cocci in clusters, Gram-positive cocci in pairs and chains, and Gram-negative rods. Less-common morphotypes (e.g., Gram-positive rods or yeast) and polymicrobial infections were excluded. To capture real-world variability, slides were not prescreened for suitability for automated microscopy or deep learning and had characteristic slide-to-slide variability in staining intensity, staining artifacts, and sample distribution. We anticipated that inherent variability would pose a real-world challenge to slide classification models.
Automated image collection.
CNN-based deep-learning models require large data sets for training, typically at least on the order of thousands of images (and ideally at least an order of magnitude more). Therefore, an automated microscopy image acquisition strategy was used. We performed image acquisition on a MetaFer Slide Scanning and Imaging platform (MetaSystems Group, Inc., Newton, MA) based on a robust Gram stain-compatible autofocus system, ability to sample multiple distributed positions on a slide to account for variations in specimen distribution, and automated slide loading capability to enable high-throughput slide scanning.
Clinically, Gram stains are read under oil immersion. However, semicontinuous addition of oil during automated microscopy was undesirable. In preliminary experiments performed with slides that were not coverslipped (data not shown), we determined that the 40× dry objective provided sufficient resolution for machine learning applications based on our prior experience (19). Therefore, we selected use of the 40× air objective for image acquisition, thus avoiding the requirement for oil immersion and allowing us to capture a larger field of view in each image.
Deep convolutional neural network training.
For CNN training, a total of 25,488 images were automatically collected from distributed locations on 180 slides. A representative image is shown in Fig. 1. This image demonstrates features typical of blood culture Gram stain smears, including (A) intense background staining; (B) a stain crystallization artifact; (C) diffuse background staining; (D) individually resolvable, high-contrast Gram-negative cells; and (E) individually resolvable, low-contrast Gram-negative cells. Of note, ubiquitous background material was often similar in color, intensity, and/or shape to bacterial cells.
FIG 1.
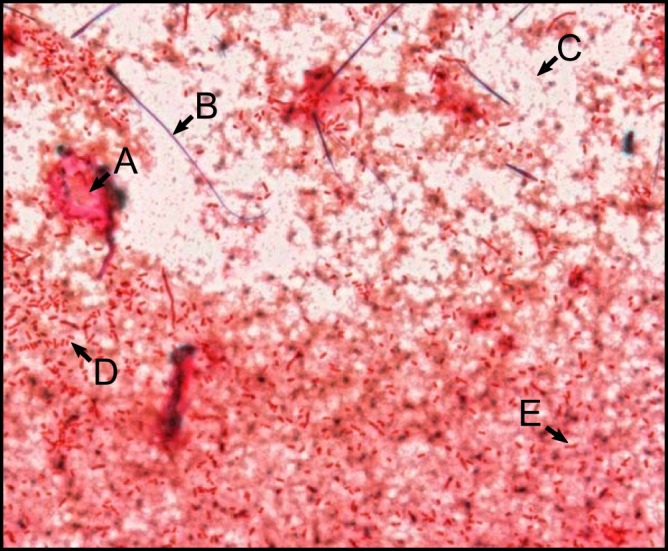
Representative image collected using our automated imaging protocol. This image shows several features characteristic of blood culture Gram stains, including (A) an area of intense background staining, (B) an artifact from stain crystallization, (C) diffuse background staining, and (D and E) individually resolved Gram-negative rods with (D) high contrast and (E) low contrast compared to background.
Highly experienced medical technologists can readily differentiate bacteria from this background. However, it is prohibitively difficult to manually define computational rules for Gram stain classification that would adequately distinguish signal from noise in highly variable Gram stain preparations. Therefore, we chose instead to use a deep-learning approach, more specifically, a CNN, for image analysis. CNNs do not interpret raw images directly. Rather, they consist of a number of layers, each of which convolutes regions of the image to detect specific features. During each step of the learning process, a subset of images is presented to the network, allowing function parameters to be changed such that the CNN identifies features important for classification based on optimization of output accuracy. The final model is defined by a set of weights and biases that control the flow of information through the network such that the most discriminatory features in the images are used for classification.
Each CNN model has a unique architecture that differs in organization, function, and number of convolutional layers (10). The model used in our analysis, Inception v3, has previously been shown to perform robustly in complex image classification tasks, including accurate classification of 1,000 different objects (11). The Inception v3 model is composed of a series of small convolutional networks termed “inception modules” and was designed to be less computationally intensive than comparable networks (20). Nevertheless, it is still a highly complex model requiring weeks to train even with state-of-the-art computational infrastructure (11). However, training the entire network is not always necessary. Many image classification tasks can be addressed using precomputed parameters from a network trained to classify an unrelated image set, a method called transfer learning (21). To this end, we used an Inception v3 model previously trained to recognize 1,000 different image classes from the 2012 ImageNet Large Scale Visual Recognition Competition data set (22) and retrained the final layer to identify our Gram stain categories of interest.
From an image analysis perspective, blood culture Gram stains are mostly background. This excessive background increases the likelihood that a CNN will learn features during training that are unrelated to bacterial Gram stain classification. This is termed “overfitting” and results in a model with high accuracy in classifying images on which it was trained (the training set) but with poor accuracy when presented with an independent validation set. Therefore, we enriched the training data through use of selected image crops rather than whole-slide images. A training crop selection tool was created using the Python programming language which allowed the trainer to select areas of an image containing bacteria with a single mouse click. This allowed us to train our model on regions of images containing bacteria without inclusion of excessive background.
For model training (Fig. 2), we used our training crop selection tool to generate a total of 100,213 manually classified image crops from 180 slides. Training accuracy and validation accuracy were indistinguishable (Fig. 2A), implying a robust ability of the model to evaluate data on which it had not previously been trained. It further confirmed success in minimizing overfitting. During training, predictions made by our model were compared to the observed data, and differences between these values were quantified using a metric called cross entropy (15). In practice, low cross entropy indicates that the model fits the observed data well. Cross entropy decreased during training and plateaued after 12,000 iterations (Fig. 2B). Additional training iterations beyond what is shown in Fig. 2 did not reduce cross entropy and therefore did not improve model accuracy.
FIG 2.

CNN model training results. (A) Training and validation accuracy increased exponentially, plateauing at ∼95%. There was no observable difference in the data with respect to training and validation accuracy, indicating negligible overfitting during training. (B) Cross entropy is a metric used for comparing model predictions to observed data. Lower cross entropy values indicate a better fit of the model to the data. During training, we observed that cross entropy decreased to a final value of ∼0.1. Cross entropy plateaued at approximately 12,000 training iterations, indicating that additional learning was not possible without increasing the number of input images, a goal of future work.
Evaluation of model performance on a per-crop basis.
Our CNN outputs the relative probabilities that an image crop belongs to each of four categories of training data, specifically, Gram-positive cocci in chains/pairs, Gram-positive cocci in clusters, Gram-negative rods, and background (i.e., no bacteria) (23). Per convention (10), the class with the highest probability is assigned as the predicted class. Using this method, we tested our model using a test set of image crops not used during model training and achieved a classification accuracy of 94.9%, providing an initial estimate of model performance. However, this metric may be impacted by the fact that the test set was not wholly independent of the training set, as it might still have contained crops from the same slide or images used in developing the training and validation sets.
Therefore, to rigorously evaluate the ability of our model to generalize to an entirely independent data set, we evaluated performance on an evaluation set of 4,000 manually classified image crops (n = 1,000 crops per class) from 59 slides that were not a component of the training, validation, or test sets. Here, we achieved a similar overall 93.1% image crop classification accuracy. Importantly, the evaluation set also allowed us to calculate sensitivity and specificity on a per-category basis. Sensitivity and specificity were 96.6% and 99.4% for Gram-positive clusters, 97.7% and 99.0% for Gram-positive chains, 80.1% and 99.4% for Gram-negative rods, and 97.4% and 93.0% for background, respectively. Calculation of the area under the receiver operating characteristic (ROC) curve (AUC) for each category (Fig. 3) further indicated a robust ability to differentiate between categories (AUC > 0.98 for all).
FIG 3.
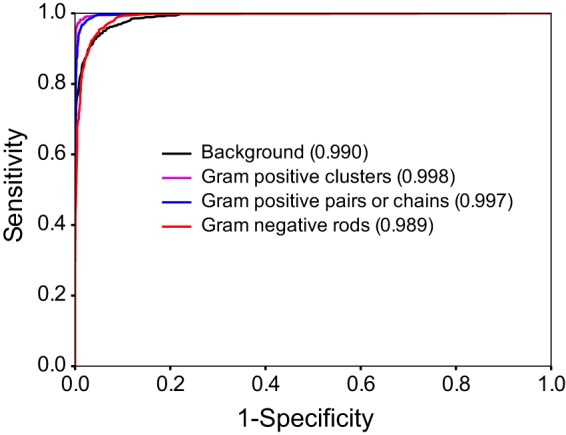
Receiver operating characteristic (ROC) curve. Curves were generated for each category by varying the threshold for positivity. Values corresponding to the area under the curve are indicated in parentheses.
Development of whole-slide classification algorithm.
To this point, we performed classifications on manually selected cropped images based on category assignment using the highest probability output from the classification. However, we hypothesized that it was not the optimal way to interpret our results for whole-slide classification. Specifically, a whole-slide classification task differs from our evaluation experiments in that it necessarily examines a much larger number of crops that are not preselected and consist only of background. Given that background may simulate bacterial cells (Fig. 1), we expected a greater likelihood of false-positive calls.
To test this possibility during whole-slide classification, we decided to set a very stringent probability cutoff (0.99) for category calls to minimize false positives at the image crop level and maximize specificity at the whole-slide level. Using this stringent cutoff, 65.6% of evaluated crops had a prediction with confidence of ≥0.99, and 99.6% of these were correctly classified. The classification accuracy levels were 99.9% for Gram-positive clusters, 100% for Gram-positive chains, and 97.4% for Gram-negative rods.
To investigate how this stringent cutoff would impact false-positive rates on a per-slide basis when applied to images cropped automatically, we collected 350 whole images containing no visible cells and that were not part of the training, validation, or evaluation data sets. Images were cropped into 192 nonoverlapping crops (n = 67,200) using a custom Python script and evaluated using our trained model with the classification threshold described above. For each category, false-positive rates were ≤0.006% on a per-image crop basis. On the basis of an assumed normal distribution of false-positive calls, we set a minimal threshold for slide classification of 6 positive crops per category in order to achieve the desired ≤0.1% false-positive whole-slide classification rate.
Our whole-slide classification algorithm was then tested on 189 slides that had been previously classified manually by a microbiologist and that were not components of the training, validation, test, or evaluation sets. Each of 54 images scanned per slide was divided into 192 nonoverlapping 146-by-146 pixel crops and evaluated using the parameters described above for a total of 10,368 crops per slide. We first qualitatively evaluated performance on automated image crops. This was achieved by writing a Python program (called “TA” for technologist assist) that would output images corresponding to crop calls by the CNN, allowing specific review. Figure 4 shows examples of correctly classified image crops corresponding to each of the four classification labels.
FIG 4.

Automatically classified crops. Each image represents a correctly classified crop that was automatically extracted from an image during whole-slide classification. Rows of images represent (A) background, (B) Gram-positive chains/pairs, (C) Gram-positive clusters, or (D) Gram-negative rods. One practical application of the platform would be to present such organism-enriched images to a technologist to expedite smear review.
We then quantitatively evaluated our whole-slide classification accuracy in comparison to manual classification by constructing a table that shows each slide's manual classification and corresponding automated prediction (Table 1). We found that bacteria were detected in 84.7% (n = 160) of slides by our automated algorithm. For those slides where bacteria were detected, we calculated classification accuracy, sensitivity, and specificity. Classification accuracy was 92.5% across all categories. Sensitivity was >97% for Gram-negative rods and Gram-positive clusters. Sensitivity was lower for Gram-positive chains, largely owing to misclassifications as Gram-positive clusters across a relatively lower overall number of slides (n = 40). Further, manual inspection of Gram-positive chains misclassified as clusters revealed that the data represented by these slides were somewhat ambiguous owing to substantial clumping of cells. Specificity for Gram-positive chains and Gram-negative rods was >96%. Specificity was slightly lower (93.2%) for Gram-positive clusters, again owing to misclassification of Gram-positive chains as clusters. Despite qualitative differences in background staining, the levels of accuracy of data based on slides from aerobic bottles (88.8%) or anaerobic bottles (92.9%) were not significantly different (Fisher's exact test, P > 0.05).
TABLE 1.
Confusion matrix of whole-slide classification results
| Human classification | Predicted classification (n) |
% sensitivity (CI)a | % specificity (CI)a | |||
|---|---|---|---|---|---|---|
| Gram negative | Gram-positive pairs or chains | Gram-positive clusters | Background | |||
| Gram negative | 51 | 1 | 0 | 17 | 98.1 (94.3–100) | 96.3 (93.7–98.9) |
| Gram-positive pairs or chains | 3 | 27 | 6 | 4 | 75.0 (60.9–89.0) | 98.4 (90.8–100) |
| Gram-positive clusters | 1 | 1 | 70 | 8 | 97.2 (93.4–100) | 93.2 (89.7–96.6) |
Data were determined based on slides where bacteria were detected. CI, 95% confidence interval.
Overall, the most common error was misclassification of slides as background, representing 70.7% (n = 29) of all misclassifications. On manual review of images from these slides, we found that 44.8% (n = 13) had insufficient crops with bacteria to make a positive call based on our preestablished thresholds. We found that an additional 48.3% (n = 14) had organisms that either were out of focus or had very low contrast, and of these, the majority (78.6%, n = 11) contained Gram-negative organisms, as expected based on superficial similarity to background material. The remaining 6.9% (n = 2) of slides contained highly elongated Gram-negative rods or minute Gram-negative coccobacilli. Neither of those morphologies was a component of our training set. Gram stain category miscalls (n = 5), other than conflation of Gram-positive cocci in chains and Gram-positive cocci in clusters, were related to a combination of poor representation of the causal organism in crops and excessive background artifact.
DISCUSSION
The Gram stain smear provides the first microbiological data to guide treatment for BSI. Notably, earlier results are correlated with positive patient outcome (6). However, interpretation of Gram stains is time intensive and strongly operator dependent, requiring a skilled technologist for interpretation. Concerningly, the most recent survey from the American Society for Clinical Pathology indicates that, as of 2014, trained microbiology technologist jobs in the United States had a vacancy rate of ∼9% and nearly 20% of technologists planned to retire in the next 5 years (8). This finding highlights the need for development of solutions to make the current work force more efficient. However, there has been relatively little progress in automation of tests requiring subjective interpretation such as the Gram stain.
Lack of progress in this area is related to technical issues with automated microscopy and need for imaging interpretation algorithms that are robust with respect to identifying rare organisms in the presence of variable background. Here, we demonstrated that the MetaFer Slide Scanning and Imaging platform provides a robust automated image acquisition system, capable of providing sufficient resolution for Gram stain analysis using a 40× dry objective. For such analysis, we chose to use a CNN based on its ability to excel in image analysis tasks with minimal human intervention. A summary of workflow for implementation, testing, and validation of our platform is provided in Fig. 5.
FIG 5.
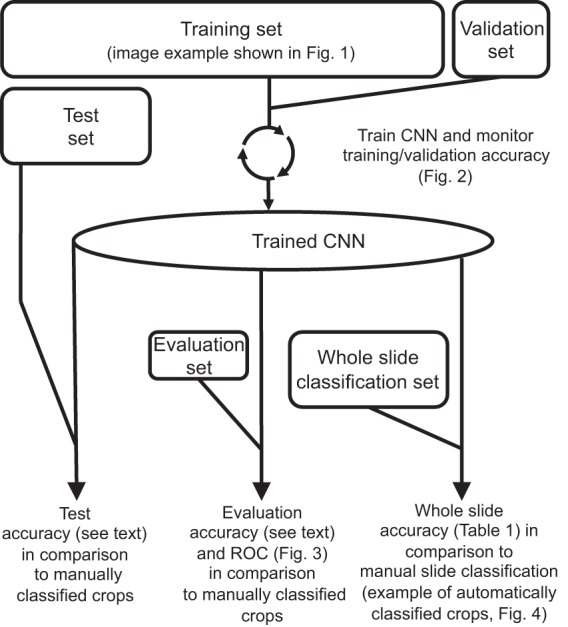
Summary of CNN training and evaluation. Prior to CNN training, we collected images using an automated microscopy protocol (image example shown in Fig. 1). For CNN training and preliminary testing, 100,213 image crops were manually selected, classified, and randomly partitioned into training, validation, and test sets. The sizes of the boxes correlate to the relative sizes of the data sets. During iterative model training, accuracy was monitored using the training and validation sets (Fig. 2). After completion of training, model accuracy was initially assessed by quantification of accuracy on the test set (as discussed in the text). However, the test set image crops came from the same slides as the training set. We therefore further assessed performance using a completely independent evaluation set to obtain a more reliable, real-world readout of image crop classification accuracy and to generate the data corresponding to the receiver operating characteristics (ROC) shown in Fig. 3. Finally, we used a second independent data set of automatically generated image crops from 189 slides to evaluate whole-slide classification accuracy. Each whole-slide classification was based on aggregate CNN categorizations of all image crops from a given slide (examples of such crops are shown in Fig. 4). Accuracy was determined in comparison to manual slide interpretation (Table 1).
This work adds to the examples of successful CNN use in several areas of image-based diagnostics. These include detection of skin cancer (24); interpretation of echocardiograms (25); and detection of metastatic cancer in lymph nodes (26) in which the combined contributions of pathologists and a CNN increased sensitivity for diagnosis (27). A CNN has also previously been used by our group for early prediction of antibiotic MICs in microscopy-based microdilution assays (19).
Importantly, CNNs improve in performance as more image data are added to the training set. Unlike other machine learning models, however, training on more data increases neither the size of a CNN model nor the complexity of model implementation. Nevertheless, training of an entire CNN model requires substantial computational infrastructure. Here, we took advantage of an existing trained CNN and retrained its final layers, a method called transfer learning (21, 24). In this way, we were able to train and implement our model using a standard office computer containing an Intel Core i7 CPU with 32 GB of RAM with no GPU (graphics processing unit [the computational workhorse for image analysis]).
Not surprisingly, implementation of the trained CNN for whole-slide analysis using this computer infrastructure was relatively slow. We therefore piloted whole-slide classification using a system containing an Nvidia GTX 1070 GPU. Though still underpowered compared to other currently available GPUs, it improved the whole-slide classification time by a factor of 6, resulting in a classification time of ∼9 min. The best available GPUs are markedly more powerful than the GTX 1070 and are expected to provide even better performance (<5 min per slide), not even considering the ability of CNN algorithms to distribute computations across multiple GPUs.
Overall, we found that our trained model performed well on whole-slide image classification. Where cells were detected, we achieved an overall classification accuracy of 92.5% and a specificity of >93% for all classification labels with no human intervention. The most common classification error from our model was misclassification of slides containing rare bacteria as background, representing the majority (70.7%) of all classification errors. In practice, these misclassifications would be flagged for direct technologist review, making these low-consequence errors. We also note that our sensitivity and specificity in whole-slide image classification accuracy were modestly lower than those seen on a per-image-crop basis. This is likely due in part to inclusion of slides with very few bacteria and therefore a higher propensity for false positives. Optimization of data collection or slide preparation would likely bring our whole-slide accuracy close to the per-image-crop accuracy.
Our study had several limitations. As a proof-of-principle examination, we included only the most common BSI pathogens and omitted several important but less-common bacterial morphologies, largely due to limitations in the availability of training data. However, given an appropriate amount of training data, these could easily be incorporated into the Inception v3 model, which can distinguish 1,000 different categories; this is a future goal. Similarly, discrimination of polymicrobial infections could be incorporated by inclusion of “mixed” categories in our algorithm.
We also recognize that there are several steps that could be taken to improve classification. Foremost, the number of slides (and therefore the number of image crops) used for training is relatively modest and could be increased to improve CNN accuracy. In addition, our whole-slide scanning protocol was based on selecting predefined positions for imaging that were invariant between slides. This contributed to inadequate sampling in a significant subset of slides, which we believe was the greatest contributor to reduction in model accuracy. This hypothesis is supported by the observation that the misclassified whole-slide calls were typically from slides with very few bacteria or poor sample spread. Notably, to address this issue, it is possible with the existing microscope platform to perform an automated rapid scan for areas of appropriate staining intensity and thereby preselect regions of the slide that are more likely to have sufficient Gram-stained sample for image acquisition.
Gram stain smear preparation is also expected to have a significant impact on automated slide imaging. Here, we used slides prepared by technologists during the course of normal laboratory operation. Slides exhibited a high degree of variability in smear area, thickness, location, and staining intensity. We anticipate that standardization of these variables will improve the ability of an automated microscope to consistently sample microscopic fields with evaluable organisms. Further, use of an automated Gram stain device for staining would also increase the reproducibility of staining characteristics and further enhance accuracy. We plan to investigate all of these areas in the future.
We envision a potential role of our technology in augmenting technologist classification. Given that manual interpretation of blood culture Gram stains by trained technologists is very accurate (28–30), our model could be used to enhance productivity by selectively presenting crops containing bacteria to local or remote technologists. This would increase the efficiency of classification by sparing the operator the need to manually locate fields of interest among a preponderance of background. This would also conceivably reduce technologist read time from minutes to seconds. Upon further development and intensive algorithm training, the platform could potentially also be used as a fully automated classification platform with no human intervention.
In the era of laboratory consolidation and limitations in the number of skilled technologists (8), we believe our system could provide enhanced opportunities for rapid Gram stain classification at the site of care or during understaffed shifts in conjunction with later analysis at a central laboratory or day shifts. We further envision extension of CNN analysis to other smear-based microbiological diagnostics in the parasitology, mycobacteriology, and mycology laboratories. We believe that this technology could form the basis of a future diagnostic platform that provides automated smear classification results and augments capabilities of clinical laboratories.
ACKNOWLEDGMENTS
We thank Andreas Plesch, Ulrich Klingbeil, and Bill Hanifin (MetaSystems Group, Inc., Newton, MA) for providing use of the MetaFer Slide Scanning and Imaging platform and Jenae Guinn (MetaSystems) for assistance in collection of image data. MetaSystems had no role in any other aspect of study design, data analysis, or decision to publish. We thank Ramy Arnaout (Beth Israel Deaconess Medical Center, Boston, MA) for critical reading of the manuscript.
This work was conducted with support from Harvard Catalyst ∣ The Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health Award UL1 TR001102) and financial contributions from Harvard University and its affiliated academic health care centers. A.D.K. was supported by the Long Term Health Education and Training Program of the United States Army as an American Society for Microbiology Committee on Postgraduate Educational Programs Fellow at Beth Israel Deaconess Medical Center. K.P.S. was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number F32 AI124590. The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health, United States Army, or Department of Defense.
Footnotes
For a commentary on this article, see https://doi.org/10.1128/JCM.01779-17.
REFERENCES
- 1.Laupland KB. 2013. Incidence of bloodstream infection: a review of population-based studies. Clin Microbiol Infect 19:492–500. doi: 10.1111/1469-0691.12144. [DOI] [PubMed] [Google Scholar]
- 2.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. 2004. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 3.Schwaber MJ, Carmeli Y. 2007. Mortality and delay in effective therapy associated with extended-spectrum beta-lactamase production in Enterobacteriaceae bacteraemia: a systematic review and meta-analysis. J Antimicrob Chemother 60:913–920. doi: 10.1093/jac/dkm318. [DOI] [PubMed] [Google Scholar]
- 4.Kang CI, Kim SH, Kim HB, Park SW, Choe YJ, Oh MD, Kim EC, Choe KW. 2003. Pseudomonas aeruginosa bacteremia: risk factors for mortality and influence of delayed receipt of effective antimicrobial therapy on clinical outcome. Clin Infect Dis 37:745–751. doi: 10.1086/377200. [DOI] [PubMed] [Google Scholar]
- 5.Wain J, Diep TS, Ho VA, Walsh AM, Nguyen TT, Parry CM, White NJ. 1998. Quantitation of bacteria in blood of typhoid fever patients and relationship between counts and clinical features, transmissibility, and antibiotic resistance. J Clin Microbiol 36:1683–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barenfanger J, Graham DR, Kolluri L, Sangwan G, Lawhorn J, Drake CA, Verhulst SJ, Peterson R, Moja LB, Ertmoed MM, Moja AB, Shevlin DW, Vautrain R, Callahan CD. 2008. Decreased mortality associated with prompt Gram staining of blood cultures. Am J Clin Pathol 130:870–876. doi: 10.1309/AJCPVMDQU2ZJDPBL. [DOI] [PubMed] [Google Scholar]
- 7.Bourbeau PP, Ledeboer NA. 2013. Automation in clinical microbiology. J Clin Microbiol 51:1658–1665. doi: 10.1128/JCM.00301-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia E, Ali AM, Soles RM, Lewis DG. 2015. The American Society for Clinical Pathology's 2014 vacancy survey of medical laboratories in the United States. Am J Clin Pathol 144:432–443. doi: 10.1309/AJCPN7G0MXMSTXCD. [DOI] [PubMed] [Google Scholar]
- 9.Meyer J, Pare G. 2015. Telepathology impacts and implementation challenges: a scoping review. Arch Pathol Lab Med 139:1550–1557. doi: 10.5858/arpa.2014-0606-RA. [DOI] [PubMed] [Google Scholar]
- 10.LeCun Y, Bengio Y, Hinton G. 2015. Deep learning. Nature 521:436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 11.Szegedy C, Vanhoucke V, Ioffe S, Shlens J, Wojna Z. 2015. Rethinking the inception architecture for computer vision. arXiv https://arxiv.org/abs/1512.00567 Accessed 12 September 2017.
- 12.Anonymous. 2017. TensorFlow: an open-source software library for machine intelligence. https://www.tensorflow.org/ Accessed 12 September 2017.
- 13.Anonymous. 2017. TensorFlow GitHub repository. https://github.com/tensorflow/tensorflow Accessed 12 September 2017.
- 14.Nesterov Y. 1983. A method of solving a convex programming problem with convergence rate O(1/sqr (k)). Soviet Mathematics Doklady 27:372–376. [Google Scholar]
- 15.de Boer P-T, Kroese D, Reuven S, Rubinstein RY. 2005. A tutorial on the cross-entropy method. Ann Oper Res 134:19–67. doi: 10.1007/s10479-005-5724-z. [DOI] [Google Scholar]
- 16.Jones E, Oliphant E, Peterson P. 2001. SciPy: open source scientific tools for Python. http://www.scipy.org/ Accessed 12 September 2017.
- 17.Hunter JD. 2007. Matplotlib: a 2D graphics environment. Comput Sci Eng 9:90–95. doi: 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 18.Stehman SV. 1997. Selecting and interpreting measures of thematic classification accuracy. Remote Sens Environ 62:77–89. doi: 10.1016/S0034-4257(97)00083-7. [DOI] [Google Scholar]
- 19.Smith KP, Richmond DL, Brennan-Krohn T, Elliott HL, Kirby JE. 2017. Development of MAST: a microscopy-based antimicrobial susceptibility testing platform. SLAS Technol 22:662–674. doi: 10.1177/2472630317727721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szegedy C, Wei L, Yangqing J, Sermanet P, Reed S, Anguelov D, Erhan D, Vanhoucke V, Rabinovich A. Going deeper with convolutions. arXiv https://arxiv.org/abs/1409.4842.
- 21.Shin H-C, Roth HR, Gao M, Lu L, Xu Z, Nogues I, Yao J, Mollura D, Summers RM. 2016. Deep convolutional neural networks for computer-aided detection: CNN architectures, dataset characteristics and transfer learning. IEEE Trans Med Imaging 35:1285–1298. doi: 10.1109/TMI.2016.2528162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russakovsky O, Deng J, Su H, Krause J, Satheesh S, Ma S, Huang Z, Karpathy A, Khosla A, Bernstein M, Berg AC, Fei-Fei L. 2015. ImageNet large scale visual recognition challenge. Int J Comput Vis 115:211–252. doi: 10.1007/s11263-015-0816-y. [DOI] [Google Scholar]
- 23.Bridle JS. 1989. Probabilistic interpretation of feedforward classification network outputs, with relationships to statistical pattern recognition, p 227–236. In Soulíe FF, H́erault J (ed), Neurocomputing. NATO ASI Series (series F: computer and systems sciences), vol 68 Springer, Berlin, Germany. doi: 10.1007/978-3-642-76153-9_28. [DOI] [Google Scholar]
- 24.Esteva A, Kuprel B, Novoa RA, Ko J, Swetter SM, Blau HM, Thrun S. 2017. Dermatologist-level classification of skin cancer with deep neural networks. Nature 542:115–118. doi: 10.1038/nature21056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madani A, Arnaout R, Mofrad M, Arnaout R. 2017. Fast and accurate classification of echocardiograms using deep learning. arXiv https://arxiv.org/ftp/arxiv/papers/1706/1706.08658.pdf Accessed 22 September 2017. [DOI] [PMC free article] [PubMed]
- 26.Litjens G, Sanchez CI, Timofeeva N, Hermsen M, Nagtegaal I, Kovacs I, Hulsbergen-van de Kaa C, Bult P, van Ginneken B, van der Laak J. 2016. Deep learning as a tool for increased accuracy and efficiency of histopathological diagnosis. Sci Rep 6:26286. doi: 10.1038/srep26286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang D, Khosla A, Gargeya R, Irshad H, Beck AH. 2016. Deep learning for identifying metastatic breast cancer. arXiv https://arxiv.org/abs/1606.05718 Accessed 12 September 2017.
- 28.Samuel LP, Balada-Llasat JM, Harrington A, Cavagnolo R. 2016. Multicenter assessment of gram stain error rates. J Clin Microbiol 54:1442–1447. doi: 10.1128/JCM.03066-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Søgaard M, Nørgaard M, Schønheyder HC. 2007. First notification of positive blood cultures and the high accuracy of the Gram stain report. J Clin Microbiol 45:1113–1117. doi: 10.1128/JCM.02523-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rand KH, Tillan M. 2006. Errors in interpretation of Gram stains from positive blood cultures. Am J Clin Pathol 126:686–690. doi: 10.1309/V4KE2FPM5T8V4552. [DOI] [PubMed] [Google Scholar]